
Formation of a Low-Density Liquid Phase during the Dissociation of Gas Hydrates in Confined Environments


 , ,
, , 
Abstract
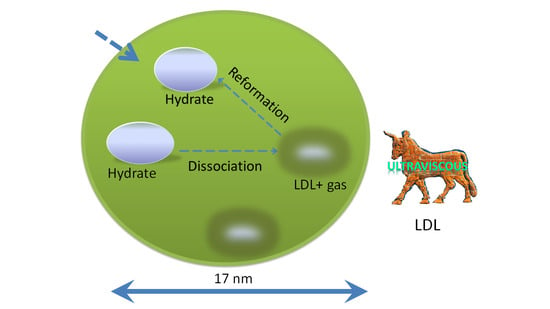
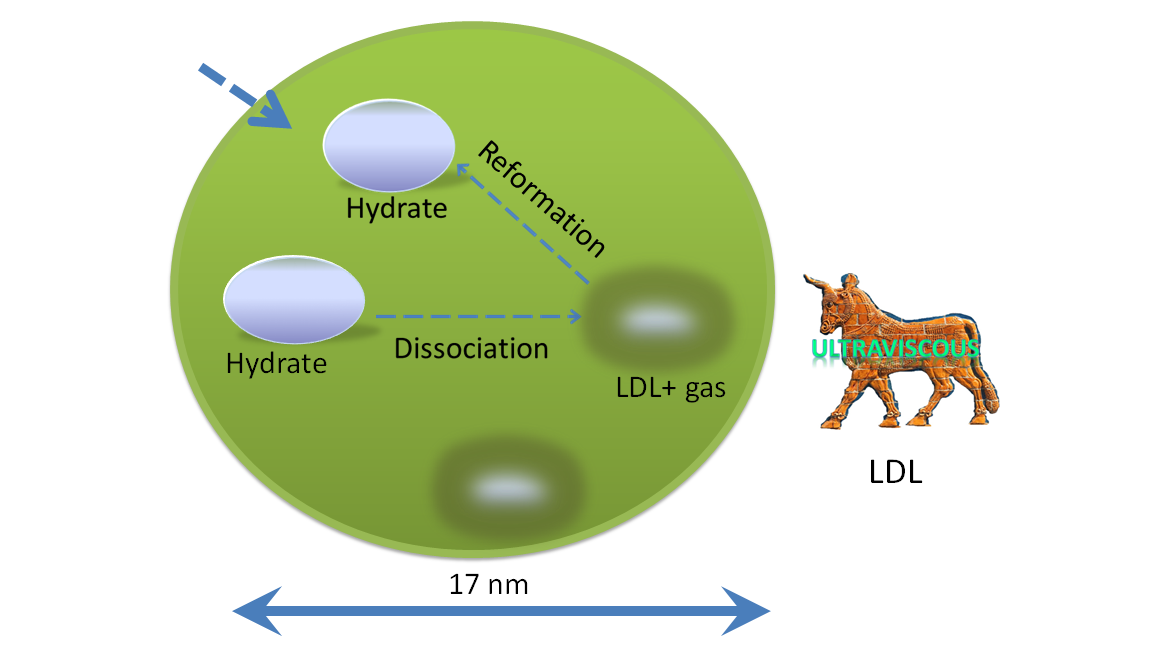{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation of Pore Water
2.2. Preparation of Nanoscale Hydrate
2.3. Experimental Methods
3. Results and Discussion
3.1. Characterization of Confined Hydrate
3.2. Phase State of Water from Decomposing Hydrate
3.3. Desorption Characteristics of Methane Molecules
3.4. Dissociation Characteristics at Temperatures under Atmospheric Pressure
3.5. Dissociation Mechanism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sloan, E.D. Clathrate Hydrates of Natural Gases; Marcel Dekker, INC.: New York, NY, USA, 1998. [Google Scholar]
- Sloan, E.D. Fundamental principles and applications of natural gas hydrates. Nature 2003, 426, 353–363. [Google Scholar] [CrossRef]
- Koh, C.A. Towards a fundamental understanding of natural gas hydrates. Chem. Soc. Rev. 2002, 31, 157–167. [Google Scholar] [CrossRef]
- Englezos, A.P.; Lee, J.D. Gas Hydrates: A cleaner source of energy and opportunity for innovative technologies. Kor. J. Chem. Eng. 2005, 22, 671–681. [Google Scholar] [CrossRef]
- Casco, M.E.; Silvestre-Albero, J.; Ramirez-Cuesta, A.J.; Rey, F.; Jorda, J.L.; Bansode, A.; Urakawa, A.; Peral, I.; Martinez-Escandell, M.; Kaneko, K.; et al. Methane hydrate formation in confined nanospace can surpass nature. Nat. Commun. 2015, 6, 6432-8. [Google Scholar] [CrossRef]
- Celzard, A.; Marêché, J.F. Optimal wetting of active carbons for methane hydrate formation. Fuel 2006, 85, 957–966. [Google Scholar] [CrossRef]
- Anderson, R.; Llamedo, M.; Tohidi, B. Characteristics of clathrate hydrate equilibria in mesopores and interpretation of experimental data. J. Phys. Chem. B 2003, 107, 3500–3506. [Google Scholar] [CrossRef]
- Borchardt, L.; Nickel, W.; Casco, M.; Senkovska, I.; Bon, V.; Wallacher, D.; Grimm, N.; Krause, S.; Silvestre-Albero, J. Illuminating solid gas storage in confined spaces-methane hydrate formation in porous model carbons. Phys. Chem. Chem. Phys. 2016, 18, 20607–20614. [Google Scholar] [CrossRef]
- Zhou, L.; Sun, Y.; Zhou, Y. Enhancement of the methane storage on wet activated carbon by preadsorbed water. AIChE J. 2002, 48, 2412–2416. [Google Scholar] [CrossRef]
- Perrin, A.; Celzard, A.; Marêché, J.F.; Furdin, G. Methane storage within dry and wet active carbons: A comparative study. Energy Fuels 2003, 17, 1283–1291. [Google Scholar] [CrossRef]
- Li, X.-S.; Zhang, Y. Study on dissociation behaviors of methane hydrate in porous media based on experiments and fractional dimension shrinking-core model. Ind. Eng. Chem. Res. 2011, 50, 8263–8271. [Google Scholar] [CrossRef]
- Liu, H.; Zhan, S.; Guo, P.; Fan, S.; Zhang, S. Understanding the characteristic of methane hydrate equilibrium in materials and its potential application. Chem. Eng. J. 2018, 349, 775–781. [Google Scholar] [CrossRef]
- Aladko, E.Y.; Dyadin, Y.A.; Fenelonov, V.B.; Larionov, E.G.; Mel’gunov, M.S.; Manakov, A.Y.; Nesterov, A.N.; Zhurko, F.V. Dissociation conditions of methane hydrate in mesoporous silica gels in wide ranges of pressure and water content. J. Phys. Chem. B 2004, 108, 16540–16547. [Google Scholar] [CrossRef]
- Uchida, T.; Ebinuma, T.; Ishizaki, T. Dissociation condition measurements of methane hydrate in confined small pores of porous glass. J. Phys. Chem. B 1999, 103, 3659–3662. [Google Scholar] [CrossRef]
- Park, T.; Lee, J.Y.; Kwon, T.-H. Effect of pore size distribution on dissociation temperature depression and phase boundary shift of gas hydrate in various fine-grained sediments. Energy Fuels 2018, 32, 5321–5330. [Google Scholar] [CrossRef]
- Handa, Y.P.; Stupin, D.Y. Thermodynamic properties and dissociation characteristics of methane and propane hydrates in 70-.ANG.-radius silica gel pores. J. Phys. Chem. A 1992, 96, 8599–8603. [Google Scholar] [CrossRef]
- Clennell, M.B.; Hovland, M.; Booth, J.S.; Henry, P.; Winters, W.J. Formation of natural gas hydrates in marine sediments conceptual model of gas hydrate growth conditioned by host sediment properties. J. Geophys. Res. 1999, 104, 23005–23022. [Google Scholar] [CrossRef]
- Seo, Y.; Lee, S.; Cha, I.; Lee, J.D.; Lee, H. Phase equilibria and thermodynamic modeling of ethane and propane hydrates in porous silica gels. J. Phys. Chem. B 2009, 113, 5487–5492. [Google Scholar] [CrossRef]
- Anderson, R.; Llamedo, M.; Tohidi, B.; Burgass, R.W. Experimental measurement of methane and carbon dioxide clathrate hydrate equilibria in mesoporous silica. J. Phys. Chem. B 2003, 107, 3507–3514. [Google Scholar] [CrossRef]
- Melnikov, V.P.; Nesterov, A.N.; Podenko, L.S.; Reshetnikov, A.M.; Shalamov, V.V. NMR evidence of supercooled water formation during gas hydrate dissociation below the melting point of ice. Chem. Eng. Sci. 2012, 71, 573–577. [Google Scholar] [CrossRef]
- Melnikov, V.P.; Nesterov, A.N.; Reshetnikov, A.M.; Istomin, V.A. Metastable states during dissociation of carbon dioxide hydrates below 273 K. Chem. Eng. Sci. 2011, 66, 73–77. [Google Scholar] [CrossRef]
- Melnikov, V.P.; Nesterov, A.N.; Reshetnikov, A.M.; Zavodovsky, A.G. Evidence of liquid water formation during methane hydrates dissociation below the ice point. Chem. Eng. Sci. 2009, 64, 1160–1166. [Google Scholar] [CrossRef]
- Takeya, S.; Ripmeester, J.A. Dissociation behavior of clathrate hydrates to ice and dependence on guest molecules. Angew. Chem. Int. Ed. 2008, 47, 1276–1279. [Google Scholar] [CrossRef]
- Stern, L.A.; Circone, S.; Kirby, S.H.; Durham, W.B. Anomalous preservation of pure methane hydrate at 1 atm. J. Phys. Chem. B 2001, 105, 1756–1762. [Google Scholar] [CrossRef]
- Takeya, S.; Shimada, W.; Kamata, Y.; Ebinuma, T.; Uchida, T.; Nagao, J.; Narita, H. In situ X-ray diffraction measurements of the self-preservation effect of CH4 hydrate. J. Phys. Chem. A 2001, 105, 9756–9759. [Google Scholar] [CrossRef]
- Takeya, S.; Ripmeester, J.A. Anomalous preservation of CH4 hydrate and its dependence on the morphology of hexagonal ice. Chem. Phys. Chem. 2010, 11, 70–73. [Google Scholar] [PubMed]
- Klauda, J.B.; Sandler, S.I. Predictions of gas hydrate phase equilibria and amounts in natural sediment porous media. Mar. Pet. Geol. 2003, 20, 459–470. [Google Scholar] [CrossRef]
- Klauda, J.B.; Sandler, S.I. Global distribution of methane hydrate in ocean sediment. Energy Fuels 2005, 19, 459–470. [Google Scholar] [CrossRef]
- Maekawa, Y.; Sasaoka, K.; Yamamoto, T. Structure of water clusters on graphene: A classical molecular dynamics approach. Jpn. J. Appl. Phys. 2018, 57, 035102-7. [Google Scholar] [CrossRef]
- Brubach, J.B.; Mermet, A.; Filabozzi, A.; Gerschel, A.; Lairez, D.; Krafft, M.P.; Roy, P. Dependence of water dynamics upon confinement size. J. Phys. Chem. B 2001, 105, 430–435. [Google Scholar] [CrossRef]
- Boissière, C.; Brubach, J.B.; Mermet, A.; de Marzi, G.; Bourgaux, C.; Prouzet, E.; Roy, P. Water confined in lamellar structures of AOT surfactants: An infrared investigation. J. Phys. Chem. B 2002, 106, 1032–1035. [Google Scholar] [CrossRef]
- Mallamace, F.; Broccio, M.; Corsaro, C.; Faraone, A.; Majolino, D.; Venuti, V.; Liu, L.; Mou, C.Y.; Chen, S.H. Evidence of the existence of the low-density liquid phase in supercooled, confined water. Proc. Natl. Acad. Sci. USA 2007, 104, 424–428. [Google Scholar] [CrossRef]
- Maréchal, Y. The molecular structure of liquid water delivered by absorption spectroscopy in the whole IR region completed with thermodynamics data. J. Mol. Struct. 2011, 1004, 146–155. [Google Scholar] [CrossRef]
- Vasylieva, A.; Doroshenko, I.; Vaskivskyi, Y.; Chernolevska, Y.; Pogorelov, V. FTIR study of condensed water structure. J. Mol. Struct. 2018, 1167, 232–238. [Google Scholar] [CrossRef]
- Bernini, M.C.; Brusau, E.V.; Narda, G.E.; Echeverria, G.; Fantoni, A.; Punte, G.; Ayala, A.P. Tapes of water hexamer clusters in the interlayer space of a 2D MOF: Structural, spectroscopic and computational insight of the confined water. Polyhedron 2012, 31, 729–737. [Google Scholar] [CrossRef]
- Johari, G.; Chew, H. O-H stretching vibrations in ice clathrate. Nature 1983, 303, 604–605. [Google Scholar] [CrossRef]
- Schicks, J.M.; Erzinger, J.; Ziemann, M.A. Raman spectra of gas hydrates-differences and analogies to ice Ih and (gas saturated) water. Spectrochim. Acta Part A 2005, 61, 2399–2403. [Google Scholar] [CrossRef]
- Minceva-Sukarova, B.; Sherman, W.; Wilkinson, G. The raman spectra of ice (Ih, II, III, V, VI and IX) as functions of pressure and temperature. J. Phys. C Solid State Phys. 1984, 17, 5833–5850. [Google Scholar] [CrossRef]
- Thomas, M.V.; Phillip, D.S.; Alejandro, D.R. Infrared spectra of gas hydrates from first-principles. J. Phys. Chem. B 2019, 123, 936–947. [Google Scholar]
- Hagen, W.; Tielens, A.; Greenberg, J.M. The infrared spectra of amorphous solid water and ice Ic between 10 and 140 K. Chem. Phys. 1981, 56, 367–379. [Google Scholar] [CrossRef]
- Sadtchenko, V.; Ewing, G.E. Interfacial melting of thin ice films: An infrared study. J. Chem. Phys. 2002, 116, 4686–4697. [Google Scholar] [CrossRef]
- Stanley, H.E.; Teixeira, J. Interpretation of the unusual behavior of H2O and D2O at low temperatures: Tests of a percolation model. J. Chem. Phys. 1980, 73, 3404–3422. [Google Scholar] [CrossRef]
- Rousset, J.L.; Duval, E.; Boukenter, A. Dynamical structure of water: Low-frequency Raman Scattering from a disordered network and aggregates. J. Chem. Phys. 1990, 92, 2150–2154. [Google Scholar] [CrossRef]
- Walrafen, G.E. Structure of Water in Hydrogen Bonded Solvent Systems; Covington, A.K., Jones, P., Eds.; Taylor & Francis Ltd.: London, UK, 1968; pp. 9–30. [Google Scholar]
- Dartois, E.; Deboffle, D. Methane clathrate hydrate FTIR spectrum. A&A 2008, 490, L19–L22. [Google Scholar]
- Dartois, E.; Duret, P.; Marboeuf, U.; Schmitt, B. Hydrogen sulfide clathrate hydrate FTIR spectroscopy: A help gas for clathrate formation in the Solar system? Icarus 2012, 220, 427–434. [Google Scholar] [CrossRef]
- Fleyfel, F.; Devlin, J.P. FT-IR Spectra of 90 K films of simple, mixed, and double clathrate hydrates of trimethylene oxide, methyl chloride, carbon dioxide, tetrahydrofuran, and ethylene oxide containing decoupled D2O. J. Phys. Chem. 1988, 92, 631–635. [Google Scholar] [CrossRef]
- Tse, J.S. Vibrations of methane in structure I clathrate hydrate-an ab initio density functional molecular dynamics study. J. Supramol. Chem. 2002, 2, 429–433. [Google Scholar] [CrossRef]
- Brubach, J.B.; Mermet, A.; Filabozzi, A.; Gerschel, A.; Roy, P. Signatures of the hydrogen bonding in the infrared bands of water. J. Chem. Phys. 2005, 122, 184509-7. [Google Scholar] [CrossRef]
- Maekawa, Y.; Sasaoka, K.; Yamamoto, T. Prediction of the infrared spectrum of water on graphene substrate using hybrid classical/quantum simulation. Jpn. J. Appl. Phys. 2019, 58, 068008-4. [Google Scholar] [CrossRef]
- Yao, Y.; Fella, V.; Huang, W.; Zhang, K.A.I.; Landfester, K.; Butt, H.-J.; Vogel, M.; Floudas, G. Crystallization and dynamics of water confined in model mesoporous silica particles: Two ice nuclei and two fractions of water. Langmuir 2019, 35, 5890–5901. [Google Scholar] [CrossRef] [PubMed]
- Selevou, A.; Papamokos, G.; Steinhart, M.; Floudas, G. 8OCB and 8CB liquid crystals confined in nanoporous alumina: Effect of confinement on the structure and dynamics. J. Phys. Chem. B 2017, 121, 7382–7394. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Kaneko, T.; Bai, J.; Francisco, J.S.; Yasuoka, K.; Zeng, X.C. Evidence of low-density and high-density liquid phases and isochore end point for water confined to carbon nanotube. Proc. Natl. Acad. Sci. USA 2017, 114, 4066–4071. [Google Scholar] [CrossRef] [PubMed]
- Mishima, O.; Stanley, H.E. Decompression-induced melting of ice IV and the liquid–liquid transition in water. Nature 1998, 392, 164–168. [Google Scholar] [CrossRef]
- Mishima, O. Liquid-liquid critical point in heavy water. Phys. Rev. Lett. 2000, 85, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Mallamace, F.; Mou, C.Y.; Broccio, M.; Corsaro, C.; Faraone, A.; Liu, L. The violation of the stokes-einstein relation in supercooled water. Proc. Natl. Acad. Sci. USA 2006, 103, 12974–12978. [Google Scholar] [CrossRef] [PubMed]
- Cupane, A.; Fomina, M.; Piazza, I.; Peters, J.; Schirò, G. Experimental evidence for a liquid-liquid crossover in deeply cooled confined water. Phys. Rev. Lett. 2014, 113, 215701-5. [Google Scholar] [CrossRef]
- Kim, C.U.; Barstow, B.; Tate, M.W.; Gruner, S.M. Evidence for liquid water during the high-density to low-density amorphous ice transition. Proc. Natl. Acad. Sci. USA 2009, 106, 4596–4600. [Google Scholar] [CrossRef]
- Kirchner, M.T.; Boese, R.; Billups, W.E.; Norman, L.R. Gas hydrate single-crystal structure analyses. J. Am. Chem. Soc. 2004, 126, 9407–9412. [Google Scholar] [CrossRef]
- Hollander, F.; Jeffrey, G.A. Neutron diffraction study of the crystal structure of ethylene oxide deuterohydrate at 80 °K. J. Chem. Phys. 1977, 66, 4699–4705. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Houssa, M.; Cummings, P.T. Molecular dynamics study of the structure and thermophysical properties of model sI clathrate hydrates. J. Phys. Chem. B 2002, 106, 442–451. [Google Scholar] [CrossRef]
- Soper, A.K.; Ricci, M.A. Structures of high-density and low-density water. Phys. Rev. Lett. 2000, 84, 2881–2884. [Google Scholar] [CrossRef]
- Shimanouchi, T. Tables of Molecular Vibrational Frequencies; National Bureau of Standards: Annapolis, MD, USA, 1972. [Google Scholar]
- Van der Waals, J.H.; Platteeuw, J.C. Clathrate solutions. Adv. Chem. Phys. 1959, 2, 1–57. [Google Scholar]
- Istomin, V.A.; Kwon, V.G.; Durov, V.A. Metastable states of gas hydrates. Gazovaya Promishlennost Gas Ind. Russ. Spec. Issue Gas Hydrates 2006, 7, 32–35. (In Russian) [Google Scholar]
- Evans, R. The nature of the liquid-vapour interface and other topics in the statistical mechanics of non-uniform, classical fluids. Adv. Phys. 1979, 28, 143–200. [Google Scholar] [CrossRef]
- Tarazona, P. Free energy density functional for hard spheres. Phys. Rev. A 1985, 31, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, P.; Marconi, U.M.B.; Evans, R. Phase equilibria of fluid interfaces and confined fluids. Mol. Phys. 1987, 60, 573–595. [Google Scholar] [CrossRef]
- Neimark, A.V.; Lin, Y.; Ravikovitch, P.I.; Thommes, M. Quenched solid density functional theory and pore size analysis of micro-mesoporous carbons. Carbon 2009, 47, 1617–1628. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Chem.Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Park, S.; Venditti, R.A.; Jameel, H.; Pawlak, J.J. Changes in pore size distribution during the drying of cellulose fibers as measured by differential scanning calorimetry. Carbohydr. Polym. 2006, 66, 97–103. [Google Scholar] [CrossRef]
- Ishikiriyama, K.; Todoki, M.; Motomura, K. Pore size distribution measurement of silica gels by means of differential scanning calorimetry. J. Colloid. Interface. Sci. 1995, 171, 92–102. [Google Scholar] [CrossRef]
- Keim, C.; Li, C.; Ladisch, C.M.; Ladisch, M. Modeling pore size distribution in cellulose rolling stationary phases. Biotechnol. Prog. 2002, 18, 317–321. [Google Scholar] [CrossRef]
- Furo, I.; Daicic, J. NMR cryoporometry: A novel method for the investigation of the pore structure of paper and paper coatings. Nord. Pulp. Paper Res. J. 1999, 14, 221–225. [Google Scholar] [CrossRef]
- Findenegg, G.H.; Jähnert, S.; Akcakayiran, D.; Schreiber, A. Freezing and melting of water confined in silica nanopores. Chem. Phys. Chem 2008, 9, 2651–2659. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, B.; Hayden, J.; Loizillon, J.; Steinbacher, S.; Grosso, D.; Lend, B. Pore size-dependent structure of confined water in mesoporous silica films from water adsorption/desorption using ATR-FTIR spectroscopy. Langmuir 2019, 35, 11986–11994. [Google Scholar] [CrossRef]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The highscore suite. Powder Diffract. 2014, 29, S13–S18. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, L.; Zang, X.; Fu, J.; Zhou, X.; Lu, J.; Guan, J.; Liang, D. Formation of a Low-Density Liquid Phase during the Dissociation of Gas Hydrates in Confined Environments. Nanomaterials 2021, 11, 590. https://doi.org/10.3390/nano11030590
Wan L, Zang X, Fu J, Zhou X, Lu J, Guan J, Liang D. Formation of a Low-Density Liquid Phase during the Dissociation of Gas Hydrates in Confined Environments. Nanomaterials. 2021; 11(3):590. https://doi.org/10.3390/nano11030590
Chicago/Turabian StyleWan, Lihua, Xiaoya Zang, Juan Fu, Xuebing Zhou, Jingsheng Lu, Jinan Guan, and Deqing Liang. 2021. "Formation of a Low-Density Liquid Phase during the Dissociation of Gas Hydrates in Confined Environments" Nanomaterials 11, no. 3: 590. https://doi.org/10.3390/nano11030590
APA StyleWan, L., Zang, X., Fu, J., Zhou, X., Lu, J., Guan, J., & Liang, D. (2021). Formation of a Low-Density Liquid Phase during the Dissociation of Gas Hydrates in Confined Environments. Nanomaterials, 11(3), 590. https://doi.org/10.3390/nano11030590