Stabilization of Calcium Oxalate Precursors during the Pre- and Post-Nucleation Stages with Poly(acrylic acid)


 ,
, 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reactants and Instruments
2.2. CaOx Crystallization
2.3. Conductivity Measurements of the Ionic Complexes of Ca:C2O4
2.4. Characterization of Pre-Nucleation Entities
2.5. Characterization of Post-Nucleation Stage Precipitates
3. Results and Discussion
3.1. Non-Classical Crystallization of CaOx
3.2. Characterization of Pre-Nucleation Products
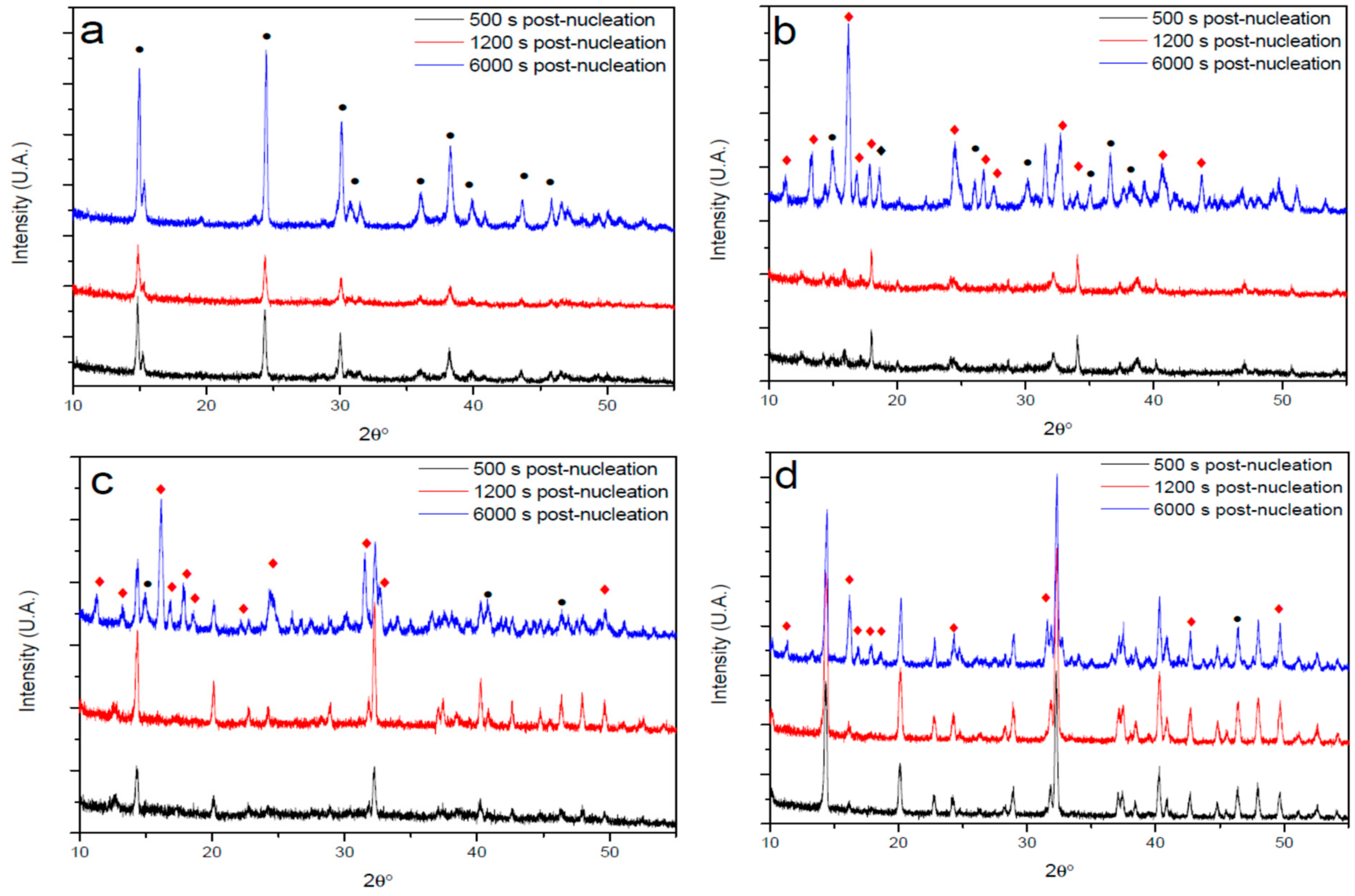
3.3. XRD Analysis of Precipitates
3.4. FTIR Analysis of Precipitates
3.5. Ultrastructural Characterization of Precipitates
3.6. Influence of PAA on CaOx Precursors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yasui, T.; Iguchi, M.; Suzuki, S.; Kohri, K. Prevalence and epidemiological characteristics of urolithiasis in Japan: National trends between 1965 and 2005. Urology 2008, 71, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Lieske, J.C.; Rule, A.D.; Krambeck, A.E.; Williams, J.C.; Bergstralh, E.J.; Mehta, R.A.; Moyer, T.P. Stone composition as a function of age and sex. Clin. J. Am. Soc. Nephrol. 2014, 9, 2141–2146. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, I.; Mamoulakis, C.; Miyazawa, K.; Rodgers, A.; Talati, J.; Lotan, Y. Epidemiology of stone disease across the world. World J. Urol. 2017, 35, 1301–1320. [Google Scholar] [CrossRef] [PubMed]
- Curhan, G.C. Epidemiology of stone disease. Urol. Clin. N. Am. 2007, 34, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Nancollas, G. Crystallization theory relating to urinary stone formation. World J. Urol. 1983, 1, 131–137. [Google Scholar] [CrossRef]
- Zuckerman, J.M.; Assimos, D.G. Hypocitraturia: Pathophysiology and medical management. Rev. Urol. 2009, 11, 134–144. [Google Scholar]
- Evan, A.P. Physiopathology and etiology of stone formation in the kidney and the urinary tract. Pediatr. Nephrol. 2010, 25, 831–841. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, C.; Ruiz-Agudo, E.; Harris, J.; Wolf, S.E. Nonclassical crystallization in vivo et in vitro (II): Nanogranular features in biomimetic minerals disclose a general colloid-mediated crystal growth mechanism. J. Struct. Biol. 2016, 196, 260–287. [Google Scholar] [CrossRef]
- Frey-Wyssling, A. Crystallography of the two hydrates of crystalline calcium oxalate in plants. Am. J. Bot. 1981, 68, 130–141. [Google Scholar] [CrossRef]
- Horner, H.T.; Zindler-Frank, E. Histochemical, spectroscopic, and X-ray diffraction identifications of the two hydration forms of calcium oxalate crystals in three legumes and Begonia. Can. J. Bot. 1982, 60, 1021–1027. [Google Scholar] [CrossRef]
- Basso, R.; Lucchetti, G.; Zefiro, L.; Palenzona, A. Caoxite, Ca (H2O)3(C2O4), a new mineral from the Cerchiara mine, northern Apennines, Italy. Neu. Jb. Mineral. 1997, 2, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Beazley, M.J.; Rickman, R.D.; Ingram, D.K.; Boutton, T.W.; Russ, J. Natural abundances of carbon isotopes (14 C, 13 C) in lichens and calcium oxalate pruina: Implications for archaeological and paleoenvironmental studies. Radiocarbon 2002, 44, 675–683. [Google Scholar] [CrossRef]
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable prenucleation calcium carbonate clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Burgos-Cara, A.; Ruiz-Agudo, C.; Ibañez-Velasco, A.; Cölfen, H.; Rodriguez-Navarro, C. A non-classical view on calcium oxalate precipitation and the role of citrate. Nat. Commun. 2017, 8, 768. [Google Scholar] [CrossRef]
- Li, S.; Tang, W.; Li, M.; Wang, L.; Yang, Y.; Gong, J. Understanding the role of citric acid on the crystallization pathways of calcium oxalate hydrates. Cryst. Growth Des. 2019, 19, 3139–3147. [Google Scholar] [CrossRef]
- Thongboonkerd, V.; Chutipongtanate, S.; Semangoen, T.; Malasit, P. Urinary trefoil factor 1 is a novel potent inhibitor of calcium oxalate crystal growth and aggregation. J. Urol. 2008, 179, 1615–1619. [Google Scholar] [CrossRef]
- Munoz, J.A.; Valiente, M. Effects of trace metals on the inhibition of calcium oxalate crystallization. Urol. Res. 2005, 33, 267–272. [Google Scholar] [CrossRef]
- Baumann, J.M.; Affolter, B. The paradoxical role of urinary macromolecules in the aggregation of calcium oxalate: A further plea to increase diuresis in stone metaphylaxis. Urolithiasis 2016, 44, 311–317. [Google Scholar] [CrossRef]
- Alamani, B.G.; Rimer, J.D. Molecular modifiers of kidney stones. Curr. Opin. Nephrol. Hypertens. 2017, 26, 256–265. [Google Scholar] [CrossRef]
- Boyce, W.H.; Garvey, F.K. The amount and nature of the organic matrix in urinary calculi: A review. J. Urol. 1956, 76, 213–227. [Google Scholar] [CrossRef]
- Mustafi, D.; Nakagawa, Y. Characterization of Ca2+-binding sites in the kidney stone inhibitor glycoprotein nephrocalcin using vanadyl ions: Different metal binding properties in strong and weak inhibitor proteins revealed by EPR and ENDOR. Biochemistry 1996, 35, 14703–14709. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y. Properties and function of nephrocalcin: Mechanism of kidney stone inhibition or promotion. Keio J. Med. 1997, 46, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fischer, V.; Landfester, K.; Muñoz-Espí, R. Stabilization of calcium oxalate metastable phases by oligo (L-glutamic acid): Effect of peptide chain length. Cryst. Growth Des. 2011, 11, 1880–1890. [Google Scholar] [CrossRef]
- Rimer, J.D.; Kolbach-Mandel, A.M.; Ward, M.D.; Wesson, J.A. The role of macromolecules in the formation of kidney stones. Urolithiasis 2017, 45, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Ihli, J.; Wang, Y.-W.; Cantaert, B.; Kim, Y.-Y.; Green, D.C.; Bomans, P.H.H.; Sommerdijk, N.A.J.M.; Meldrum, F.C. Precipitation of amorphous calcium oxalate in aqueous solution. Chem. Mater. 2015, 27, 3999–4007. [Google Scholar] [CrossRef]
- Rieger, J.; Frechen, T.; Cox, G.; Heckmann, W.; Schmidt, C.; Thieme, J. Precursor structures in the crystallization/precipitation processes of CaCO3 and control of particle formation by polyelectrolytes. Faraday Discuss. 2007, 136, 265–277. [Google Scholar] [CrossRef]
- Xu, Y.; Tijssen, K.C.H.; Bomans, P.H.H.; Akiva, A.; Friedrich, H.; Kentgens, A.P.M.; Sommerdijk, N.A.J.M. Microscopic structure of the polymer-induced liquid precursor for calcium carbonate. Nat. Commun. 2018, 9, 2582. [Google Scholar] [CrossRef]
- Yao, S.; Lin, X.; Xu, Y.; Chen, Y.; Qiu, P.; Shao, C.; Jin, B.; Mu, Z.; Sommerdijk, N.A.J.M.; Tang, R. Osteoporotic bone recovery by a highly bone-Inductive calcium phosphate polymer-induced liquid-precursor. Adv. Sci. 2019, 6, 1900683. [Google Scholar] [CrossRef]
- Gal, J.Y.; Bollinger, J.C.; Tolosa, H.; Gache, N. Calcium carbonate solubility: A reappraisal of scale formation and inhibition. Talanta 1996, 43, 1497–1509. [Google Scholar] [CrossRef]
- Wolf, S.E.; Leiterer, J.; Kappl, M.; Emmerling, F.; Tremel, W. Early homogenous amorphous precursor stages of calcium carbonate and subsequent crystal growth in levitated droplets. J. Am. Chem. Soc. 2008, 130, 12342–12347. [Google Scholar] [CrossRef]
- Jia, C.; Wu, L.; Chen, Q.; Ke, P.; De Yoreo, J.J.; Guan, B. Structural evolution of amorphous calcium sulfate nanoparticles into crystalline gypsum phase structural evolution of amorphous calcium sulfate nanoparticles into crystalline gypsum phase. CrystEngComm 2020, 22, 6805–6810. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, C.; Kudłacz, K.; Cizer, Ö.; Ruiz-Agudo, E. Formation of amorphous calcium carbonate and its transformation into mesostructured calcite. CrystEngComm 2015, 17, 58–72. [Google Scholar] [CrossRef]
- Gower, L.B. Biomimetic model systems for investigating the amorphous precursor pathway and its role in biomineralization. Chem. Rev. 2008, 108, 4551–4627. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Navarro, C.; Burgos Cara, A.; Elert, K.; Putnis, C.V.; Ruiz-Agudo, E. Direct nanoscale imaging reveals the growth of calcite crystals via amorphous nanoparticles. Cryst. Growth Des. 2016, 16, 1850–1860. [Google Scholar] [CrossRef]
- Dai, L.; Douglas, E.P.; Gower, L.B. Compositional analysis of a polymer-induced liquid-precursor (PILP) amorphous CaCO3 phase. J. Non-Cryst. Solids 2008, 354, 1845–1854. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Soler, F.; Rodriguez-Navarro, C.; Ruiz-Agudo, E.; Neira-Carrillo, A. Stabilization of Calcium Oxalate Precursors during the Pre- and Post-Nucleation Stages with Poly(acrylic acid). Nanomaterials 2021, 11, 235. https://doi.org/10.3390/nano11010235
Díaz-Soler F, Rodriguez-Navarro C, Ruiz-Agudo E, Neira-Carrillo A. Stabilization of Calcium Oxalate Precursors during the Pre- and Post-Nucleation Stages with Poly(acrylic acid). Nanomaterials. 2021; 11(1):235. https://doi.org/10.3390/nano11010235
Chicago/Turabian StyleDíaz-Soler, Felipe, Carlos Rodriguez-Navarro, Encarnación Ruiz-Agudo, and Andrónico Neira-Carrillo. 2021. "Stabilization of Calcium Oxalate Precursors during the Pre- and Post-Nucleation Stages with Poly(acrylic acid)" Nanomaterials 11, no. 1: 235. https://doi.org/10.3390/nano11010235
APA StyleDíaz-Soler, F., Rodriguez-Navarro, C., Ruiz-Agudo, E., & Neira-Carrillo, A. (2021). Stabilization of Calcium Oxalate Precursors during the Pre- and Post-Nucleation Stages with Poly(acrylic acid). Nanomaterials, 11(1), 235. https://doi.org/10.3390/nano11010235