
A Chemosensor Based on Gold Nanoparticles and Dithiothreitol (DTT) for Acrylamide Electroanalysis

 ,
,  ,
,  ,
,  and
and
Abstract
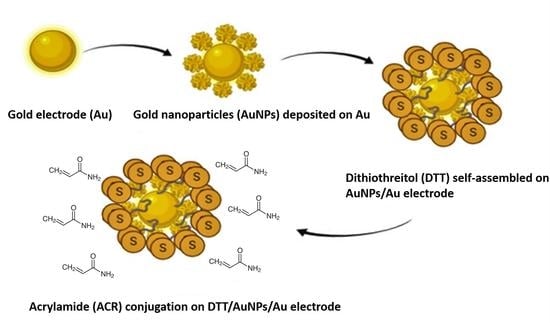
1. Introduction
2. Materials and Methods
2.1. Chemicals and Instruments
2.2. Cleaning and Preparation of the Working Electrode
2.3. Fabrication of the Working Electrode
2.4. Extraction of ACR from Food Samples and Its Quantification Using HPLC
2.5. Surface Plasmon Resonance Interaction Studies
2.6. Electrochemical Studies of Electrode and Detection of ACR Using the Au/AuNP-Modified Electrode
2.7. Interference Study
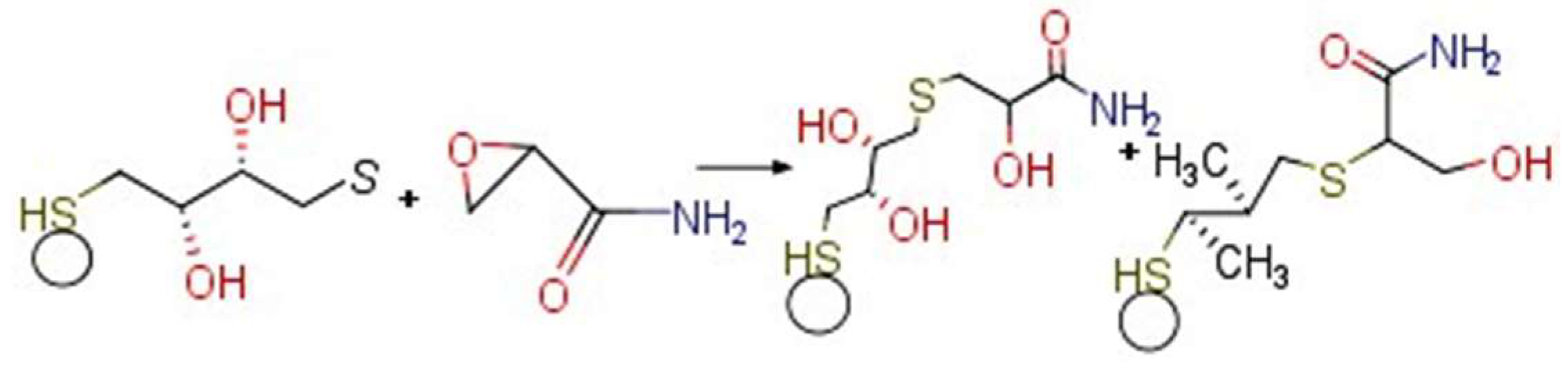
3. Results and Discussions
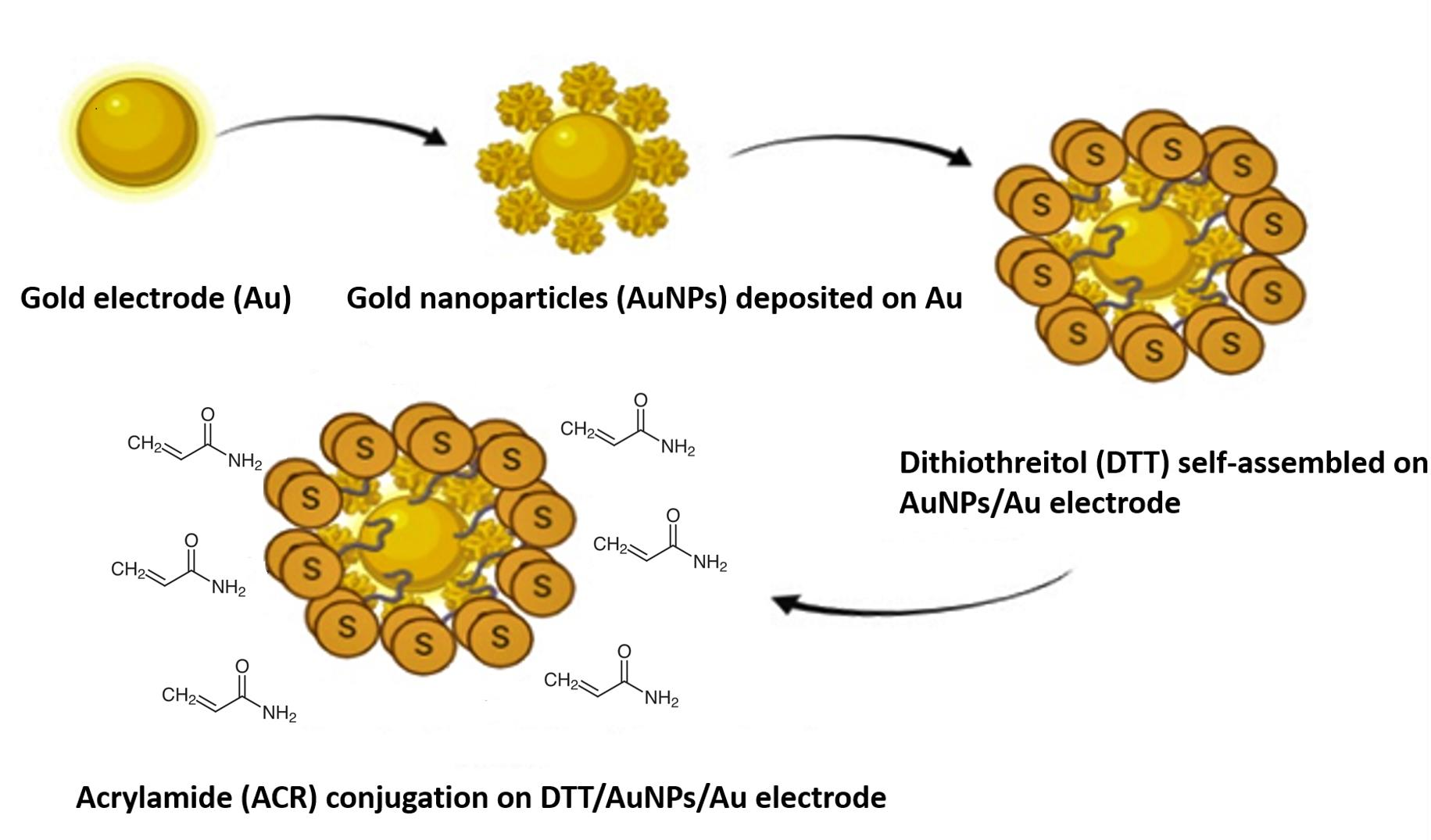
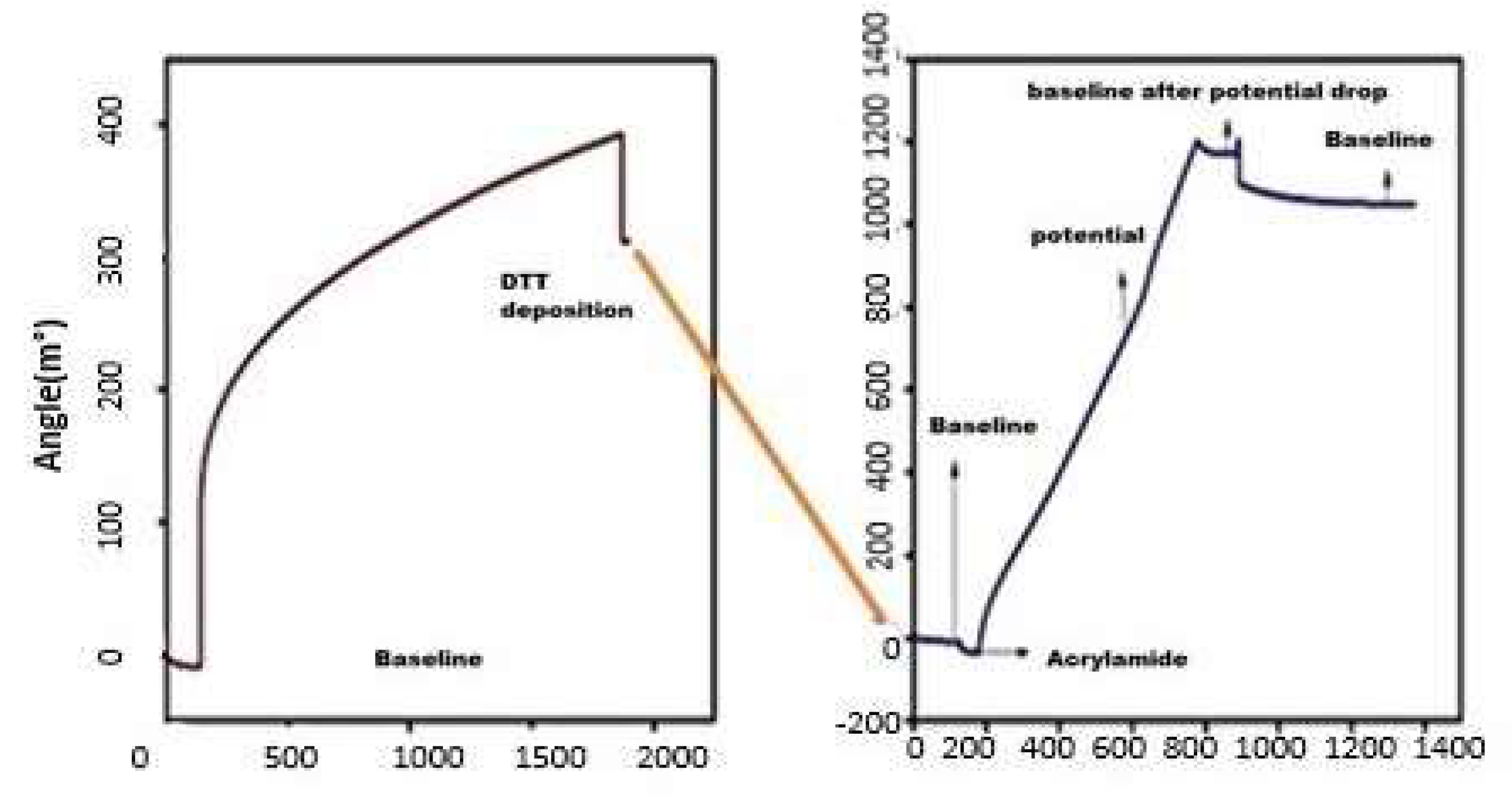
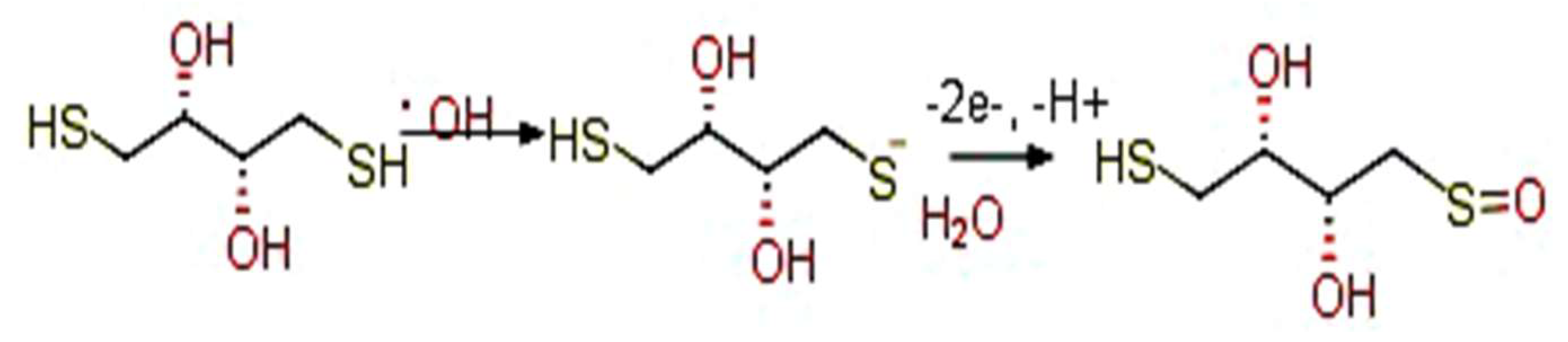
3.1. CV Analysis of Deposition of Gold Nanoparticles (AuNPs) and DTT on Bare Au Electrode
3.2. Characteristics of the Bare Au Electrode
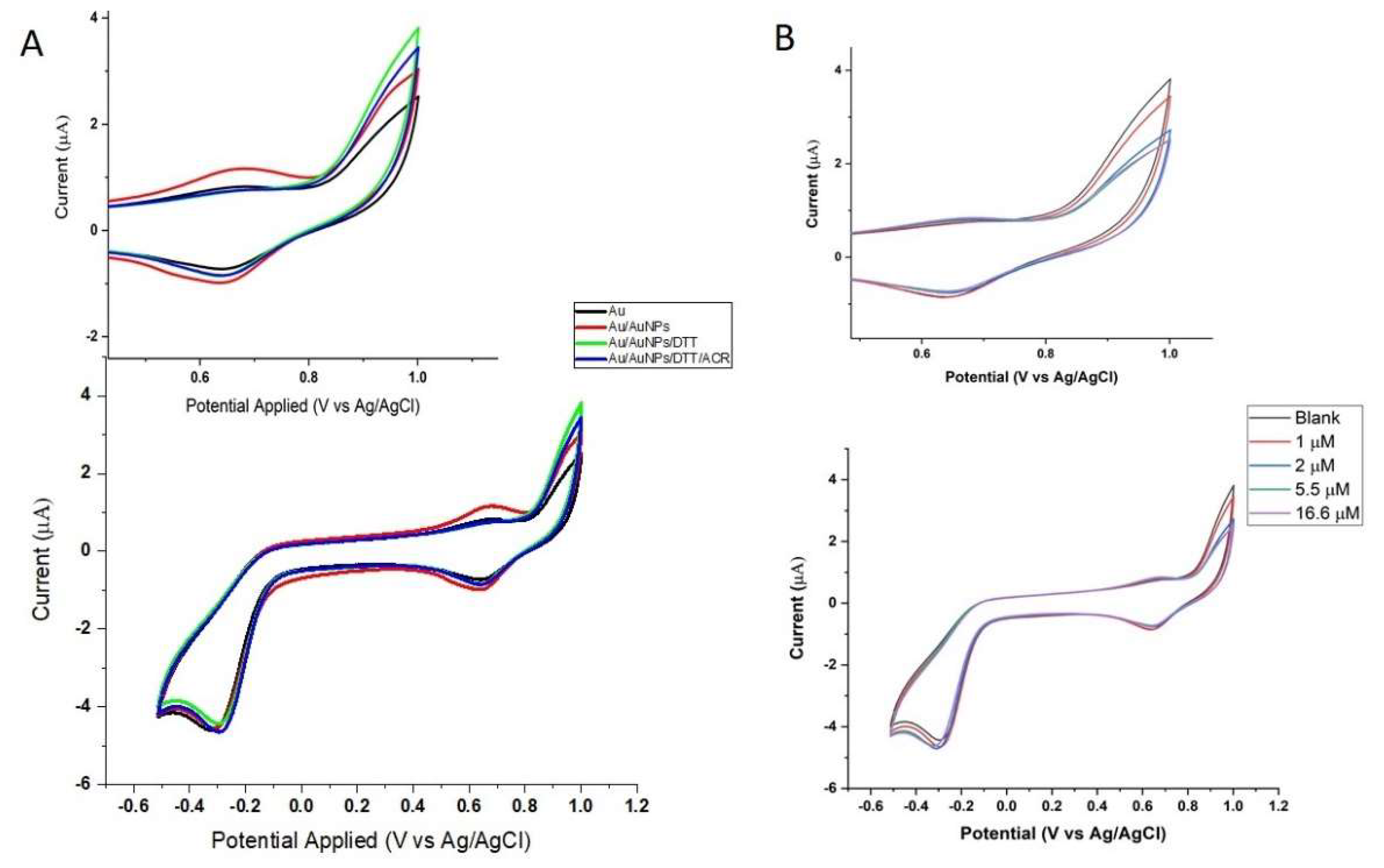
3.3. Characteristics of the Au Electrode Modified with Gold Nanoparticles (AuNPs)
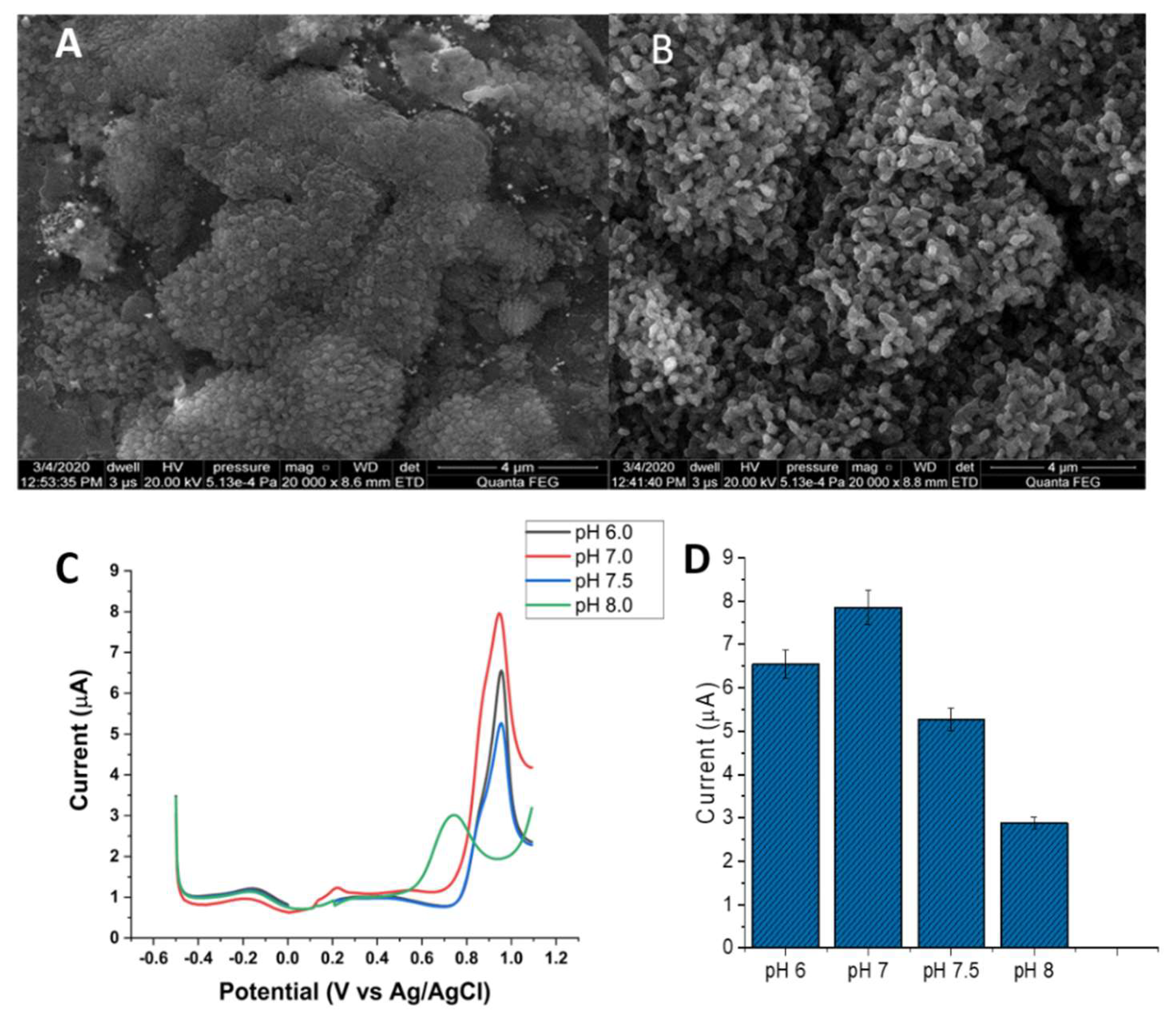
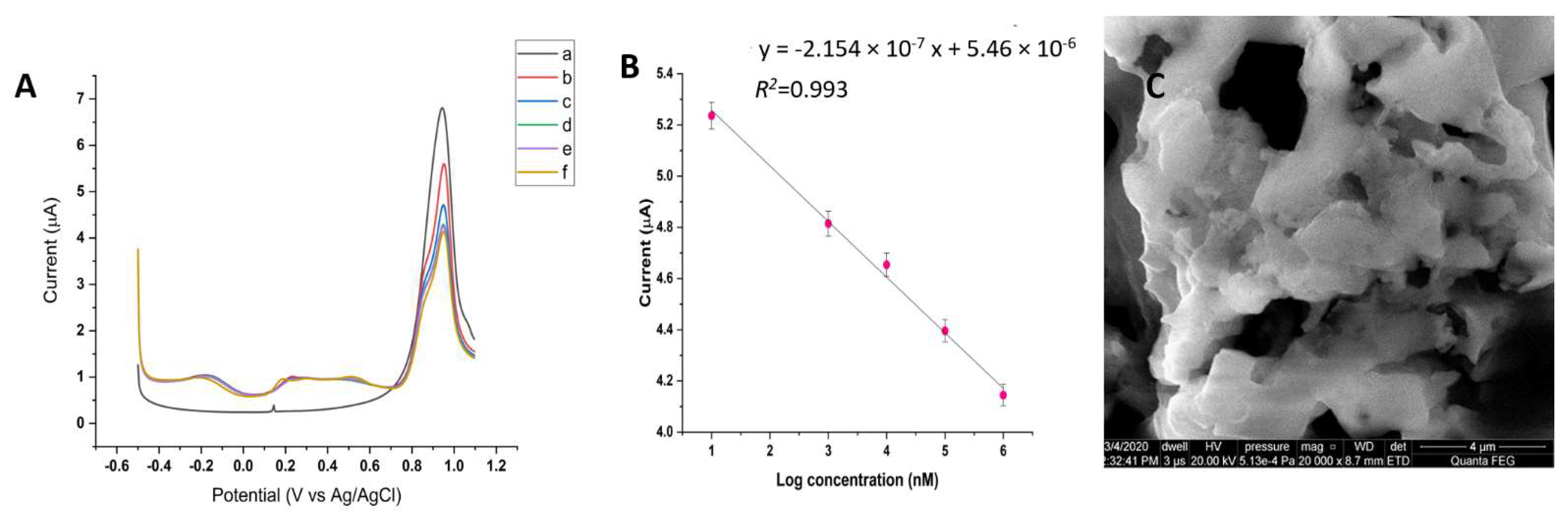
3.4. Electrochemistry of the Au/AuNPs/DTT in the Presence of ACR
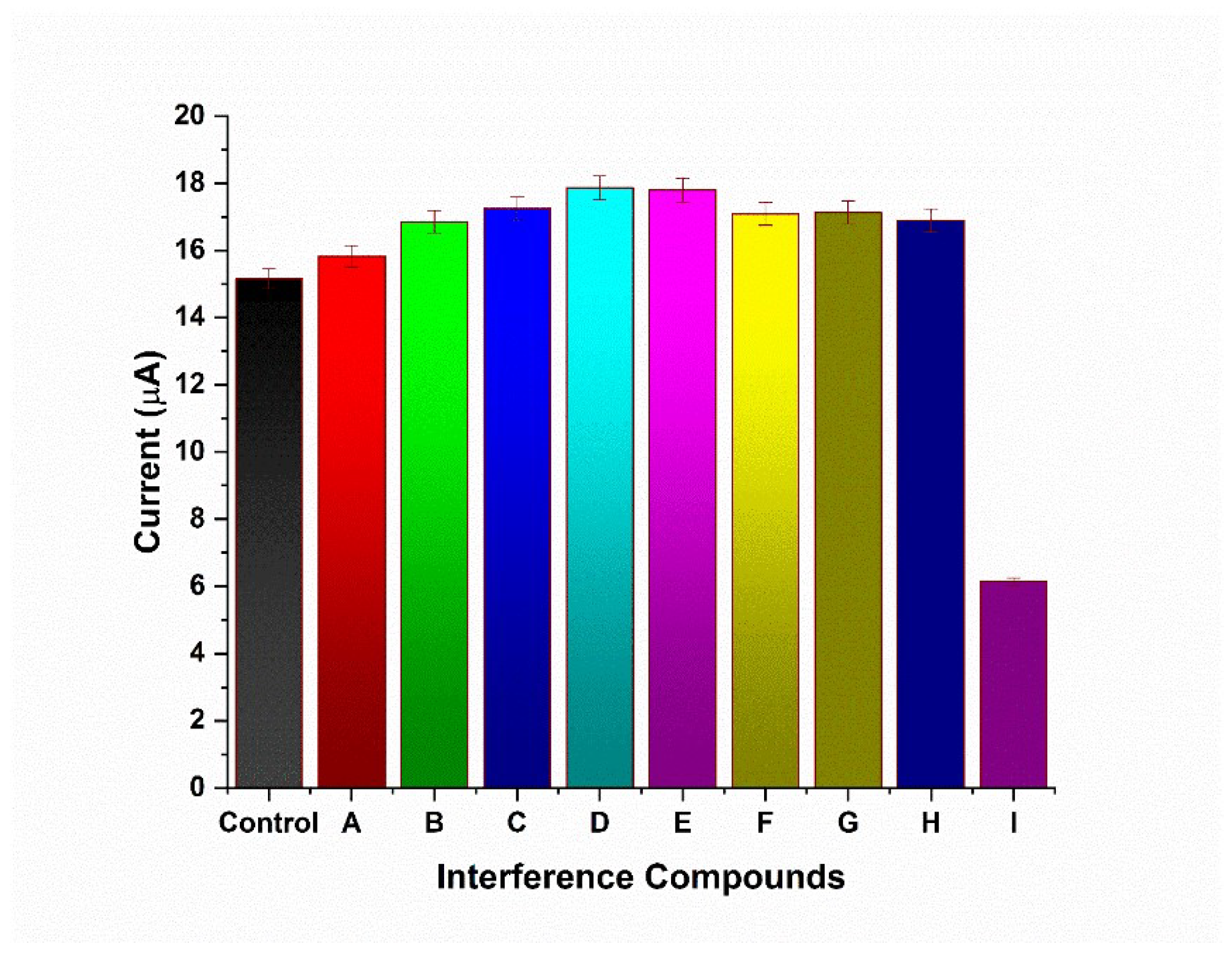
3.5. Interference Study
3.6. Surface Plasmon Resonance (SPR) Analysis
3.7. Sensing of ACR from Food Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tareke, E.; Rydberg, P.; Karlsson, P.; Eriksson, S.; Törnqvist, M. Acrylamide: A cooking carcinogen? Chem. Res. 2000, 13, 517–522. [Google Scholar] [CrossRef]
- Alam, S.; Ahmad, R.; Pranaw, K.; Mishra, P.; Khare, S.K. Asparaginase conjugated magnetic nanoparticles used for reducing acrylamide formation in food model system. Biores. Technol. 2018, 269, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Pranaw, K.; Tiwari, R.; Khare, S.K. Recent development in the uses of asparaginase as food enzyme. In Green Bio-Processes; Springer: Berlin/Heidelberg, Germany, 2019; pp. 55–81. [Google Scholar]
- Zyzak, D.V.; Sanders, R.A.; Stojanovic, M.; Tallmadge, D.H.; Eberhart, B.L.; Ewald, D.K.; Gruber, D.C.; Morsch, T.R.; Strothers, M.A.; Rizzi, G.P. Acrylamide formation mechanism in heated foods. J. Agric. Food. Chem. 2003, 51, 4782–4787. [Google Scholar] [CrossRef] [PubMed]
- Juncker, J. Commission Regulation (EU) 2017/2158 of 20 November 2017 establishing mitigation measures and benchmark levels for the reduction of the presence of acrylamide in food. Off. J. Eur. Union 2017, 204, 24–44. [Google Scholar]
- Zamani, E.; Shokrzadeh, M.; Fallah, M.; Shaki, F. A review of acrylamide toxicity and its mechanism. Pharm. Biomed. Res. 2017, 3, 1–7. [Google Scholar] [CrossRef]
- Dybing, E.; Sanner, T.J.T.S. Risk assessment of acrylamide in foods. Toxicol. Sci. 2003, 75, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Bandarra, S.; Fernandes, A.S.; Magro, I.; Guerreiro, P.S.; Pingarilho, M.; Churchwell, M.I.; Gil, O.M.; Batinić-Haberle, I.; Gonçalves, S.; Rueff, J. Mechanistic insights into the cytotoxicity and genotoxicity induced by glycidamide in human mammary cells. Mutagenesis 2013, 28, 721–729. [Google Scholar] [CrossRef][Green Version]
- Kacar, S.; Vejselova, D.; Kutlu, H.; Sahinturk, V. Acrylamide-derived cytotoxic, anti-proliferative, and apoptotic effects on A549 cells. Hum. Exp. Toxicol. 2018, 37, 468–474. [Google Scholar] [CrossRef]
- Becalski, A.; Lau, B.P.-Y.; Lewis, D.; Seaman, S.W. Acrylamide in foods: Occurrence, sources, and modeling. J. Agric. Food Chem. 2003, 51, 802–808. [Google Scholar] [CrossRef]
- Robarge, T.; Phillips, E.; Conoley, M. Analysis of acrylamide in food by GC/MS. LC GC N. Am. 2003, 21, 45. [Google Scholar]
- Wang, H.; Feng, F.; Guo, Y.; Shuang, S.; Choi, M.M. HPLC-UV quantitative analysis of acrylamide in baked and deep-fried Chinese foods. J. Food Compos. Anal. 2013, 31, 7–11. [Google Scholar] [CrossRef]
- Bortolomeazzi, R.; Munari, M.; Anese, M.; Verardo, G. Rapid mixed mode solid phase extraction method for the determination of acrylamide in roasted coffee by HPLC–MS/MS. Food Chem. 2012, 135, 2687–2693. [Google Scholar] [CrossRef] [PubMed]
- Galani, J.H.; Patel, N.J.; Talati, J.G. Acrylamide-forming potential of cereals, legumes and roots and tubers analyzed by UPLC-UV. Food Chem. Toxicol. 2017, 108, 244–248. [Google Scholar] [CrossRef]
- Kafouris, D.; Stavroulakis, G.; Christofidou, M.; Iakovou, X.; Christou, E.; Paikousis, L.; Christodoulidou, M.; Ioannou-Kakouri, E.; Yiannopoulos, S. Determination of acrylamide in food using a UPLC–MS/MS method: Results of the official control and dietary exposure assessment in Cyprus. Food. Addit. Contam. Part A 2018, 35, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, A.; Radecki, J.; Radecka, H. A voltammetric biosensor based on glassy carbon electrodes modified with single-walled carbon nanotubes/hemoglobin for detection of acrylamide in water extracts from potato crisps. Sensors 2008, 8, 5832–5844. [Google Scholar] [CrossRef]
- Batra, B.; Lata, S.; Pundir, C. Construction of an improved amperometric acrylamide biosensor based on hemoglobin immobilized onto carboxylated multi-walled carbon nanotubes/iron oxide nanoparticles/chitosan composite film. Bioproc. Biosyst. Eng. 2013, 36, 1591–1599. [Google Scholar] [CrossRef]
- Umam, K.; Saepudin, E.; Ivandini, T. Modification of nitrogen-terminated boron-doped diamond electrodes with gold nanoparticles and hemoglobin for acrylamide biosensors. AIP Conf. Proc. 2018, 2023, 020108. [Google Scholar] [CrossRef]
- Yadav, N.; Chhillar, A.K.; Pundir, C.S. Preparation, characterization and application of haemoglobin nanoparticles for detection of acrylamide in processed foods. Int. J. Biol. Macromol. 2018, 107, 1000–1013. [Google Scholar] [CrossRef]
- Lau, P.Y.; Ng, K.L.; Yusof, N.A.; Liu, G.; Alias, Y.; Khor, S.M. A sample pre-treatment-free electrochemical immunosensor with negative electro-pulsion for the quantitative detection of acrylamide in coffee, cocoa and prune juice. Anal. Methods 2019, 11, 4299–4313. [Google Scholar] [CrossRef]
- Wu, M.-F.; Wang, Y.; Li, S.; Dong, X.-X.; Yang, J.-Y.; Shen, Y.-D.; Wang, H.; Sun, Y.-M.; Lei, H.-T.; Xu, Z.-L. Ultrasensitive immunosensor for acrylamide based on chitosan/SnO2-SiC hollow sphere nanochains/gold nanomaterial as signal amplification. Anal. Chim. Acta 2019, 1049, 188–195. [Google Scholar] [CrossRef]
- AlShamaileh, E.; Saadeh, H.; Favry, V. Modification of gold surface with gold nanoparticles and cyclohexyl dithiocarbamate as a selective sensor for cysteine. J. Chem. 2013, 2013, 383921. [Google Scholar] [CrossRef]
- Liang, P.; Canoura, J.; Yu, H.; Alkhamis, O.; Xiao, Y. Dithiothreitol-regulated coverage of oligonucleotide-modified gold nanoparticles to achieve optimized biosensor performance. ACS Appl. Mater. Interfaces 2018, 10, 4233–4242. [Google Scholar] [CrossRef]
- Hamzalıoğlu, A.; Gökmen, V. Investigation of the reactions of acrylamide during in vitro multistep enzymatic digestion of thermally processed foods. Food Func. 2015, 6, 108–113. [Google Scholar] [CrossRef]
- Lim, H.-H.; Shin, H.-S. A new derivatization approach with d-cysteine for the sensitive and simple analysis of acrylamide in foods by liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2014, 1361, 117–124. [Google Scholar] [CrossRef]
- Horcas, I.; Fernández, R.; Gomez-Rodriguez, J.; Colchero, J.; Gómez-Herrero, J.; Baro, A. WSXM: A software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. 2007, 78, 013705. [Google Scholar] [CrossRef] [PubMed]
- Elgrishi, N.; Rountree, K.J.; McCarthy, B.D.; Rountree, E.S.; Eisenhart, T.T.; Dempsey, J.L. A practical beginner’s guide to cyclic voltammetry. J. Chem. Educ. 2018, 95, 197–206. [Google Scholar] [CrossRef]
- Merli, D.; Ferrari, C.; Cabrini, E.; Dacarro, G.; Pallavicini, P.; Profumo, A. A gold nanoparticle chemically modified gold electrode for the determination of surfactants. RSC Adv. 2016, 6, 106500–106507. [Google Scholar] [CrossRef]
- Asnaashari, M.; Kenari, R.E.; Farahmandfar, R.; Taghdisi, S.M.; Abnous, K. Fluorescence quenching biosensor for acrylamide detection in food products based on double-stranded DNA and gold nanoparticles. Sens. Actuators B-Chem. 2018, 265, 339–345. [Google Scholar] [CrossRef]
- Xu, X.; Makaraviciute, A.; Pettersson, J.; Zhang, S.-L.; Nyholm, L.; Zhang, Z. Revisiting the factors influencing gold electrodes prepared using cyclic voltammetry. Sens. Actuators B Chem. 2019, 283, 146–153. [Google Scholar] [CrossRef]
- Juodkazis, K.; Juodkazyt, J.; Jasulaitien, V.; Lukinskas, A.; Šebeka, B. XPS studies on the gold oxide surface layer formation. Electrochem. Commun. 2000, 2, 503–507. [Google Scholar] [CrossRef]
- Bessegato, G.; Cooke, M.; Christensen, P.; Wood, D.; Zanoni, M. Synthesis and electrochemical characterization of Si/TiO2/Au composite anode: Efficient oxygen evolution and hydroxyl radicals generation. Electrochim. Acta 2021, 370, 137742. [Google Scholar] [CrossRef]
- Cleland, W.W. Dithiothreitol, a new protective reagent for SH groups. J. Biochem. 1964, 3, 480–482. [Google Scholar] [CrossRef]
- Zahler, W.; Cleland, W. A specific and sensitive assay for disulfides. J. Biol. Chem. 1968, 243, 716–719. [Google Scholar] [CrossRef]
- Zhong, Z.; Male, K.B.; Luong, J.H. More recent progress in the preparation of Au nanostructures, properties, and applications. Anal. Lett. 2003, 36, 3097–3118. [Google Scholar] [CrossRef]
- Li, C.-Z.; Liu, Y.; Luong, J.H. Impedance sensing of DNA binding drugs using gold substrates modified with gold nanoparticles. Anal. Chem. 2005, 77, 478–485. [Google Scholar] [CrossRef]
- Poirier, G.; Pylant, E. The self-assembly mechanism of alkanethiols on Au(111). Science 1996, 272, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Matthiesen, J.E.; Jose, D.; Sorensen, C.M.; Klabunde, K.J. Loss of hydrogen upon exposure of thiol to gold clusters at low temperature. J. Am. Chem. Soc. 2012, 134, 9376–9379. [Google Scholar] [CrossRef]
- Xue, Y.; Li, X.; Li, H.; Zhang, W. Quantifying thiol–gold interactions towards the efficient strength control. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rouhana, L.L.; Moussallem, M.D.; Schlenoff, J.B. Adsorption of short-chain thiols and disulfides onto gold under defined mass transport conditions: Coverage, kinetics, and mechanism. J. Am. Chem. Soc. 2011, 133, 16080–16091. [Google Scholar] [CrossRef]
- Krüger, D.; Rousseau, R.; Fuchs, H.; Marx, D. Towards “mechanochemistry”: Mechanically induced isomerizations of thiolate–gold clusters. Angew. Chem. Int. 2003, 42, 2251–2253. [Google Scholar] [CrossRef]
- Xu, B.; Xiao, X.; Tao, N.J. Measurements of single-molecule electromechanical properties. J. Am. Chem. Soc. 2003, 125, 16164–16165. [Google Scholar] [CrossRef] [PubMed]
- Grönbeck, H.; Curioni, A.; Andreoni, W. Thiols and disulfides on the Au(111) surface: The headgroup—Gold interaction. J. Am. Chem. Soc. 2000, 122, 3839–3842. [Google Scholar] [CrossRef]
- Basch, H.; Cohen, R.; Ratner, M.A. Interface geometry and molecular junction conductance: Geometric fluctuation and stochastic switching. Nano Lett. 2005, 5, 1668–1675. [Google Scholar] [CrossRef] [PubMed]
- Santarino, I.B.; Oliveira, S.C.B.; Oliveira-Brett, A.M. Protein reducing agents dithiothreitol and tris(2-carboxyethyl) phosphine anodic oxidation. Electrochem. Commun. 2012, 23, 114–117. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Wu, B.; Raymond, J.E.; Zeng, Y.; Fang, X.; Wooley, K.L.; Liu, W.R. A genetically encoded acrylamide functionality. ACS Chem. Biol. 2013, 8, 1664–1670. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kurra, Y.; Liu, W.R. Phospha-Michael addition as a new click reaction for protein functionalization. ChemBioChem 2016, 17, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yuan, Z.; Huang, L.; Kang, J.; Jiang, R.; Zhong, H. Titanium dioxide photocatalytic polymerization of acrylamide for gel electrophoresis (TIP PAGE) of proteins and structural identification by mass spectrometry. Sci. Rep. 2016, 6, 20981. [Google Scholar] [CrossRef]
- Sumner, S.C.; Fennell, T.R.; Moore, T.A.; Chanas, B.; Gonzalez, F.; Ghanayem, B.I. Role of cytochrome P450 2E1 in the metabolism of acrylamide and acrylonitrile in mice. Chem. Res. Toxicol. 1999, 12, 1110–1116. [Google Scholar] [CrossRef]
- Sun, J.; Schnackenberg, L.K.; Pence, L.; Bhattacharyya, S.; Doerge, D.R.; Bowyer, J.F.; Beger, R.D. Metabolomic analysis of urine from rats chronically dosed with acrylamide using NMR and LC/MS. Metabolomics 2010, 6, 550–563. [Google Scholar] [CrossRef]
- Friedman, M. Chemistry, biochemistry, and safety of acrylamide. A review. J. Agric. Food Chem. 2003, 51, 4504–4526. [Google Scholar] [CrossRef]
- Sumner, S.C.; Selvaraj, L.; Nauhaus, S.K.; Fennell, T.R. Urinary metabolites from F344 rats and B6C3F1 mice coadministered acrylamide and acrylonitrile for 1 or 5 days. Chem. Res. Toxicol. 1997, 10, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Sumner, S.C.; MacNeela, J.P.; Fennell, T.R. Characterization and quantitation of urinary metabolites of [1, 2, 3-13C] acrylamide in rats and mice using carbon-13 nuclear magnetic resonance spectroscopy. Chem. Res. Toxicol. 1992, 5, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Mülazimoğlu, A.; Sağir, S.; Durmuş, A.; Mülazimoğlu, I. A comparative study of electrochemical behaviors of acrylamide on glassy carbon, platinum and gold electrode surfaces by cyclic voltammetry. Eurasian J. Anal. Chem. 2017, 12, 15–21. [Google Scholar]
- Ishizuka, M.; Fujioka, K.; Shibamoto, T. Analysis of acrylamide in a complex matrix of polyacrylamide solutions treated by heat and ultraviolet light. J. Agric. Food Chem. 2008, 56, 6093–6096. [Google Scholar] [CrossRef] [PubMed]
- Asnaashari, M.; Kenari, R.E.; Farahmandfar, R.; Abnous, K.; Taghdisi, S.M. An electrochemical biosensor based on hemoglobin-oligonucleotides-modified electrode for detection of acrylamide in potato fries. Food Chem. 2019, 271, 54–61. [Google Scholar] [CrossRef]
- Bucur, M.-P.; Bucur, B.; Radu, G.-L. Simple, selective and fast detection of acrylamide based on glutathione S-transferase. RSC Adv. 2018, 8, 23931–23936. [Google Scholar] [CrossRef]
- González-Fuentes, F.J.; Manríquez, J.; Godínez, L.A.; Escarpa, A.; Mendoza, S. Electrochemical analysis of acrylamide using screen-printed carboxylated single-walled carbon nanotube electrodes. Electroanalytical 2014, 26, 1039–1044. [Google Scholar] [CrossRef]
- Huang, S.; Lu, S.; Huang, C.; Sheng, J.; Zhang, L.; Su, W.; Xiao, Q. An electrochemical biosensor based on single-stranded DNA modified gold electrode for acrylamide determination. Sensor. Actuators B-Chem. 2016, 224, 22–30. [Google Scholar] [CrossRef]
- Bhadani, S.N.; Prasad, Y.K.; Kundu, S. Electrochemical and chemical polymerization of acrylamide. J. Polym. Sci. Pol. Chem. 1980, 18, 1459–1469. [Google Scholar] [CrossRef]
- Casella, I.G.; Pierri, M.; Contursi, M. Determination of acrylamide and acrylic acid by isocratic liquid chromatography with pulsed electrochemical detection. J. Chromatogr. A 2006, 1107, 198–203. [Google Scholar] [CrossRef]
- Singh, G.; Brady, B.; Koerner, T.; Becalski, A.; Zhao, T.; Feng, S.; Godefroy, S.B.; Huet, A.-C.; Delahaut, P. Development of a highly sensitive competitive indirect enzyme-linked immunosorbent assay for detection of acrylamide in foods and water. Food Anal. Method 2014, 7, 1298–1304. [Google Scholar] [CrossRef]
- Wu, J.; Shen, Y.-D.; Lei, H.-T.; Sun, Y.-M.; Yang, J.-Y.; Xiao, Z.-L.; Wang, H.; Xu, Z.-L. Hapten synthesis and development of a competitive indirect enzyme-linked immunosorbent assay for acrylamide in food samples. J. Agric. Food Chem. 2014, 62, 7078–7084. [Google Scholar] [CrossRef]
- Zhu, Y.; Song, S.; Liu, L.; Kuang, H.; Xu, C. An indirect competitive enzyme-linked immunosorbent assay for acrylamide detection based on a monoclonal antibody. Food Agric. Immunol. 2016, 27, 796–805. [Google Scholar] [CrossRef]
- Wulandari, R.; Ivandini, T.A.; Irkham, S.E.; Einaga, Y. Modification of boron-doped diamond electrodes with platinum to increase the stability and sensitivity of haemoglobin-based acrylamide sensors. Sens. Mater. 2019, 31, 1105–1117. [Google Scholar] [CrossRef]
- Xiong, B.; Loss, R.D.; Shields, D.; Pawlik, T.; Hochreiter, R.; Zydney, A.L.; Kumar, M. Polyacrylamide degradation and its implications in environmental systems. NPJ Clean Water 2018, 1, 1–9. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scheme | Electrode Composition | LOD | Sample Type | Reference |
|---|---|---|---|---|
| 1 | Double-stranded DNA (dsDNA)/(Hb) modified screen-printed gold electrode | 0.15 µM | Potato fries | [56] |
| 2 | Electrode with modified cobalt-phthalocyanine with GSH enzyme coupling | 50 nM | Bread and potato chips | [57] |
| 3 | Carboxylic-modified single-walled carbon nanotube screen-printed electrodes | 30 nM | Fried potatoes | [58] |
| 4 | Gold nanoparticles (AuNPs) and FAM-labelled double-stranded DNA (FAM-dsDNA) | 10 nM | Tap water and potato chips | [29] |
| 5 | Single-stranded DNA on a gold electrode | 8.1 nM | Tap water and potato chips | [59] |
| 6 | Gold electrode/Gold nanoparticles/DTT | 3.11 nM | Potato chips and coffee | Present study |
| Food Sample | Concentration Added (nM) | Concentration Detected (nM) | Recovery (%) |
|---|---|---|---|
| Chips | 10 | 9.55 | 95.5 |
| Chips | 15 | 12.11 | 81 |
| Coffee | 25 | 28.54 | 114.17 |
| Coffee | 50 | 46.77 | 93.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, S.; Augustine, S.; Narayan, T.; Luong, J.H.T.; Malhotra, B.D.; Khare, S.K. A Chemosensor Based on Gold Nanoparticles and Dithiothreitol (DTT) for Acrylamide Electroanalysis. Nanomaterials 2021, 11, 2610. https://doi.org/10.3390/nano11102610
Alam S, Augustine S, Narayan T, Luong JHT, Malhotra BD, Khare SK. A Chemosensor Based on Gold Nanoparticles and Dithiothreitol (DTT) for Acrylamide Electroanalysis. Nanomaterials. 2021; 11(10):2610. https://doi.org/10.3390/nano11102610
Chicago/Turabian StyleAlam, Shahenvaz, Shine Augustine, Tarun Narayan, John H. T. Luong, Bansi Dhar Malhotra, and Sunil K. Khare. 2021. "A Chemosensor Based on Gold Nanoparticles and Dithiothreitol (DTT) for Acrylamide Electroanalysis" Nanomaterials 11, no. 10: 2610. https://doi.org/10.3390/nano11102610
APA StyleAlam, S., Augustine, S., Narayan, T., Luong, J. H. T., Malhotra, B. D., & Khare, S. K. (2021). A Chemosensor Based on Gold Nanoparticles and Dithiothreitol (DTT) for Acrylamide Electroanalysis. Nanomaterials, 11(10), 2610. https://doi.org/10.3390/nano11102610