
First-Principles Study of n*AlN/n*ScN Superlattices with High Dielectric Capacity for Energy Storage



Abstract
:1. Introduction
2. Theoretical Methodology
3. Results and Discussion
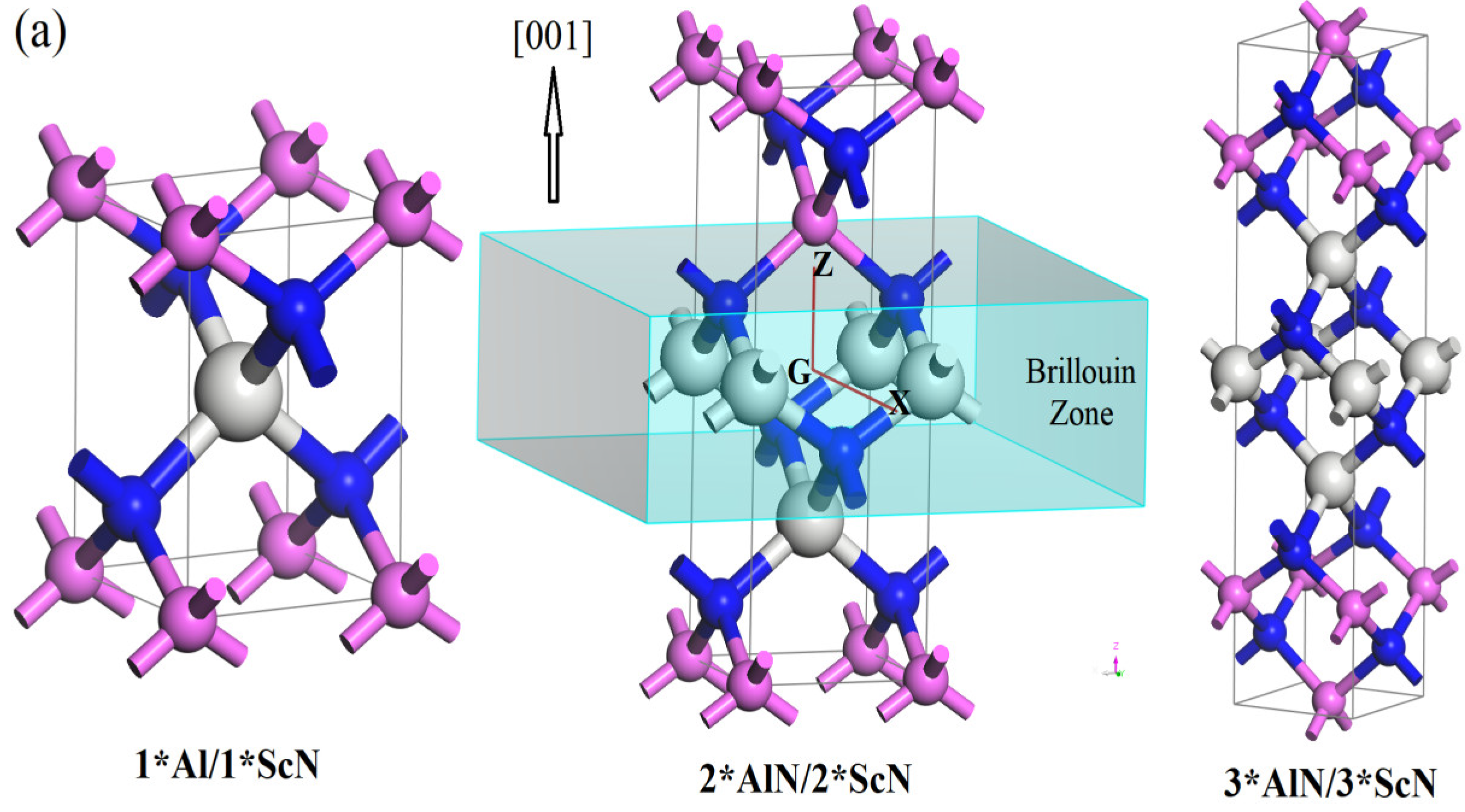
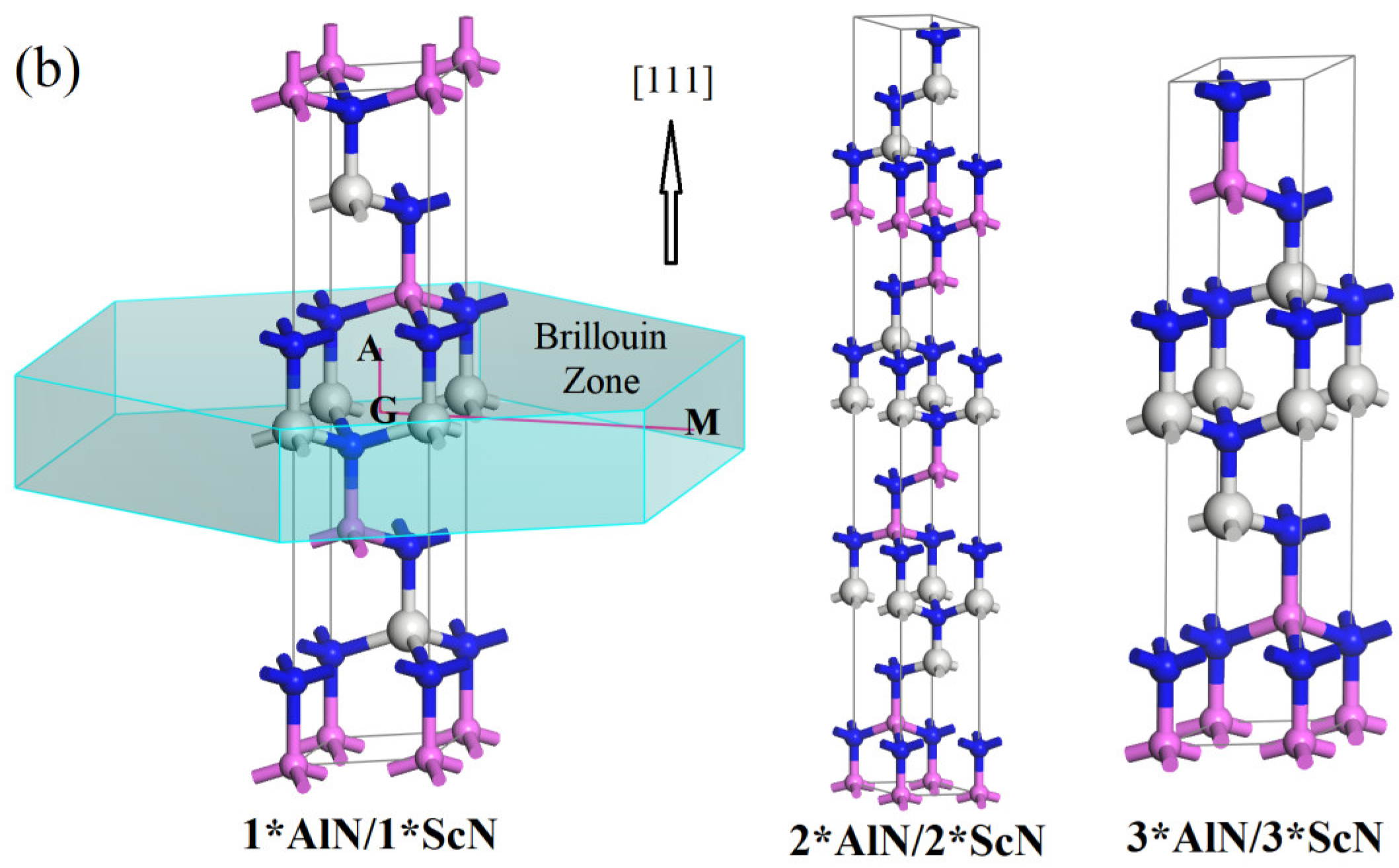
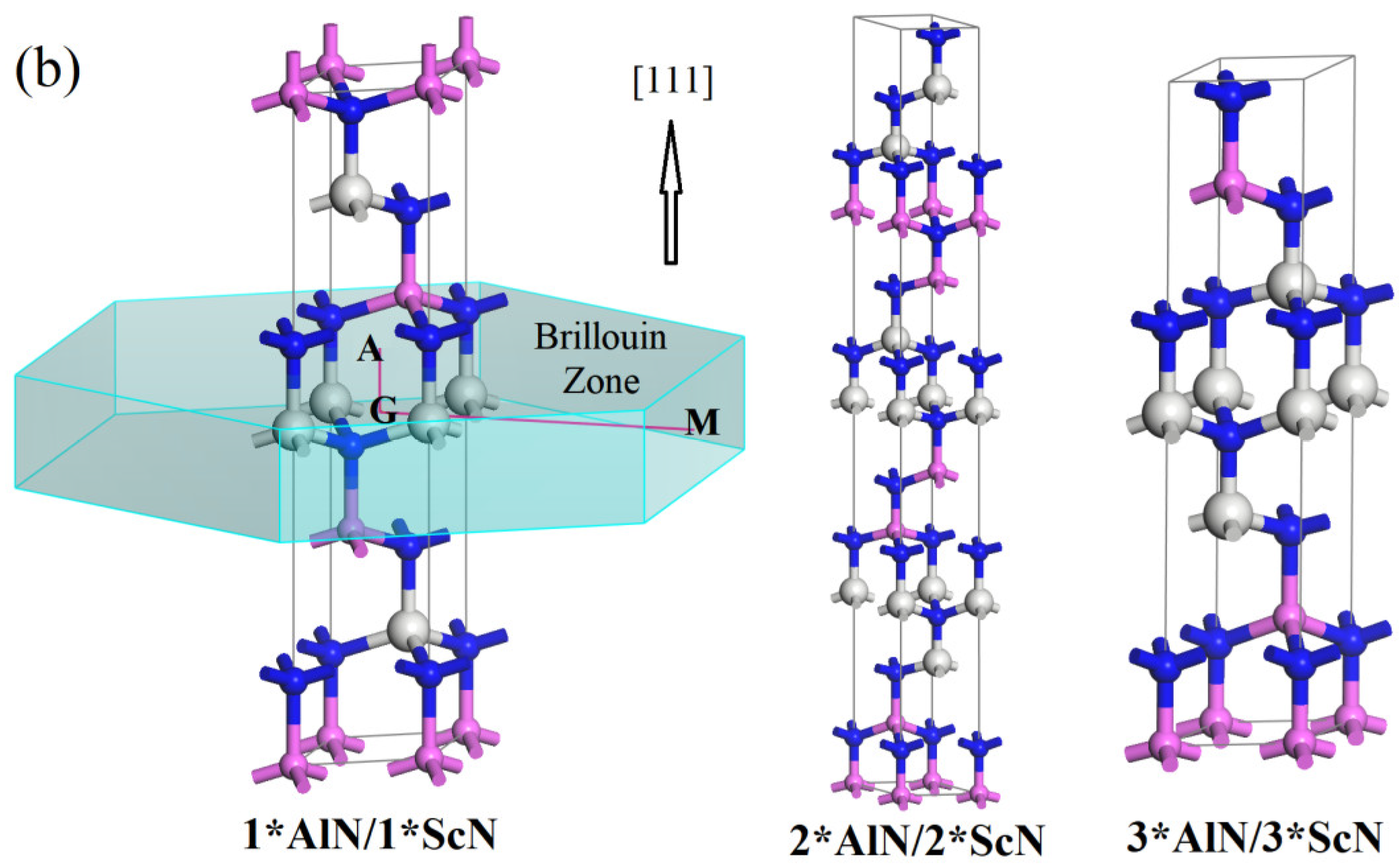
3.1. Crystal Structure
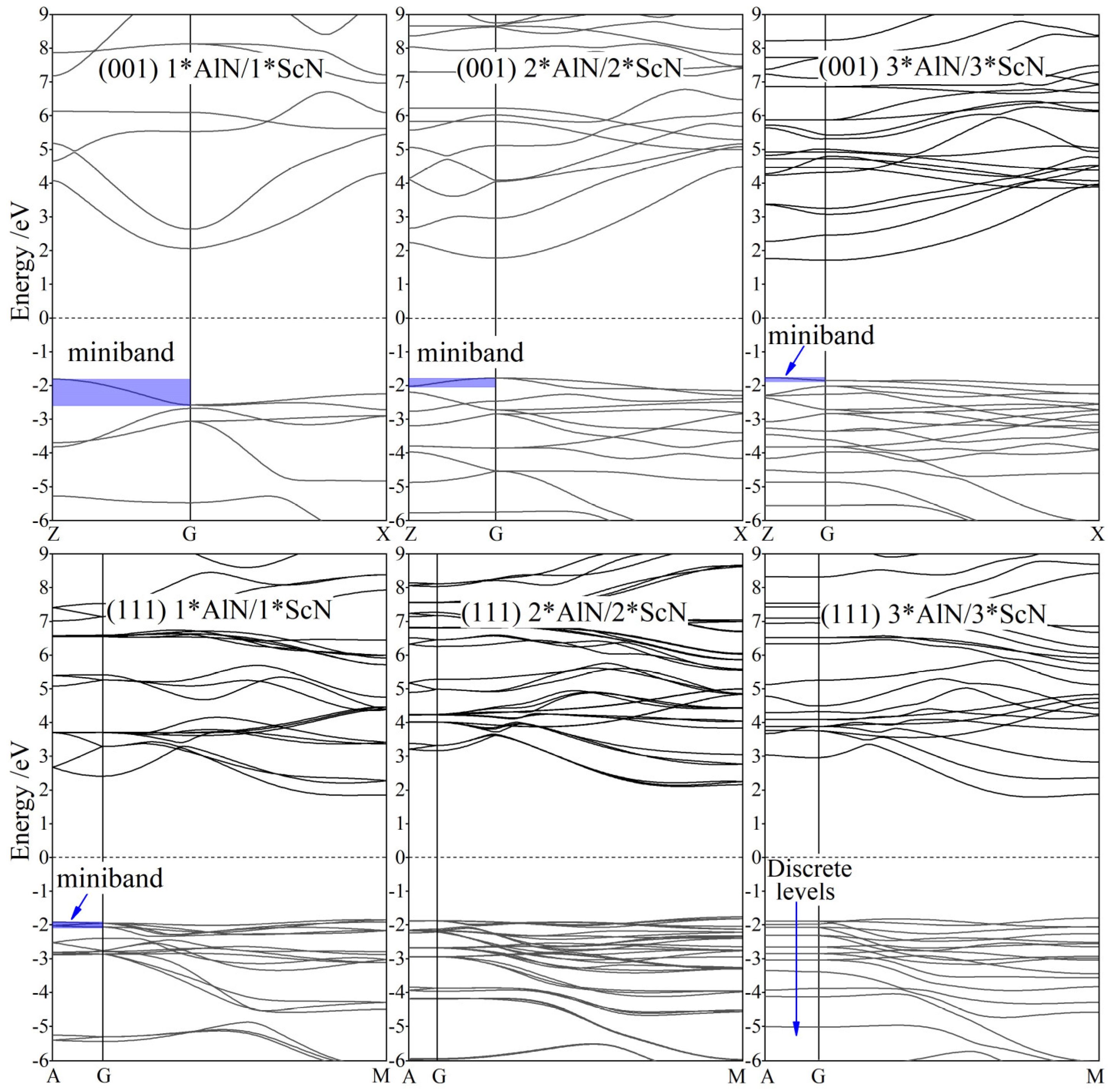
3.2. Band Structure
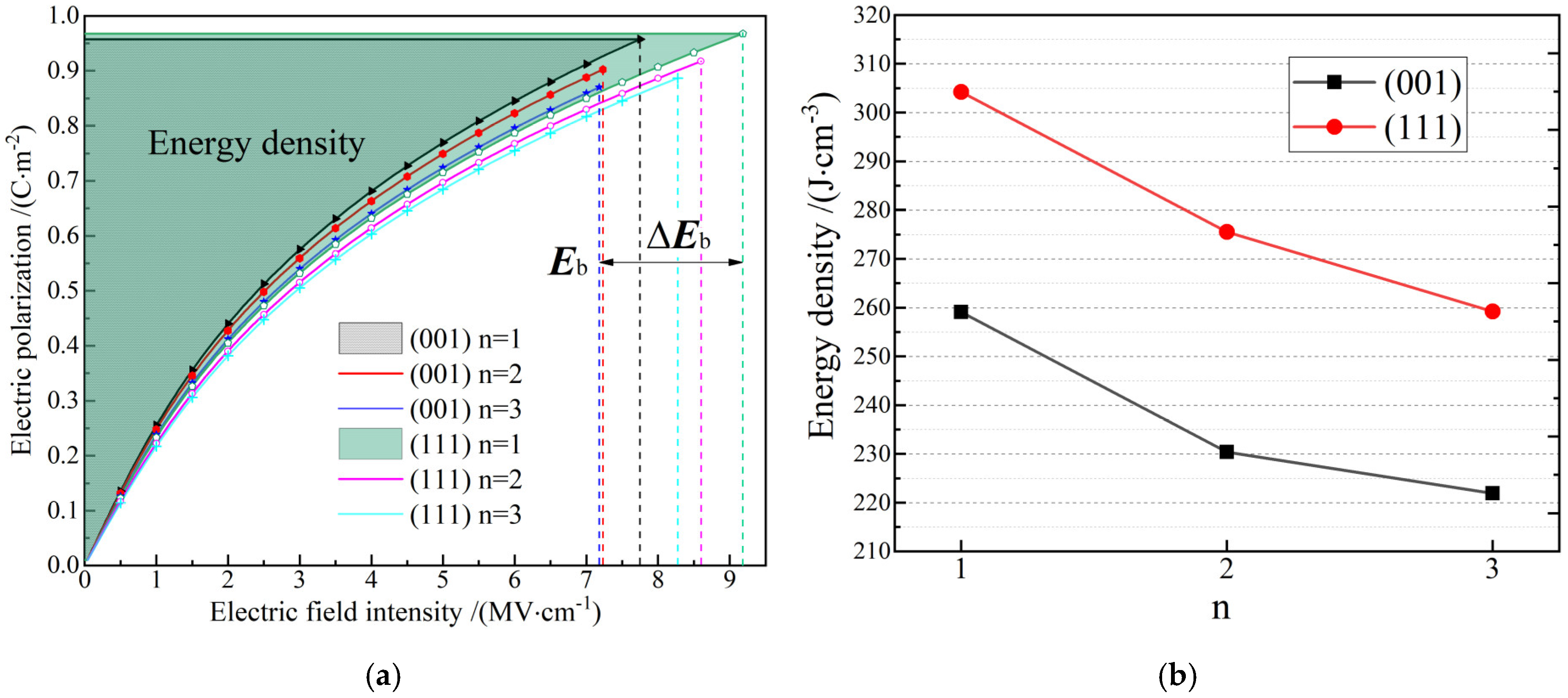
3.3. Dielectric Polarization and Energy Density
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gross, R.; Leach, M.; Bauen, A. Progress in renewable energy. Environ. Int. 2003, 29, 105–122. [Google Scholar] [CrossRef]
- Hao, H. A review on the dielectric materials for high energystorage application. J. Adv. Dielectr. 2013, 3, 1330001. [Google Scholar] [CrossRef]
- Chu, B.; Zhou, X.; Ren, K.; Neese, B.; Lin, M.; Wang, Q.; Bauer, F.; Zhang, Q.M. A Dielectric polymer with high electric energy density and fast discharge speed. Science 2006, 313, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Chauhan, A.; Vaish, R. Enhancing electrical energy storage density in anti-ferroelectric ceramics using ferroelastic domain switching. Mater. Res. Express 2014, 1, 045502. [Google Scholar] [CrossRef]
- Chauhan, A.; Patel, S.; Vaish, R.; Bowen, C.R. Anti-ferroelectric ceramics for high energy density capacitors. Materials 2015, 8, 8009–8031. [Google Scholar] [CrossRef] [Green Version]
- Prateek; Thakur, V.K.; Gupta, R.K. Recent progress on ferroelectric polymer-based nanocomposites for high energy density capacitors: Synthesis, dielectric properties, and future aspects. Chem. Rev. 2016, 116, 4260. [Google Scholar] [CrossRef]
- Peng, B.; Zhang, Q.; Li, X.; Sun, T.; Fan, H.; Ke, S.; Ye, M.; Wang, Y.; Lu, W.; Niu, H.; et al. Giant electric energy density in epitaxial lead-free thin films with coexistence of ferroelectrics and antiferroelectrics. Adv. Electron. Mater. 2015, 1, 1500052. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Z.; Lin, J.L.; Bai, Y.; Li, Y.X.; Zhang, Z.D.; Wang, Z.J. Ultrahigh-energy storage properties of (PbCa)ZrO3 antiferroelectric thin films via constructing a pyrochlore nanocrystalline structure. ACS Nano 2020, 14, 6857. [Google Scholar] [CrossRef]
- Hao, X.; Wang, Y.; Zhang, L.; Zhang, L.; An, S. Composition-dependent dielectric and energy-storage properties of (Pb,La)(Zr,Sn,Ti)O3 antiferroelectric thick films. Appl. Phys. Lett. 2013, 102, 163903. [Google Scholar] [CrossRef]
- Li, Q.; Chen, L.; Gadinski, M.R.; Zhang, S.; Zhang, G.; Li, H.U.; Iagodkine, E.; Haque, A.; Chen, L.Q.; Jackson, T.N.; et al. Flexible high-temperature dielectric materials from polymer nanocomposites. Nature 2015, 523, 576. [Google Scholar] [CrossRef]
- Emery, S.B.; Cheng, C.J.; Kan, D.; Rueckert, F.J.; Alpay, S.P.; Nagarajan, V.; Takeuchi, I.; Wells, B.O. Phase coexistence near a morphotropic phase boundary in Sm-doped BiFeO3 films. Appl. Phys. Lett. 2010, 97, 152902. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Wang, D.; Iniguez, J.; Bellaiche, L. Finite-temperature properties of rare-earth-substituted BiFeO3 multiferroic solid solutions. Adv. Funct. Mater. 2015, 25, 552–558. [Google Scholar] [CrossRef]
- Peddigari, M.; Palneedi, H.; Hwang, G.T.; Ryu, J. Linear and nonlinear dielectric ceramics for high-power energy storage capacitor applications. J. Korean Ceram. Soc. 2019, 56, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.Y.; Liu, X.Q.; Zhu, X.L.; Chen, X.M. CaTiO3 linear dielectric ceramics with greatly enhanced dielectric strength and energy storage density. J. Am. Ceram. Soc. 2018, 101, 1999. [Google Scholar] [CrossRef]
- Tasnádi, F.; Alling, B.; Häglund, C.; Wingqvist, G.; Birch, J.; Hultman, L.; Abrikosov, I.A. Origin of the Anomalous Piezoelectric Response in Wurtzite ScxAl1−xN Alloys. Phys. Rev. Lett. 2010, 104, 137601. [Google Scholar] [CrossRef] [Green Version]
- Yazawa, K.; Drury, D.; Zakutayev, A.; Brennecka, G.L. Reduced coercive field in epitaxial thin film of ferroelectric wurtzite Al0.7Sc0.3N. Appl. Phys. Lett. 2021, 118, 162903. [Google Scholar] [CrossRef]
- Jiang, Z.; Paillard, C.; Vanderbilt, D.; Xiang, H.; Bellaiche, L. Designing Multifunctionality via Assembling Dissimilar Materials: Epitaxial AlN/ScN Superlattices. Phys. Rev. Lett. 2019, 123, 096801. [Google Scholar] [CrossRef]
- Xu, B.; Wang, D.; Zhao, H.J.; Íñiguez, J.; Chen, X.M.; Bellaiche, L. Hybrid Improper ferroelectricity in multiferroic superlattices: Finite-temperature properties and electric-field-driven switching of polarization and magnetization. Adv. Funct. Mater. 2015, 25, 3626. [Google Scholar] [CrossRef]
- Fichtner, S.; Wolff, N.; Lofink, F.; Kienle, L.; Wagner, B. AlScN: A III-V semiconductor based ferroelectric. J. Appl. Phys. 2019, 125, 114103. [Google Scholar] [CrossRef]
- Akiyama, M.; Kamohara, T.; Kano, K.; Teshigahara, A.; Takeuchi, Y.; Kawahara, N. Enhancement of piezoelectric response in scandium aluminum nitride alloy thin films prepared by dual reactive cosputtering. Adv. Mater. 2009, 21, 593. [Google Scholar] [CrossRef]
- Yasuoka, S.; Shimizu, T.; Tateyama, A.; Uehara, M.; Yamada, H.; Akiyama, M.; Hiranaga, Y.; Cho, Y.; Funakubo, H. Effects of deposition conditions on the ferroelectric properties of (Al1−xScx)N thin films. J. Appl. Phys. 2020, 128, 114103. [Google Scholar] [CrossRef]
- Wu, Z.; Cohen, R.E. More accurate generalized gradient approximation for solids. Phys. Rev. B 2006, 73, 235116. [Google Scholar] [CrossRef] [Green Version]
- Lejaeghere, K.; Van Speybroeck, V.; van Oost, G.; Cottenier, S. Error estimates for solid-state density-functional theory predictions: An overview by means of the ground-state elemental crystals. Crit. Rev. Solid State Mater. Sci. 2014, 39, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative minimization techniques for ab initio total energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations-A reply. Phys. Rev. B 1977, 16, 1748. [Google Scholar]
- Milman, V.; Lee, M.H.; Payne, M.C. Ground-state properties of CoSi2 determined by a total-energy pseudopotential method. Phys. Rev. B 1994, 49, 16300. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Cote, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-newton method. J. Comput. Phys. 1997, 131, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Baroni, S.; de Gironcoli, S.; dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.M. Relationship between intrinsic breakdown field and bandgap of materials. In Proceedings of the 25th International Conference on Microelectronics, Belgrade, Serbia, 14–17 May 2006; pp. 576–579. [Google Scholar]
- Wang, N.; Cao, D.; Wang, J.; Liang, P.; Chen, X.; Shu, H. Semiconducting edges and flake-shape evolution of monolayer GaSe: Role of edge reconstructions. Nanoscale 2018, 10, 12133–12140. [Google Scholar] [CrossRef]
- Xu, X.D.; Sun, W.F. First-principles investigation of GaInSe2 monolayer as a janus material. J. Chin. Ceram. Soc. 2020, 48, 507–513. [Google Scholar]
- Lv, M.H.; Li, C.M.; Sun, W.F. Spin-orbit coupling and spin-polarized electronic structures of janus vanadium-dichalcogenide monolayers: First-principles calculations. Nanomaterials 2022, 12, 382. [Google Scholar] [CrossRef] [PubMed]
- Rani, J.R.; Thangavel, R.; Oh, S.I.; Lee, Y.S.; Jang, J.H. An ultra-high-energy density supercapacitor; fabrication based on Thiol-functionalized graphene oxide scrolls. Nanomaterials 2019, 9, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.P.; Silva, J.M.; Oliveira, M.J.; Weingärtner, T.; Sekhar, K.C.; Pereira, M.; Gomes, M.J. High-performance ferroelectric-dielectric multilayered thin films for energy storage capacitors. Adv. Funct. Mater. 2019, 29, 1807196. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Orientations | Superlattices | Space Groups | a = b/Å | c/Å | hAlN/Å | hScN/Å | Ecoh/(eV/atom) | Eg/eV | Eb/(MV·cm−1) |
|---|---|---|---|---|---|---|---|---|---|
| (001) | 1*AlN/1*ScN | P-4M2 | 3.2608 | 4.6528 | 2.0997 | 2.5531 | 7.9224 | 3.815 | 7.75 |
| 2*AlN/2*ScN | PMM2 | 3.2525 | 9.3081 | 4.1715 | 5.1366 | 7.9542 | 3.559 | 7.23 | |
| 3*AlN/3*ScN | P-4M2 | 3.2507 | 13.9629 | 6.2454 | 7.7175 | 7.9642 | 3.535 | 7.18 | |
| (111) | 1*AlN/1*ScN | R3M | 3.2551 | 16.1767 | 2.4945 | 2.8978 | 7.9126 | 4.519 | 9.18 |
| 2*AlN/2*ScN | R3M | 3.2492 | 32.4172 | 4.9897 | 5.81603 | 7.9324 | 4.231 | 8.59 | |
| 3*AlN/3*ScN | P3M1 | 3.2487 | 16.2102 | 7.4845 | 8.7257 | 7.9380 | 4.072 | 8.27 |
| Material | Energy Density/J·cm−3 | Method or Process |
|---|---|---|
| (001) 3*AlN/3*ScN superlattice | 259 | First-principles calculation |
| (111) 3*AlN/3*ScN superlattice | 304 | First-principles calculation |
| Nitrogen-Thiol-rGO Scrolls [34] | 215 | Nitrogen-doped thiol-functionalization |
| Pt(111)/Ti/SiO2/Si [35] | 99.8 | Solid-state reaction |
| rGO-based EDLC [36] | 142 | Hydrazine reduction |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.-C.; Wu, H.; Sun, W.-F.; Zhang, Z.-P. First-Principles Study of n*AlN/n*ScN Superlattices with High Dielectric Capacity for Energy Storage. Nanomaterials 2022, 12, 1966. https://doi.org/10.3390/nano12121966
Zhang W-C, Wu H, Sun W-F, Zhang Z-P. First-Principles Study of n*AlN/n*ScN Superlattices with High Dielectric Capacity for Energy Storage. Nanomaterials. 2022; 12(12):1966. https://doi.org/10.3390/nano12121966
Chicago/Turabian StyleZhang, Wei-Chao, Hao Wu, Wei-Feng Sun, and Zhen-Peng Zhang. 2022. "First-Principles Study of n*AlN/n*ScN Superlattices with High Dielectric Capacity for Energy Storage" Nanomaterials 12, no. 12: 1966. https://doi.org/10.3390/nano12121966
APA StyleZhang, W.-C., Wu, H., Sun, W.-F., & Zhang, Z.-P. (2022). First-Principles Study of n*AlN/n*ScN Superlattices with High Dielectric Capacity for Energy Storage. Nanomaterials, 12(12), 1966. https://doi.org/10.3390/nano12121966