Proteomic Analysis Identifies Markers of Exposure to Cadmium Sulphide Quantum Dots (CdS QDs)

 ,
,  , ,
, ,  ,
, 

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Standards
2.2. Synthesis and Characterisation of the CdS QDs
2.3. Yeast strains and Growth Conditions
2.4. Exposure of Yeast Cells to Different CdS QD Concentrations
2.5. Determination of ROS and Cell Viability by Flow Cytometry
2.6. Protein Extraction and Quantification
2.7. Separation of Proteins by 2D-PAGE and Identification by MALDI-TOF-TOF MS
2.8. Trypsin Hydrolysis and iTRAQ Labelling
2.9. Strong Cation Exchange (SCX) Fractionation of the iTRAQ-Labelled Peptides
2.10. Nano-LC-MS-MS Analysis of the Strong Cation Exchange Fractions
2.11. Data Processing, Protein Identification, and Quantification
2.12. Glyceraldehyde 3-Phosphate Dehydrogenase (GAPDH) Activity Assay
2.13. Data Mining and Analysis
3. Results and Discussion
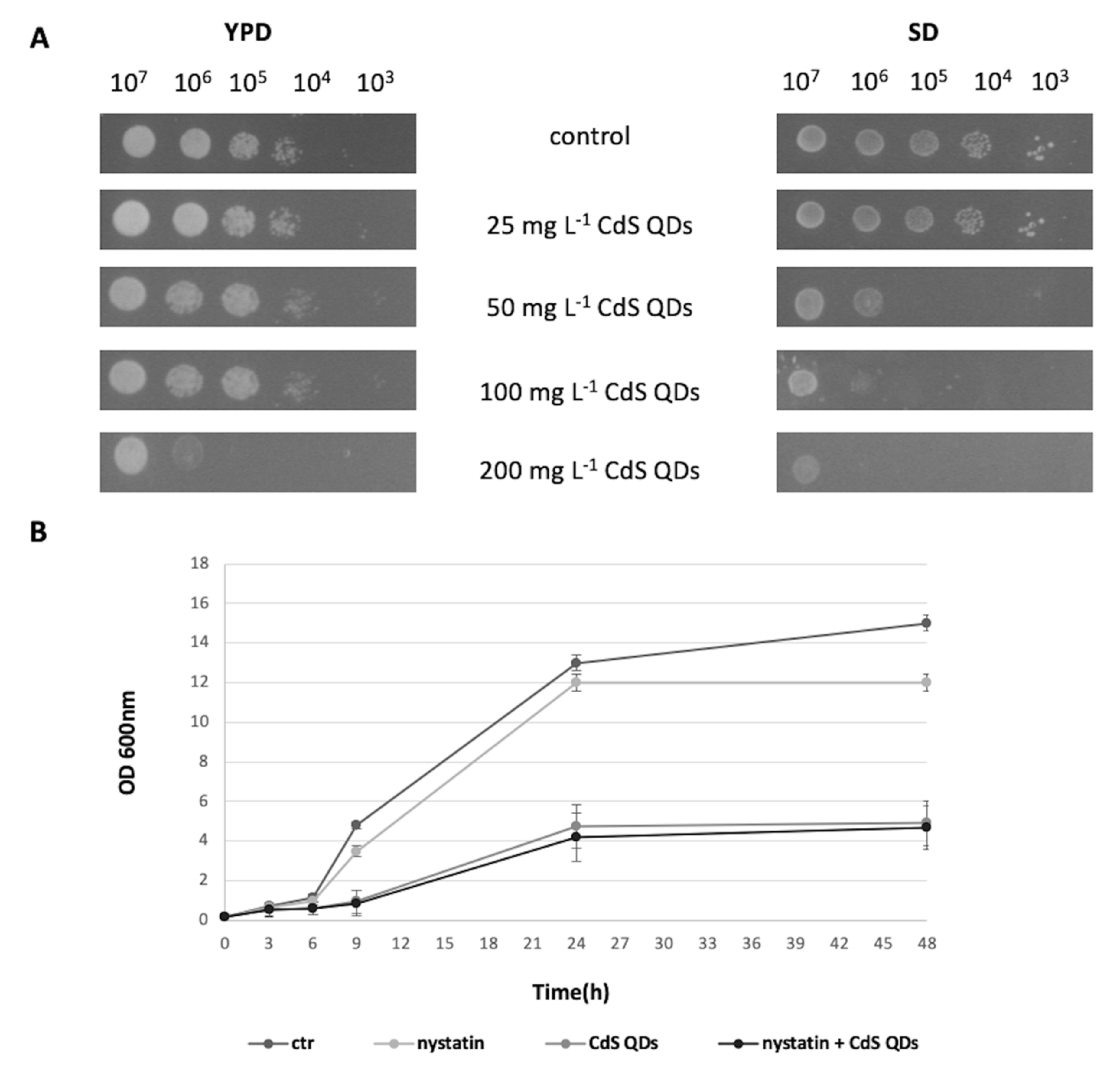
3.1. Cell Growth in the Different Conditions Tested
3.2. Effect of CdS QDs on ROS Generation and Cell Integrity in S. cerevisiae
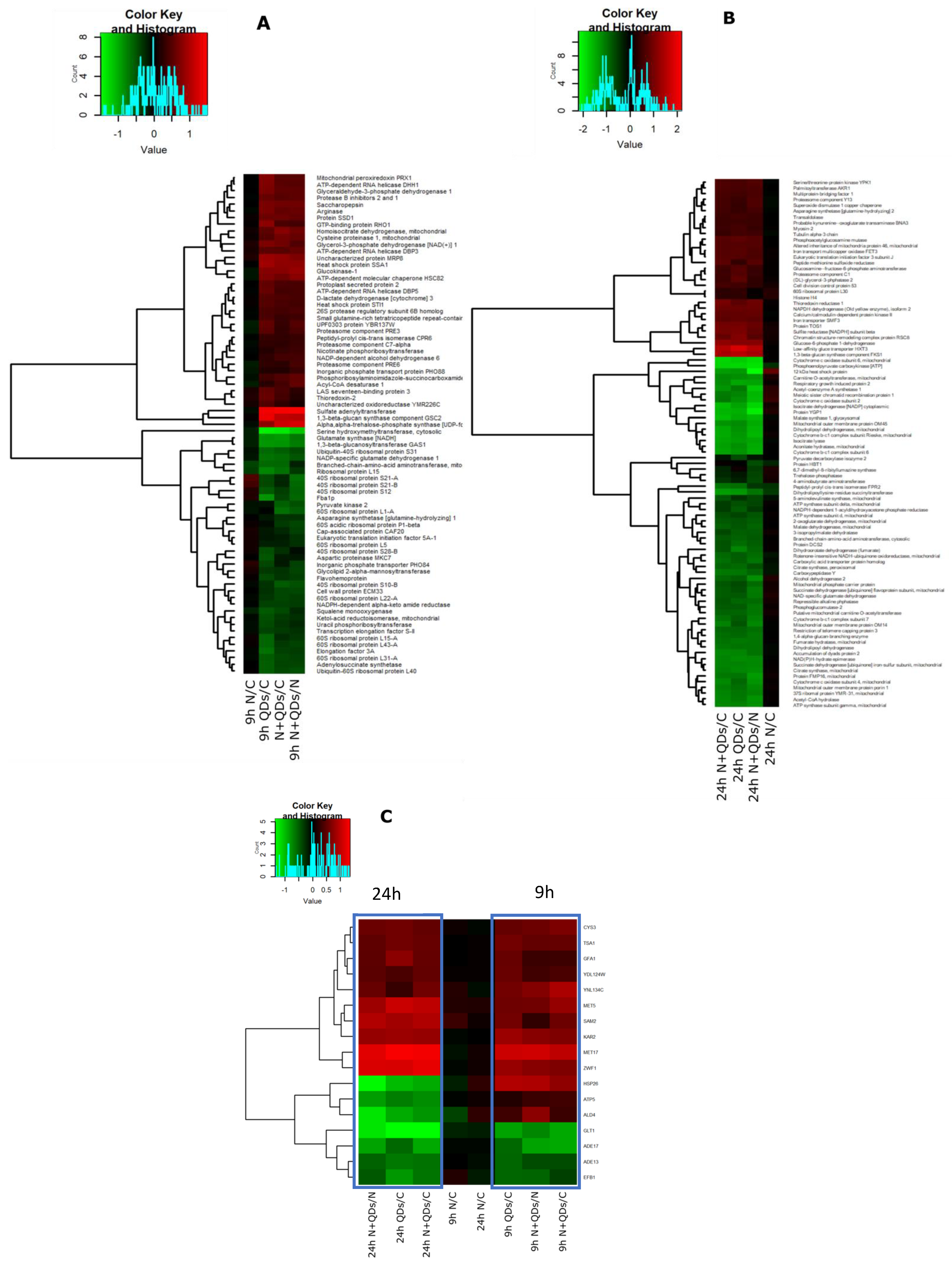
3.3. Proteomic Response to CdS QDs Exposure
3.4. Ontology Analysis of the Identified Proteins
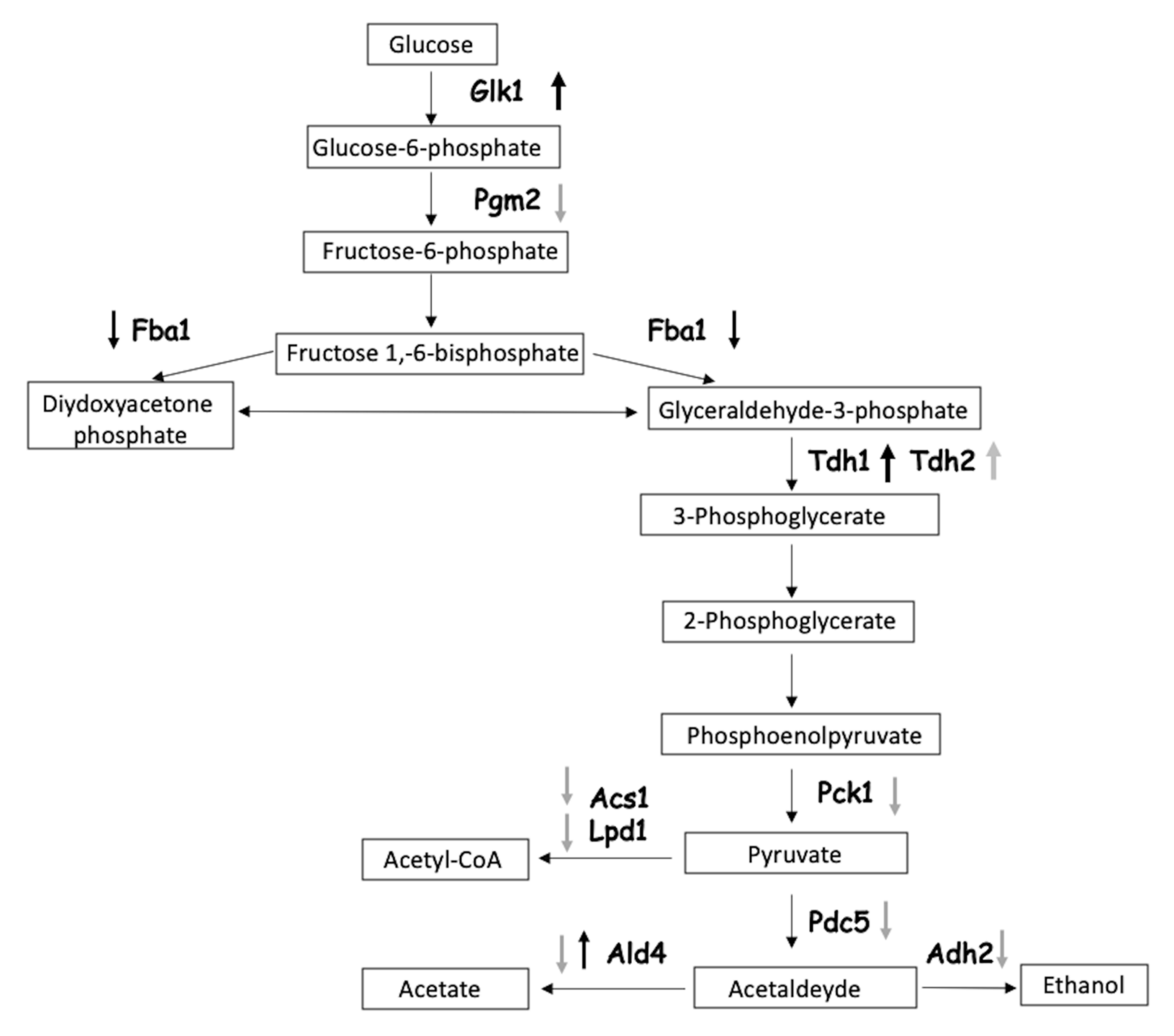
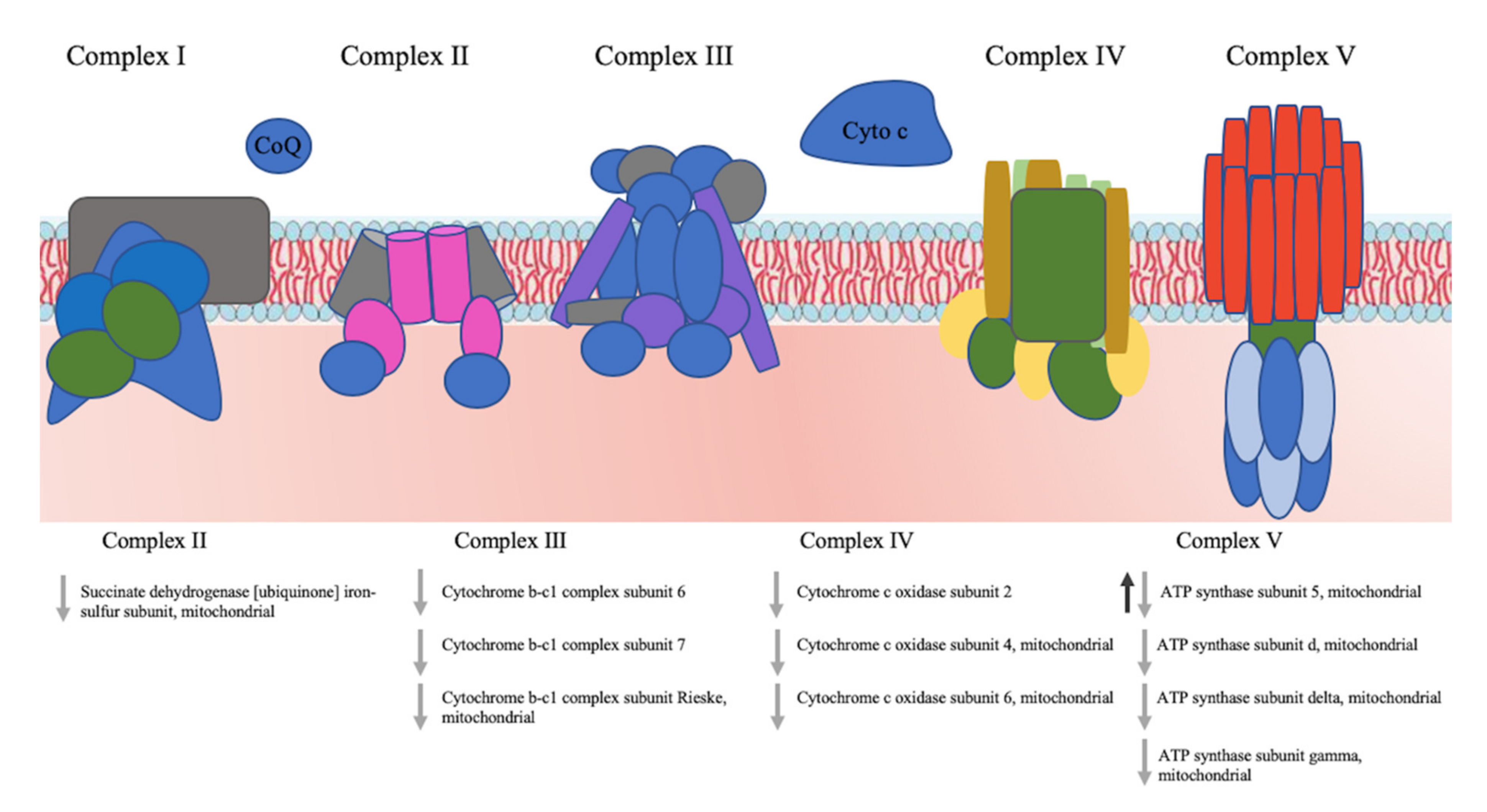
3.5. Pathway Analysis of the Identified Proteins
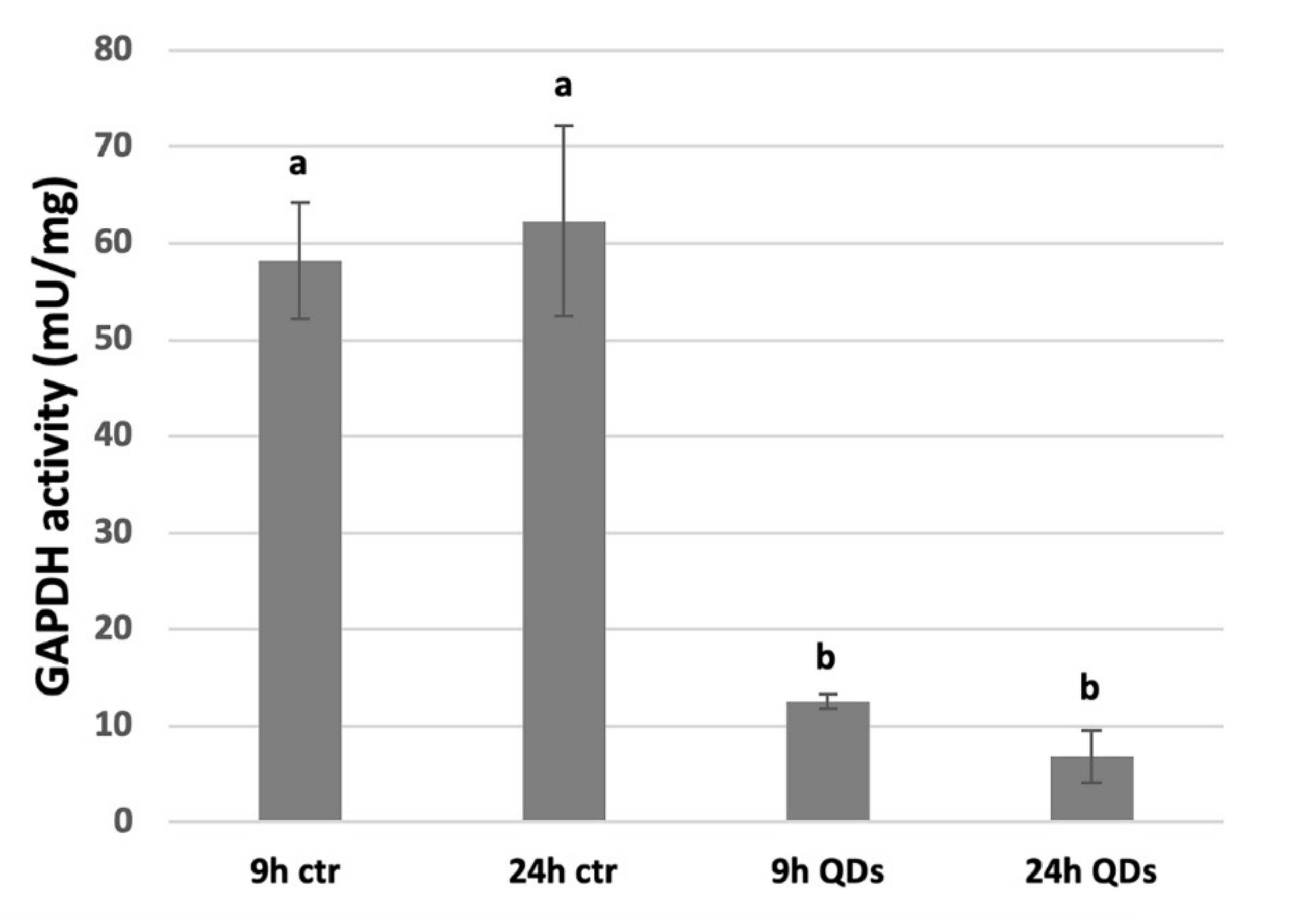
3.6. Inhibition of GAPDH Activity by CdS QDs
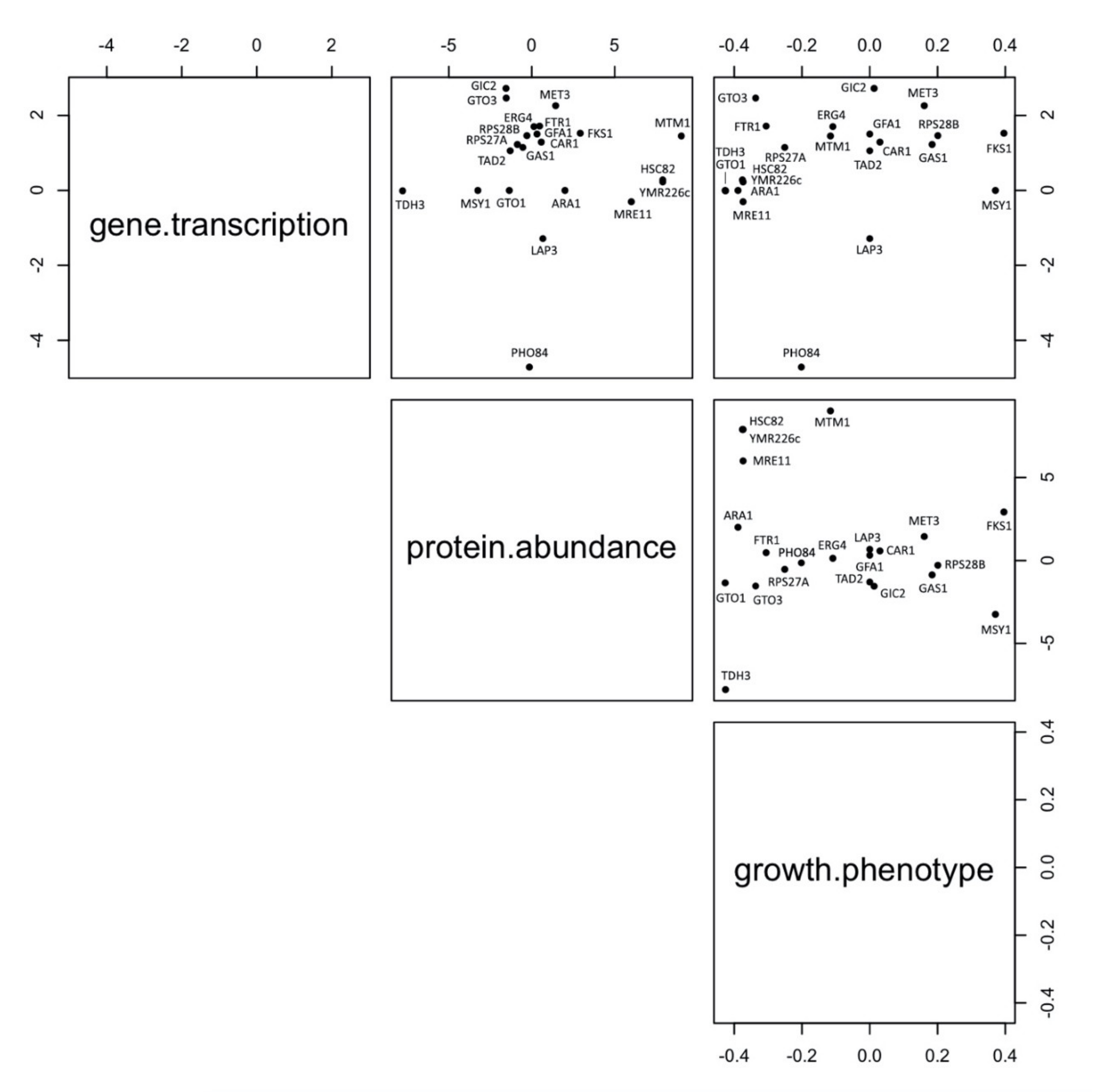
3.7. Robustness of Markers Identification Using Multiomic Approaches
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brumfield, G. Consumer products leap aboard the nano bandwagon. Nature 2006, 440, 262. [Google Scholar] [CrossRef] [PubMed]
- Kagan, C.R.; Fernandez, L.E.; Gogotsi, Y.; Hammond, P.T.; Hersam, M.C.; Nel, A.E.; Penner, R.M.; Willson, C.G.; Weiss, P. Nano Day: Celebrating the next decade of Nanoscience and Nanotechnology. ACS Nano 2016, 10, 9093–9103. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, A. The Maturing Nanotechnology Market: Products and Applications; BCC Research: Wellesley, MA, USA, 2016; pp. 8–20. [Google Scholar]
- Lai, Y.; Wang, L.; Liu, Y.; Yang, G.; Tang, C.; Deng, Y.; Li, S. Immunosensors based on nanomaterials for detection of tumor markers. J. Biomed. Nanotechnol. 2018, 14, 44–65. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.A.; Randall, J.N.; Aggarwal, R.J.; Matyi, R.J.; Moore, T.M.; Wetsel, A.E. Observation of discrete electronic states in a zero-dimensional semiconductor nanostructure. Phys. Rev. Lett. 1988, 60, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Bruchez, M.; Moronne, M.; Gin, P.; Weiss, S.; Alivisatos, A.P. Semiconductor nanocrystals as fluorescent biological labels. Science 1998, 218, 2013–2016. [Google Scholar] [CrossRef]
- Frecker, T.; Bailey, D.; Arzeta-Ferrer, X.; McBride, J.; Rosenthal, S.J. Review-Quantum Dots and their application in lighting, displays, and biology. J. Solid State Technol. 2016, 5, 3019–3031. [Google Scholar] [CrossRef]
- Jun, H.K.; Careem, M.A.; Arof, A.K. Quantum dot-sensitized solar cells-perspective and recent developments: A review of Cd chalcogenide quantum dots as sensitizers. Adv. Mater. Res. Switz. 2013, 22, 48–167. [Google Scholar] [CrossRef]
- Zhai, T.; Fang, X.; Li, L.; Bando, Y.; Golberg, D. One-dimensional CdS nanostructures: Synthesis, properties, and applications. Nanoscale 2010, 2, 168–187. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Kumagai, N.; Iwamoto, S.; Ota, Y.; Arakawa, Y. Laser oscillation in a strongly coupled single-quantum-dot–nanocavity system. Nat. Phys. 2010, 6, 279–283. [Google Scholar] [CrossRef]
- Padmanabhan, P.; Kumar, A.; Kumar, S.; Chaudhary, R.K.; Gulyás, B. Nanoparticles in practice for molecular-imaging applications: An overview. Acta Biomater. 2016, 41, 1–16. [Google Scholar] [CrossRef]
- Benayas, A.; Ren, F.; Carrasco, E.; Marzal, V.; del Rosal, B.; Gonfa, B.A.; Juarranz, A.; Sanz-Rodriguez, F.; Jaque, D.; Garcia-Sole, J.; et al. PbS/CdS/ZnS quantum dots: A multifunctional platform for in vivo nearinfrared low-dose fluorescence imaging. Adv. Funct. Mater. 2015, 25, 6650–6659. [Google Scholar] [CrossRef]
- Foda, M.F.; Huang, L.; Shao, F.; Han, H.Y. Biocompatible and highly luminescent near-infrared CuInS2/ZnS quantum dots embedded silica beads for cancer cell imaging. ACS Appl. Mater. Interfaces 2014, 6, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Volkov, Y. Quantum dots in nanomedicine: Recent trends, advances and unresolved issues. Biochem. Biophys. Res. Commun. 2015, 468, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Gladkovskaya, O.; Gunko, Y.K.; O’Connor, G.M.; Gogvadze, V.; Rochev, Y. In one harness: The interplay of cellular responses and subsequent cell fate after quantum dot uptake. Nanomedicine 2016, 11, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.E.; Smith, A.M.; Nie, S. Quantum dots in biology and medicine. Phys. E Low-Dimens. Syst. Nanostruct. 2004, 25, 1–12. [Google Scholar] [CrossRef]
- Tayo, L.L. Stimuli-responsive nanocarriers for intracellular delivery. Biophys. Rev. 2017, 9, 931–940. [Google Scholar] [CrossRef]
- Ni, X.; Jia, S.; Duan, X.; Ding, D.; Li, K. Fluorescent nanoparticles for noninvasive stem cell tracking in regenerative medicine. J. Biomed. Nanotechnol. 2018, 14, 240–256. [Google Scholar] [CrossRef]
- Oliveira, W.F.; Cabrera, M.P.; Santos, N.R.M.; Napoleão, T.H.; Paiva, P.M.G.; Neves, R.P.; Silva, M.V.; Santos, B.S.; Coelho, L.C.B.B.; Cabral Filho, P.E.; et al. Evaluating glucose and mannose profiles in Candida species using quantum dots conjugated with Cramoll lectin as fluorescent nanoprobes. Microbiol. Res. 2020, 230, 126330. [Google Scholar] [CrossRef]
- Pagano, L.; Caldara, M.; Villani, M.; Zappettini, A.; Marmiroli, N.; Marmiroli, M. In vivo-in vitro comparative toxicology of cadmium sulphide quantum dots in the model organism Saccharomyces cerevisiae. Nanomaterials 2019, 9, 512. [Google Scholar] [CrossRef]
- Paesano, L.; Perotti, A.; Buschini, A.; Carubbi, C.; Marmiroli, M.; Maestri, E.; Iannotta, S.; Marmiroli, N. Markers for toxicity to HepG2 exposed to cadmium sulphide quantum dots; damage to mitochondria. Toxicology 2016, 374, 18–28. [Google Scholar] [CrossRef]
- Majumdar, S.; Ma, C.; Villani, M.; Zuverza-Mena, N.; Pagano, L.; Huangb, Y.; Zappettini, A.; Keller, A.A.; Marmiroli, N.; Parkash Dhankher, O.; et al. Surface coating determines the response of soybean plants to cadmium sulfide quantum dots. Nanoimpact 2019, 14, 100–151. [Google Scholar] [CrossRef]
- Marmiroli, M.; Pagano, L.; Pasquali, F.; Zappettini, A.; Tosato, V.; Bruschi, C.V.; Marmiroli, N. A genome-wide nanotoxicology screen of Saccharomyces cerevisiae mutants reveals the basis for cadmium sulphidequantum dot tolerance and sensitivity. Nanotoxicology 2016, 10, 84–93. [Google Scholar] [PubMed]
- Pasquali, F.; Agrimonti, C.; Pagano, L.; Zappettini, A.; Villani, M.; Marmiroli, M.; White, J.C.; Marmiroli, N. Nucleo-mitochondrial interaction of yeast in response to cadmium sulfide quantum dot exposure. J. Hazard. Mater. 2017, 324, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Tang, M.; Kong, L.; Ying, J.; Wu, T.; Xue, Y.; Pu, Y. Liver toxicity of cadmium telluride quantum dots (CdTe QDs) due to oxidative stress in vitro and in vivo. Int. J. Mol. Sci. 2015, 16, 23279–23299. [Google Scholar] [CrossRef]
- Fan, J.; Sun, Y.; Wang, S.; Li, Y.; Zeng, X.; Cao, Z.; Yang, P.; Song, P.; Wang, Z.; Xian, Z.; et al. Inhibition of autophagy overcomes the nanotoxicity elicited by cadmium-based quantum dots. Biomaterials 2016, 78, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, S.C.; Sá-Correia, I. Yeast toxicogenomics: Lessons from a eukaryotic cell model and cell factory. Curr. Opin. Biotechnol. 2015, 33, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Malloy, T.F. Policy reforms to update chemical safety testing. Science 2017, 355, 1016–1018. [Google Scholar] [CrossRef]
- Zhang, T.; Gaffrey, M.J.; Thrall, B.D.; Qian, W.J. Mass spectrometry-based proteomics for system-level characterization of biological responses to engineered nanomaterials. Anal. Bioanal. Chem. 2018, 410, 6067–6077. [Google Scholar] [CrossRef]
- Scala, G.; Kinareta, P.; Marwah, V.; Sund, J.; Fortino, V.; Greco, D. Multi-omics analysis of ten carbon nanomaterials effects highlights cell type specific patterns of molecular regulation and adaptation. Nanoimpact 2018, 11, 99–108. [Google Scholar] [CrossRef]
- Weng, S.; Dong, Q.; Balakrishnan, R.; Christie, K.; Costanzo, M.; Dolinski, K.; Dwight, S.S.; Engel, S.; Fisk, D.G.; Hong, E.; et al. Saccharomyces Genome Database (SGD) provides biochemical and structural information for budding yeast proteins. Nucleic Acids Res. 2003, 31, 216–218. [Google Scholar] [CrossRef]
- Simon, E.S. Labeling yeast peptides with the iTRAQ reagent. Cold Spring Harb. Protoc. 2011, 2011, 676–680. [Google Scholar] [CrossRef]
- Giaver, G.; Nislow, C. The yeast deletion collection: A decade of functional genomics. Genetics 2014, 197, 451–465. [Google Scholar] [CrossRef]
- Slibinskas, R.; Ražanskas, R.; Zinkevičiūtė, R.; Čiplys, E. Comparison of first dimension IPG and NEPHGE techniques in two-dimensional gel electrophoresis experiment with cytosolic unfolded protein response in Saccharomyces cerevisiae. Proteome Sci. 2013, 11, 2736–2742. [Google Scholar] [CrossRef]
- Ramagli, L.S.; Rodriguez, L.V. Quantitation of microgram amounts of protein in SDS-mercaptoethanol-tris electrophoresis sample buffer. Electrophoresis 1985, 6, 559–563. [Google Scholar] [CrossRef]
- Srivastava, V.; Malm, E.; Sundqvist, G.; Bulone, V. Quantitative proteomics reveals that plasma membrane microdomains from poplar cell suspension cultures are enriched in markers of signal transduction, molecular transport, and callose biosynthesis. Mol. Cell. Proteom. 2013, 12, 3874–3885. [Google Scholar] [CrossRef] [PubMed]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Tabb, D.L.; Fernando, C.G.; Chambers, M.C. MyriMatch: Highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007, 8, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Beavis, R.C. TANDEM: Matching proteins with mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef]
- Shteynberg, D.; Deutsch, E.W.; Lam, H.; Eng, J.K.; Sun, Z.; Tasman, N.; Mendoza, L.; Moritz, R.L.; Aebersold, R.; Nesvizhskii, A.I. iProphet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. ProteinProphet: A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Ross, P.L.; Huang, Y.N.; Marchese, J.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef]
- Ipe, B.I.; Lehnig, M.; Niemeyer, C.M. On the generation of free radical species from quantum dots. Small 2005, 1, 706–709. [Google Scholar] [CrossRef]
- Chen, S.; Ren, Q.; Zhang, J.; Ye, Y.; Zhang, Z.; Xu, Y.; Guo, M.; Ji, H.; Xu, C.; Gu, C.; et al. N-acetyl-L-cysteine protects against cadmium-induced neuronal apoptosis by inhibiting ROS-dependent activation of Akt/mTOR pathway in mouse brain. Neuropathol. Appl. Neurobiol. 2014, 40, 759–777. [Google Scholar] [CrossRef]
- Han, P.P.; Shen, S.G.; Guo, R.J.; Zhao, D.X.; Lin, Y.H.; Jia, S.R.; Yan, R.R.; Wu, Y.K. ROS is a factor regulating the increased polysaccharide production by light quality in the edible Cyanobacterium Nostoc flagelliforme. J. Agric. Food Chem. 2018, 67, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Li, W.; Zhou, Y.; Liu, X.; Zhang, L.; Wang, L.; Li, Y.F.; Iida, A.; Tang, Z.; Zhao, Y.; et al. Full assessment of fate and physiological behavior of Quantum Dots utilizing Caenorhabditis elegans as a model organism. Nano Lett. 2011, 11, 3174–3183. [Google Scholar] [CrossRef] [PubMed]
- Insensera, M.R.; Hernáezb, M.L.; Nombelaa, C.; Molina, M.; Moleroa, G.; Gila, C. Gel and gel-free proteomics to identify Saccharomyces cerevisiae cell surface proteins. J. Proteom. 2010, 73, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, D.J.H.; Geladaki, A.; Breckels, L.M.; Oliver, S.G.; Lilley, K.S. The subcellular organisation of Saccharomyces cerevisiae. Curr. Opin. Chem. Biol. 2019, 48, 86–95. [Google Scholar]
- Pütz, S.M.; Boehm, A.M.; Stiewe, T.; Sickmann, A. iTRAQ Analysis of a cell culture model for malignant transformation, including comparison with 2D-PAGE and SILAC. J. Proteome Res. 2012, 11, 2140–2153. [Google Scholar] [CrossRef]
- Tafelmeyer, P.; Laurent, C.; Lenormand, P.; Rousselle, J.-C.; Marsollier, L.; Reysset, G.; Zhang, R.; Sickmann, A.; Stinear, T.P.; Namane, A.; et al. Comprehensive proteome analysis of Mycobacterium ulceransand quantitative comparison of mycolactone biosynthesis. Proteomics 2008, 8, 3124–3138. [Google Scholar] [CrossRef]
- Radosevich, T.J.; Reinhardt, T.A.; Lippolis, J.D.; Bannantine, J.P.; Stabel, J.R. Proteome and differential expression analysis of membrane and cytosolic proteins from Mycobacterium avium subsp. paratuberculosis strains K-10 and 187. J. Bacteriol. 2007, 189, 1109–1117. [Google Scholar] [CrossRef][Green Version]
- Lee, Y.J.; Jang, J.W.; Kim, K.J.; Maeng, P.J. TCA cycle-independent acetate metabolism via the glyoxylate cycle in Saccharomyces cerevisiae. Yeast 2011, 28, 153–166. [Google Scholar] [CrossRef]
- Kaplan, R.S.; Mayor, J.A.; Kakhniashvili, D.; Gremse, D.A.; Wood, D.O.; Nelson, D.R. Deletion of the nuclear gene encoding the mitochondrial citrate transport protein from Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1996, 226, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Huo, D.; Li, H.; Zhang, B.; Li, Y.; Liang, A.; Wang, H.; Yu, Q.; Li, M. ROS-independent toxicity of Fe3O4 nanoparticles to yeast cells: Involvement of mitochondrial dysfunction. Chem. Biol. Interact. 2018, 287, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.A.; Soares, H.V.M.; Soares, E.V. Nickel Oxide (NiO) Nanoparticles induce loss of cell viability in yeast mediated by oxidative stress. Chem. Res. Toxicol. 2018, 31, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.; Sengupta, J.; Datta, P.; Ghosh, S.; Gomes, A. Physiological interactions of nanoparticles in energy metabolism, immune function and their biosafety: A review. J. Nanosci. Nanotechnol. 2016, 16, 92–116. [Google Scholar] [CrossRef]
- Horstmann, C.; Kim, D.S.; Campbell, C.; Kim, K. Transcriptome Profile alteration with cadmium selenide/Zinc sulfide quantum dots in Saccharomyces cerevisiae. Biomolecules 2019, 9, 653. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Ray, M.; Das, M.R.; Chakrabarti, A.; Khan, A.H.; Sarma, D.D.; Acharya, S. Modulation of glyceraldehyde-3-phosphate dehydrogenase activity by surface functionalized quantum dots. Phys. Chem. Chem. Phys. 2014, 16, 5276–5283. [Google Scholar] [CrossRef]
- Ruotolo, R.; Pira, G.; Villani, M.; Zappettini, A.; Marmiroli, N. Ring-shaped corona proteins influence the toxicity of engineered nanoparticles to yeast. Environ. Sci. Nano 2018, 5, 1428–1440. [Google Scholar] [CrossRef]
- Willmund, F.; del Alamo, M.; Pechmann, S.; Chen, T.; Albanese, V.; Dammer, E.B. The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell 2013, 152, 196–209. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Albanese, V.; Frydman, J.; Xia, Y.; McClellan, A.J. Heterozygous yeast deletion collection screens reveal essential targets of Hsp90. PLoS ONE 2011, 6, e28211. [Google Scholar] [CrossRef]
- Sundarraj, K.; Raghunath, A.; Panneerselvam, L.; Perumal, E. Iron oxide nanoparticles modulate heat shock proteins and organ specific markers expression in mice male accessory organs. Toxicol. Appl. Pharmacol. 2017, 317, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Wang, Y.; Zewei, L.Z.; Li, Y.; Duan, Y. New findings of silica nanoparticles induced ER autophagy in human colon cancer cell. Sci. Rep. 2017, 7, 42591. [Google Scholar] [CrossRef]
- Schütz, I.; Lopez-Hernandez, T.; Gao, Q.; Puchkov, D.; Jabs, S.; Nordmeyer, D.; Schmudde, M.; Rühl, E.; Graf, C.M.; Haucke, V. Lysosomal dysfunction caused by cellular accumulation of silica nanoparticles. J. Biol. Chem. 2016, 291, 14170–14184. [Google Scholar] [CrossRef]
- Lin, L.; Xu, M.; Mu, H.; Wang, W.; Sun, J.; He, J.; Qiu, J.W.; Luan, T. Quantitative proteomic analysis to understand the mechanisms of Zinc Oxide Nanoparticle toxicity to Daphnia pulex (Crustacea: Daphniidae): Comparing with Bulk Zinc Oxide and Zinc salt. Environ. Sci. Technol. 2019, 7, 5436–5444. [Google Scholar] [CrossRef] [PubMed]
- Paesano, L.; Marmiroli, M.; Bianchi, M.G.; White, J.C.; Bussolati, O.; Zappettini, A.; Villani, M.; Marmiroli, N. Differences in toxicity, mitochondrial function and miRNome in human cells exposed in vitro to Cd as CdS quantum dots or ionic Cd. J. Hazard. Mater. 2020, 393, 122430. [Google Scholar] [CrossRef]
- Cho, S.J.; Maysinger, D.; Jain, M.; Roder, B.; Hackbarth, S.; Winnik, F.M. Long-term exposure to CdTe quantum dots causes functional impairments in live cells. Langmuir 2007, 23, 1974–1980. [Google Scholar] [CrossRef] [PubMed]
- Katubi, K.M.; Alzahrani, F.M.; Ali, D.; Alarifi, S. Dose- and duration-dependent cytotoxicity and genotoxicity in human hepato carcinoma cells due to CdTe QDs exposure. Hum. Exp. Toxicol. 2019, 38, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Lopes Rocha, T.; Gomes, T.; Serrão Sousa Nélia, V.; Mestre, C.; João Bebianno, M. Ecotoxicological impact of engineered nanomaterials in bivalve molluscs: An overview. Mar. Environ. Res. 2015, 111, 74–88. [Google Scholar] [CrossRef]
- Pereira, L.C.; Pazin, M.; Franco-Bernardes, M.F.; Martins, A.D.C.; Barcelos, G.R.M.; Pereira, M.C.; Mesquita, J.P.; Rodrigues, J.L.; Barbosa, F.; Dorta, D.J. A perspective of mitochondrial dysfunction in rats treated with silver and titanium nanoparticles (AgNPs and TiNPs). J. Trace Elem. Med. Biol. 2018, 47, 63–69. [Google Scholar] [CrossRef]
- Jiang, L.; Gulanski, B.I.; De Feyter, H.M.; Weinzimer, S.A.; Pittman, B.; Guidone, E.; Koretski, J.; Harman, S.; Petrakis, I.L.; Krystal, J.H.; et al. Increased brain uptake and oxidation of acetate in heavy drinkers. J. Clin. Investig. 2013, 123, 1605–1614. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Simões, A.M.; Duarte, F.V.; Rolo, A.P.; Murdoch, R.C.; Hussain, S.M.; Palmeira, C.M. Assessment of the toxicity of silver nanoparticles in vitro: A mitochondrial perspective. Toxicol. In Vitro 2011, 25, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Jarak, I.; Carrola, J.; Barros, A.S.; Gil, A.M.; Pereira, M.D.L.; Corvo, M.L.; Duarte, I.F. From the cover: Metabolism modulation in different organs by silver nanoparticles: An NMR metabolomics study of a mouse model. Toxicol. Sci. 2017, 159, 422–435. [Google Scholar] [CrossRef] [PubMed]
- McShan, D.; Ray, P.C.; Yu, H. Molecular toxicity mechanism of nanosilver. J. Food Drug Anal. 2014, 22, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Márquez, I.G.; Ghiyasvand, M.; Massarsky, A.; Babu, M.; Samanfar, B.; Omidi, K.; Moon, T.W.; Smith, M.L.; Golshani, A. Zinc oxide and silver nanoparticles toxicity in the baker’s yeast, Saccharomyces cerevisiae. PLoS ONE 2018, 13, e0193111. [Google Scholar] [CrossRef]
- Marmiroli, M.; Imperiale, D.; Pagano, L.; Villani, M.; Zappettini, A.; Marmiroli, N. The proteomic response of Arabidopsis thaliana to cadmium sulfide quantum dots, and its correlation with the transcriptomic response. Front. Plant Sci. 2015, 6, 1104. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallo, V.; Srivastava, V.; Bulone, V.; Zappettini, A.; Villani, M.; Marmiroli, N.; Marmiroli, M. Proteomic Analysis Identifies Markers of Exposure to Cadmium Sulphide Quantum Dots (CdS QDs). Nanomaterials 2020, 10, 1214. https://doi.org/10.3390/nano10061214
Gallo V, Srivastava V, Bulone V, Zappettini A, Villani M, Marmiroli N, Marmiroli M. Proteomic Analysis Identifies Markers of Exposure to Cadmium Sulphide Quantum Dots (CdS QDs). Nanomaterials. 2020; 10(6):1214. https://doi.org/10.3390/nano10061214
Chicago/Turabian StyleGallo, Valentina, Vaibhav Srivastava, Vincent Bulone, Andrea Zappettini, Marco Villani, Nelson Marmiroli, and Marta Marmiroli. 2020. "Proteomic Analysis Identifies Markers of Exposure to Cadmium Sulphide Quantum Dots (CdS QDs)" Nanomaterials 10, no. 6: 1214. https://doi.org/10.3390/nano10061214
APA StyleGallo, V., Srivastava, V., Bulone, V., Zappettini, A., Villani, M., Marmiroli, N., & Marmiroli, M. (2020). Proteomic Analysis Identifies Markers of Exposure to Cadmium Sulphide Quantum Dots (CdS QDs). Nanomaterials, 10(6), 1214. https://doi.org/10.3390/nano10061214