CXCL12-PLGA/Pluronic Nanoparticle Internalization Abrogates CXCR4-Mediated Cell Migration

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Nanoparticle Preparation and Characterization
2.1.1. Materials
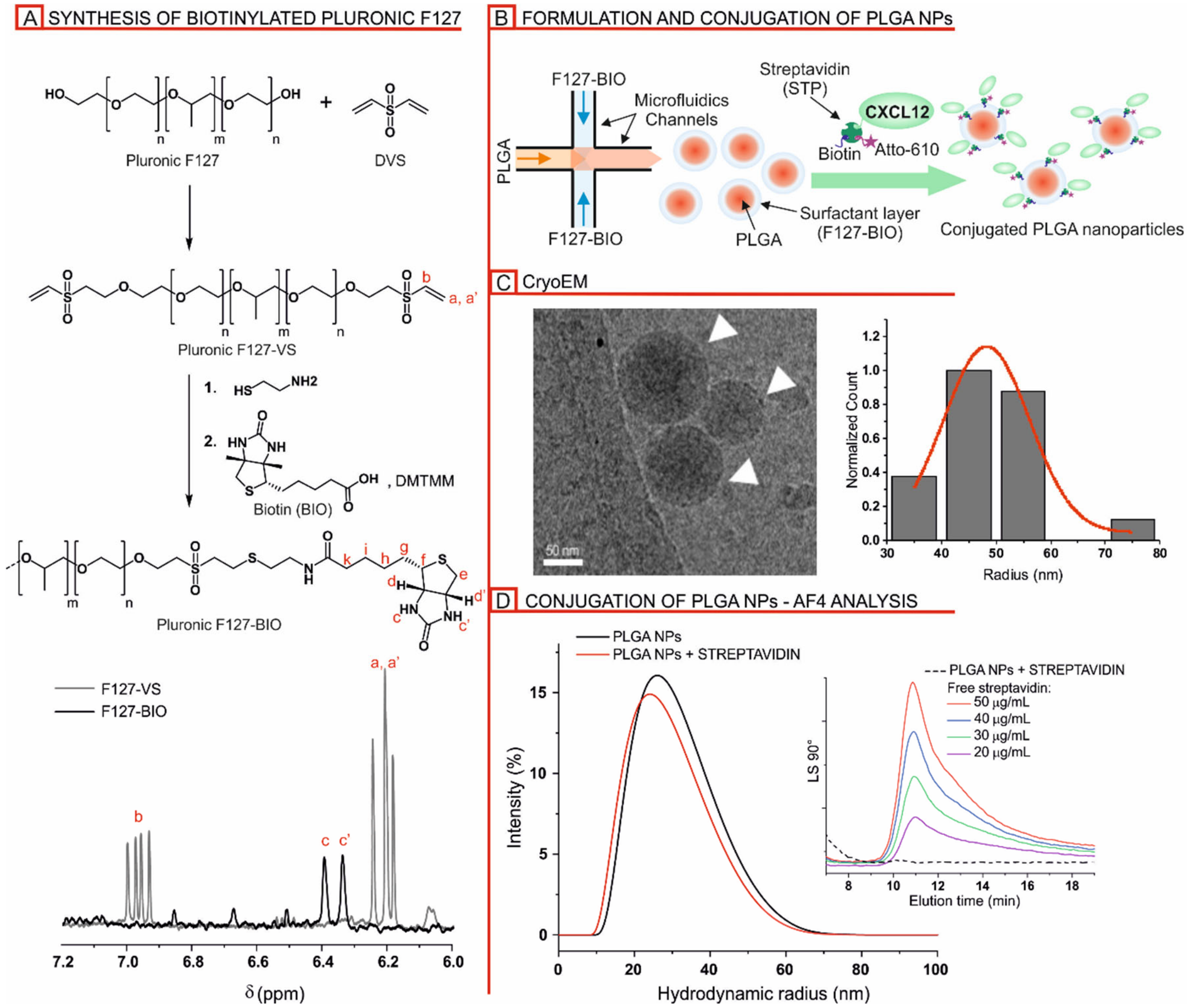
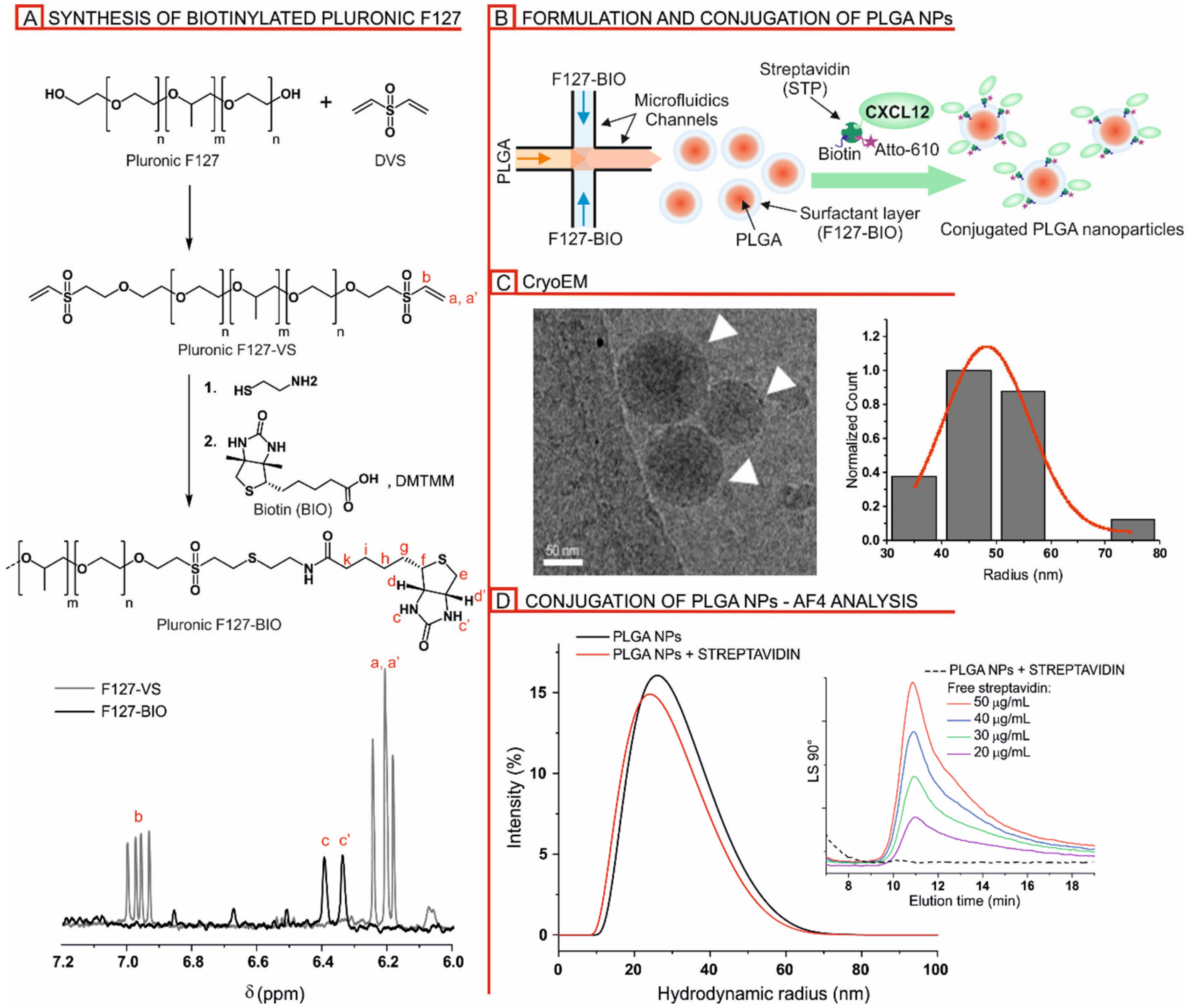
2.1.2. Preparation of Pluronic F127-Biotin (F127-BIO)
2.1.3. Nanoparticle Preparation
2.1.4. Nanoparticle Decoration
- (a)
- Control NPs—10 μL of a 100 μM solution of Atto 610-biotin (Sigma-Aldrich, Saint Louis, MO, USA) in DMSO (equivalent to 1 nmol) was mixed with 290 µL of 7 μM solution of biotin in MilliQ water (equivalent to 2 nmol), followed by 50 μL of streptavidin reconstituted at 1 mg/mL, corresponding to 1 nmol of protein units. The procedure was allowed to proceed for 15 min. The mixture was then added to 650 µL of the 0.052 wt.% nanoparticle dispersion (biotin content = 2 nmol), vortexed for 10 s and allowed to rest for 15 min.
- (b)
- CXCL12-decorated NPs—10 μL of a 100 μM solution of Atto 610-biotin in DMSO (equivalent to 1 nmol) was mixed with 280 µL of 7 μM solution of biotin in MilliQ water (equivalent to 1.96 nmol), and 10 μL of a 10 μM solution of biotinylated human CXCL12 in MilliQ water (equivalent to 0.1 nmol) (Chemotactics, San Diego, CA, USA), followed by 50 μL of streptavidin (Prospec, Rehovot, Israel) reconstituted at 1 mg/mL, corresponding to 1 nmol of protein units. The procedure was allowed to proceed for 15 min. The mixture was then added to 650 µL of the 0.052 wt.% nanoparticle dispersion (biotin content = 2 nmol), vortexed for 10 s and allowed to rest for 15 min.
2.1.5. Cryogenic Transmission Electron Microscopy (Cryo-EM)
2.1.6. Asymmetric Flow Field-Flow Fractionation (AF4)
2.1.7. Dynamic Light Scattering (DLS)
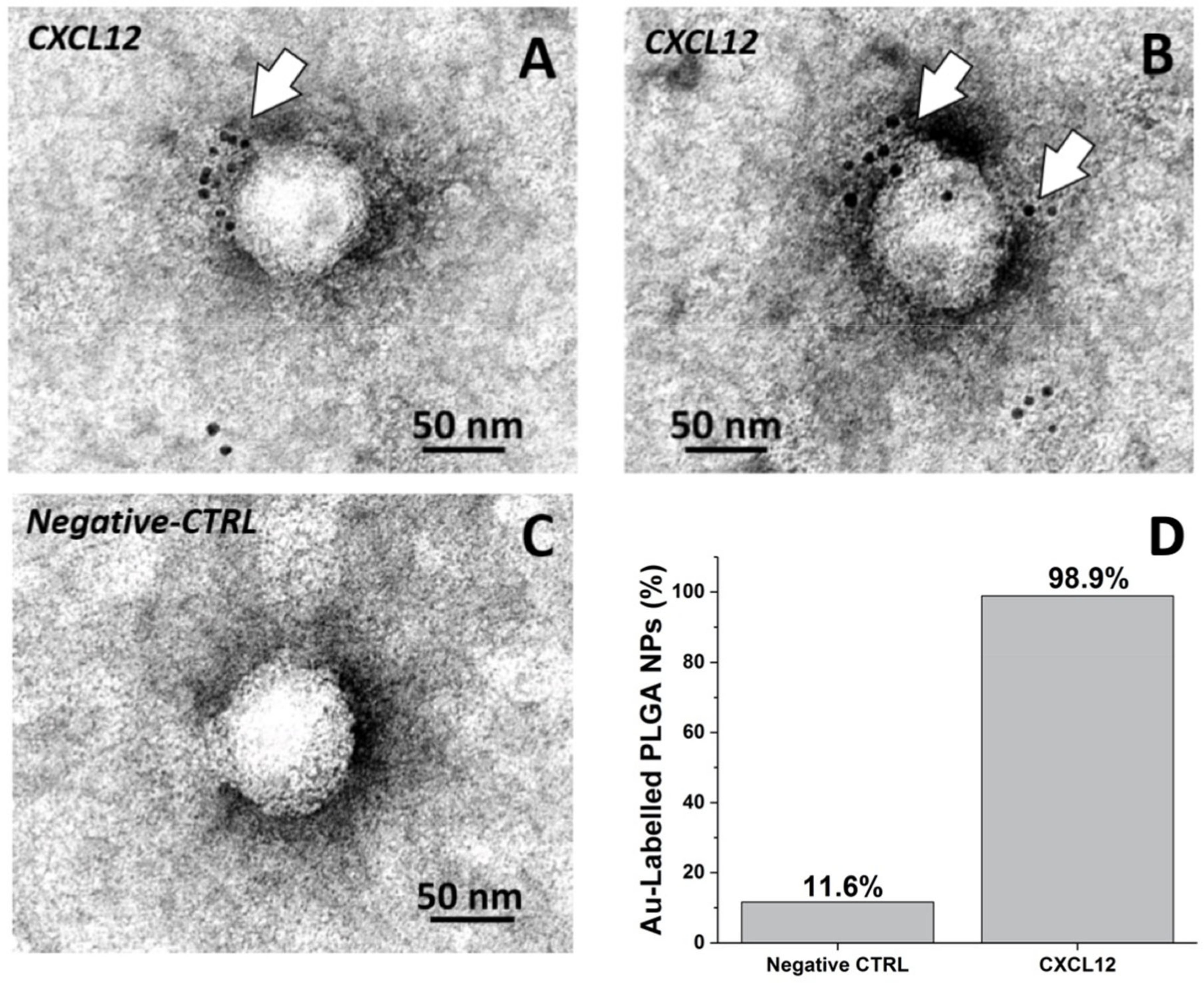
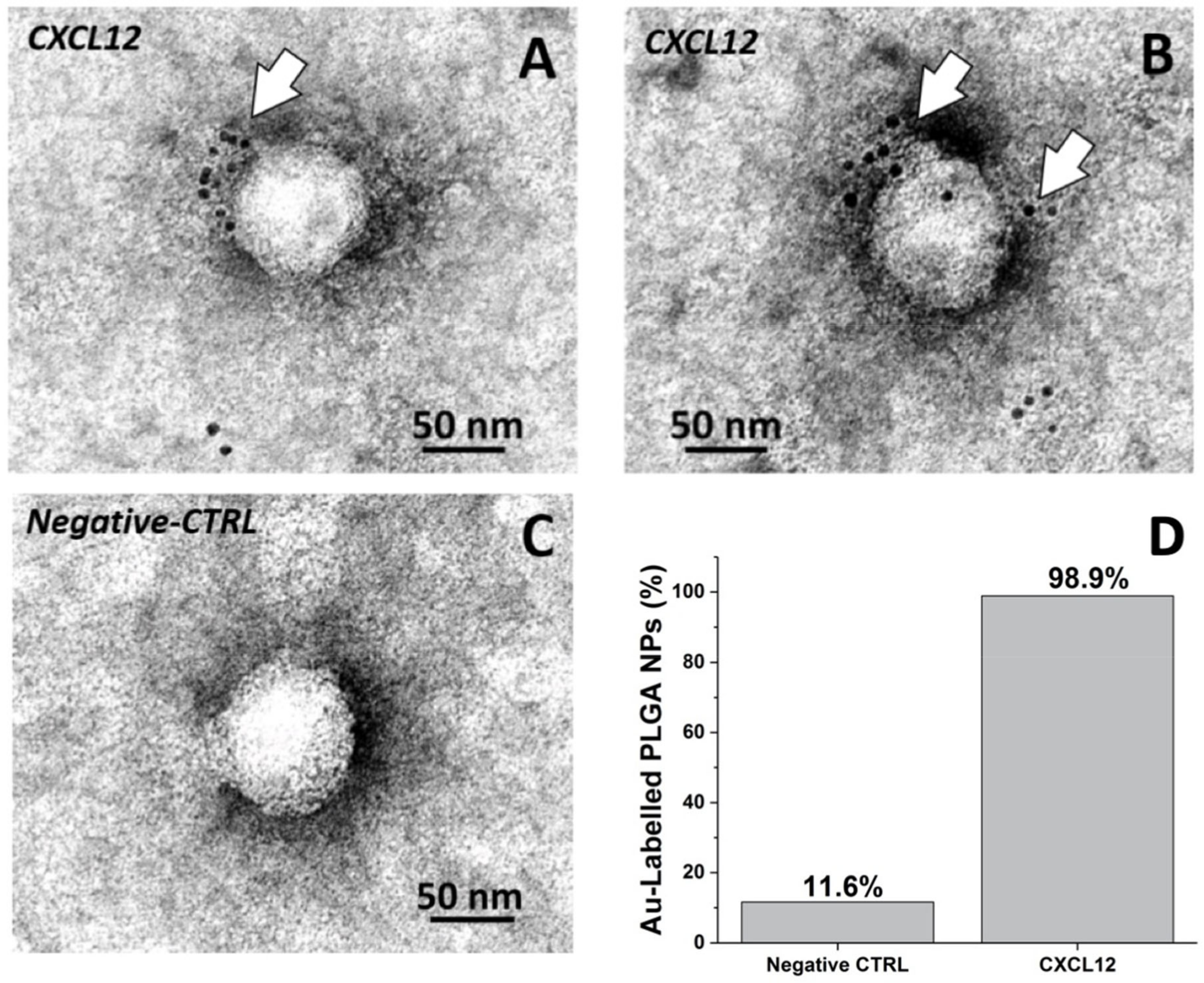
2.1.8. Negative Staining EM Immunolabeling
2.2. Cell Culture
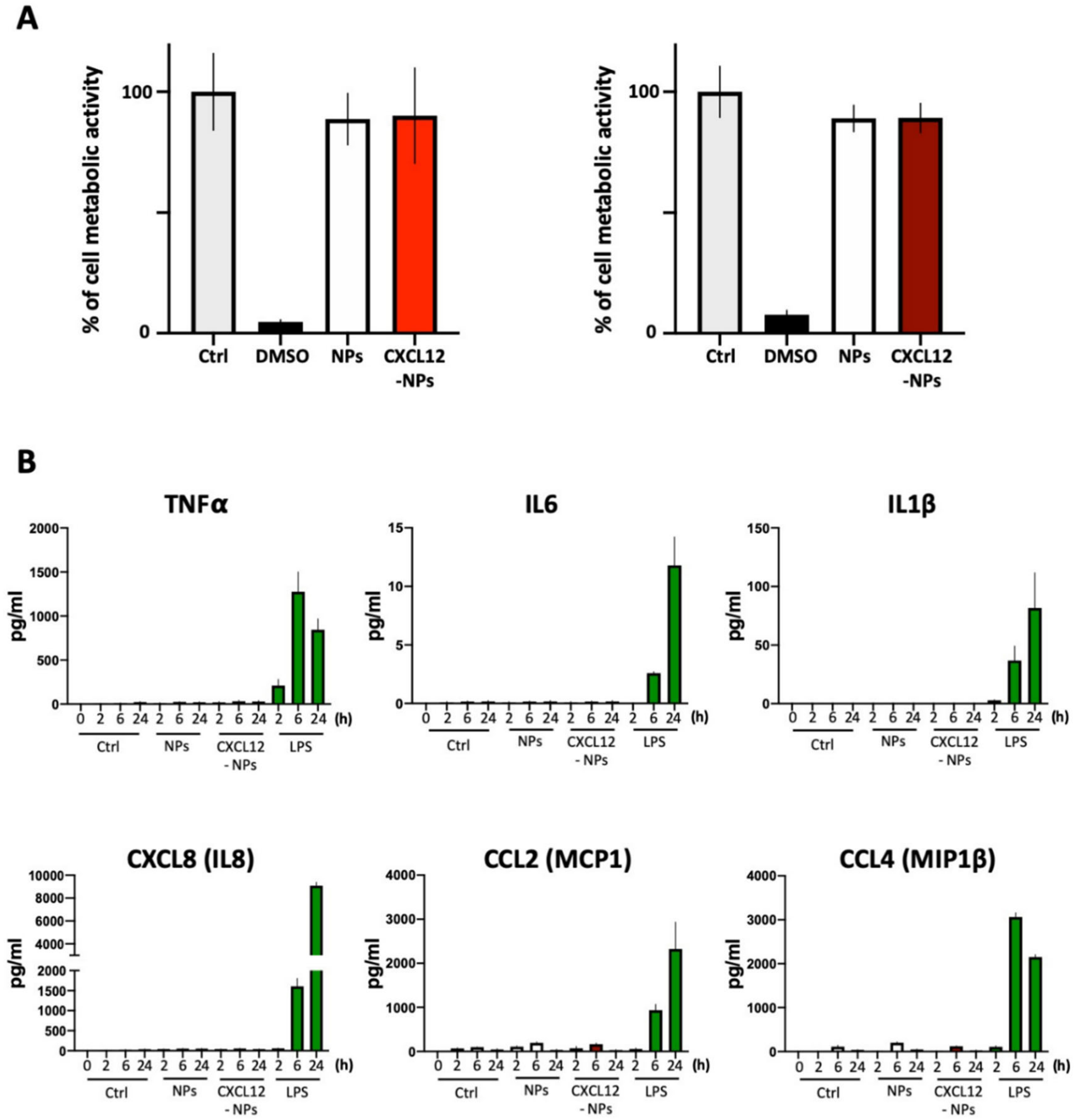
2.3. Toxicity Assay
2.4. Cytokine Release
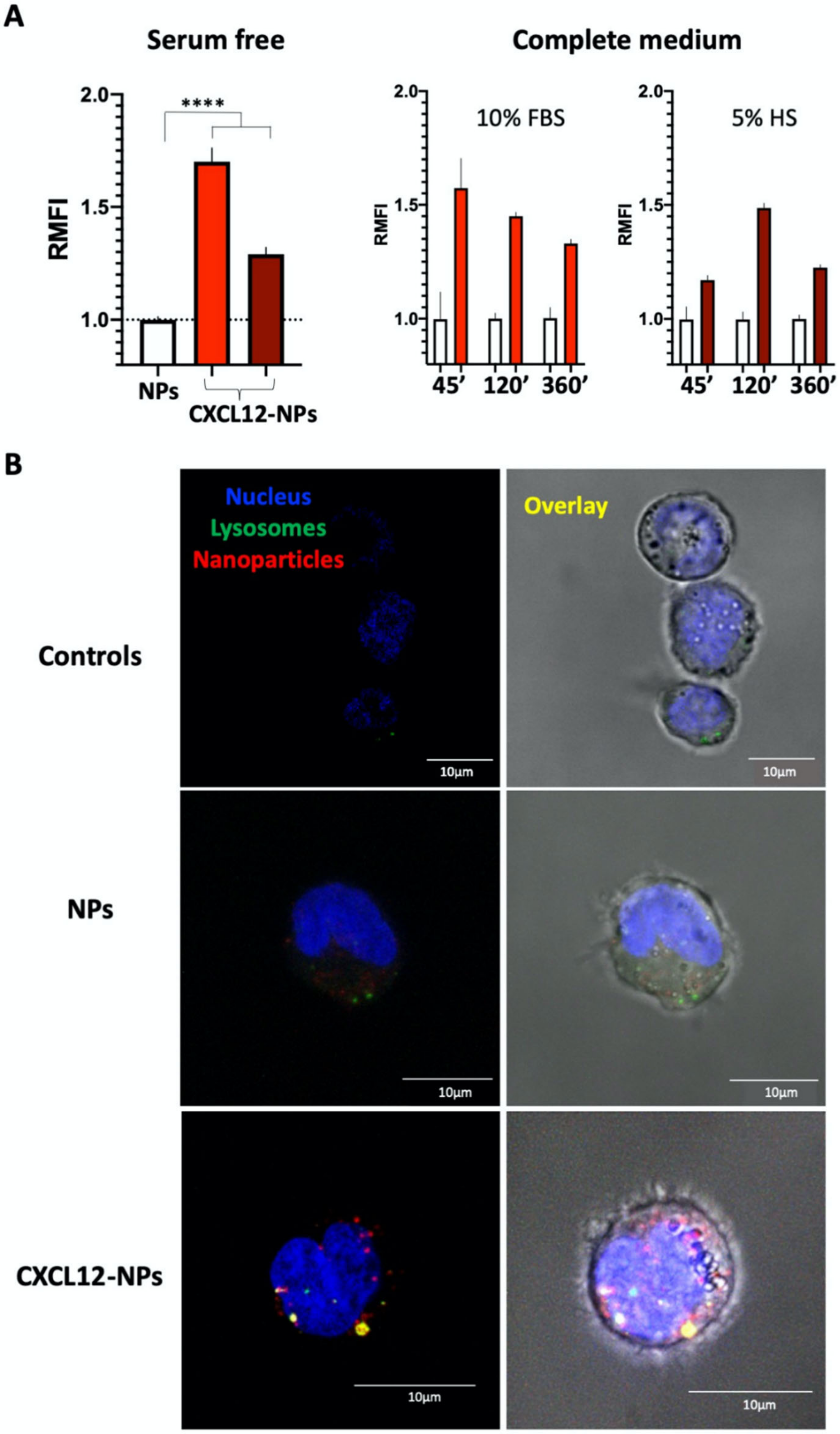
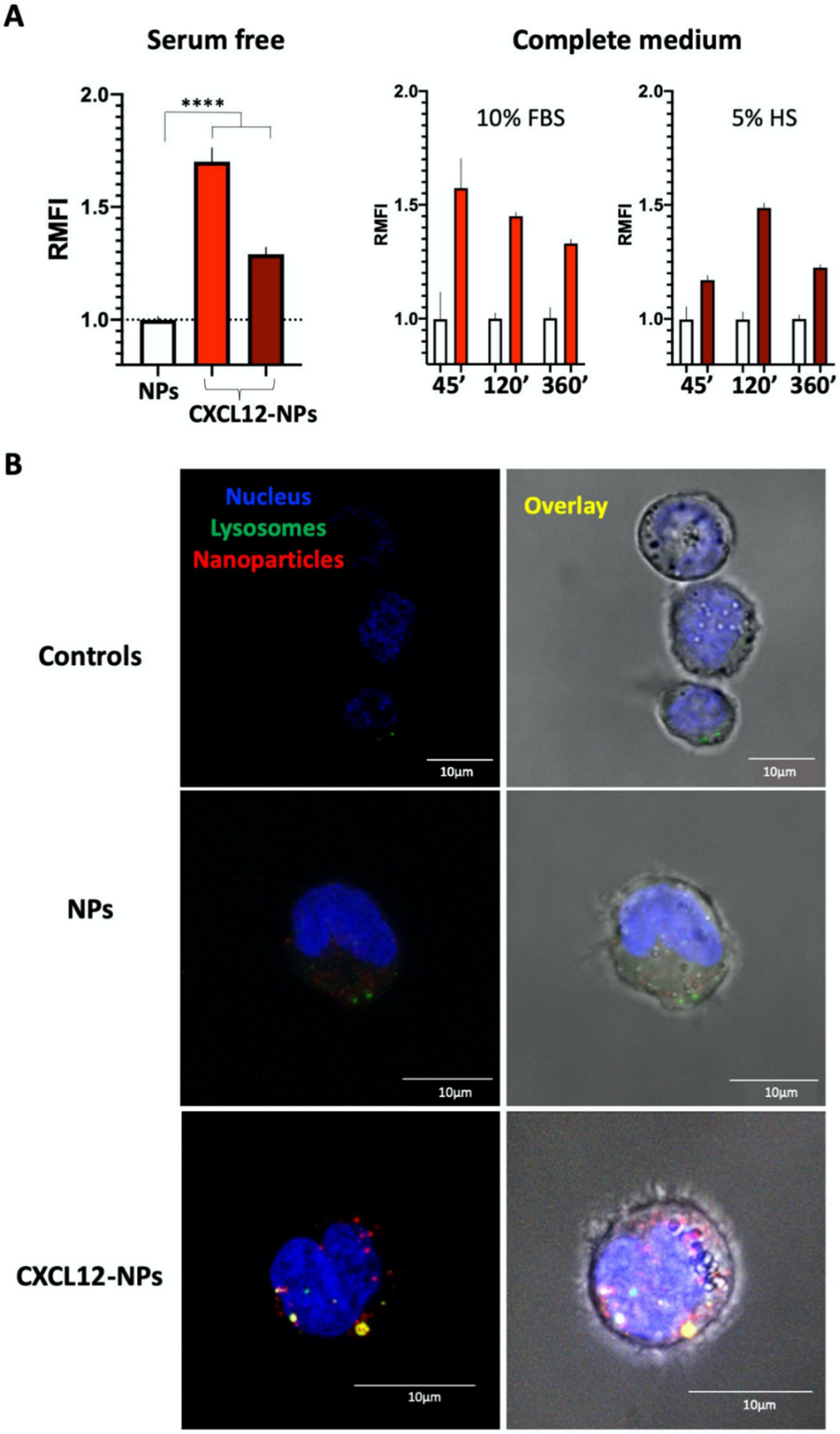
2.5. Cellular Uptake of Nanoparticles
2.5.1. CXCL12 NPs Internalization in THP-1 Cells Analysis
2.5.2. CXCR4 Expression
2.5.3. Confocal Microscopy
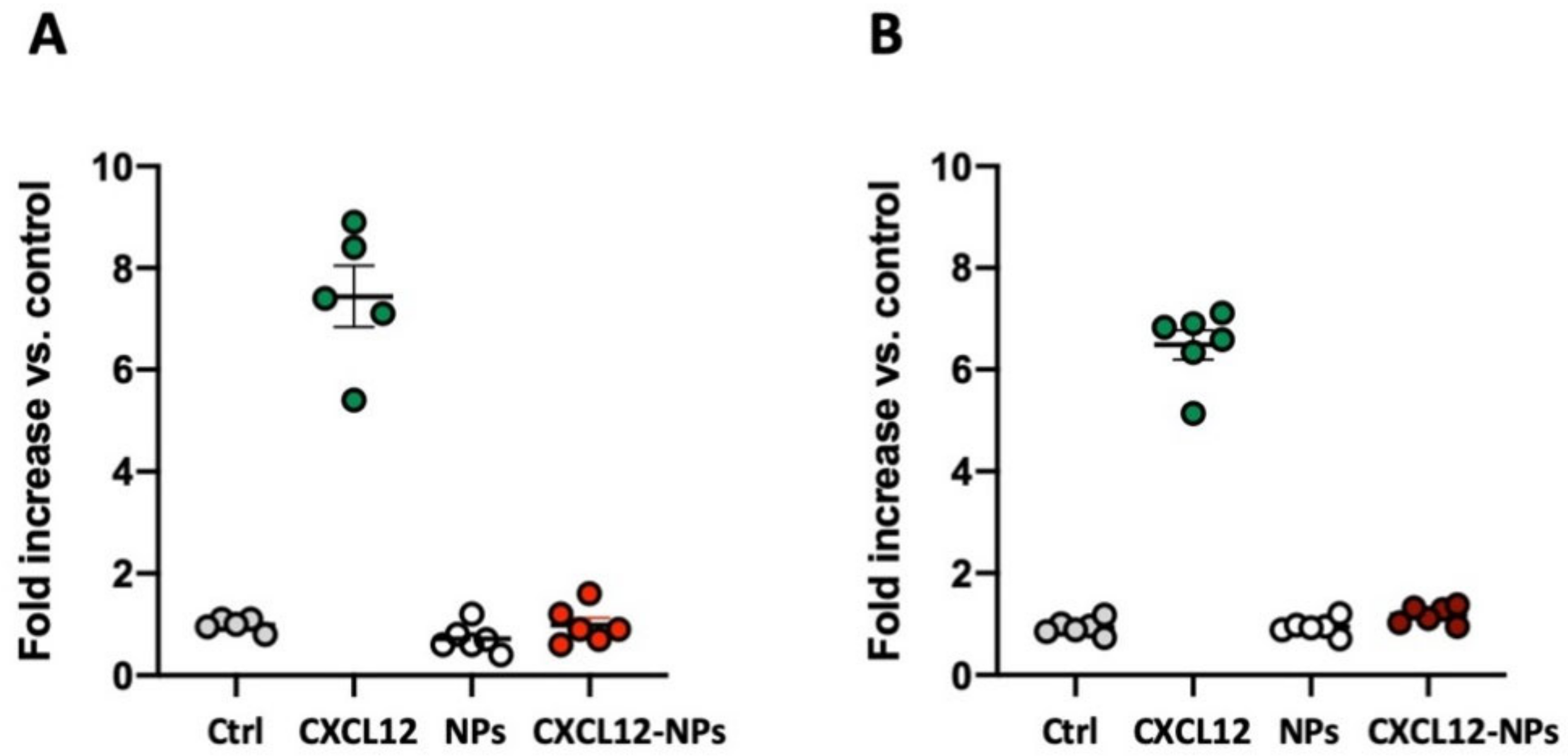
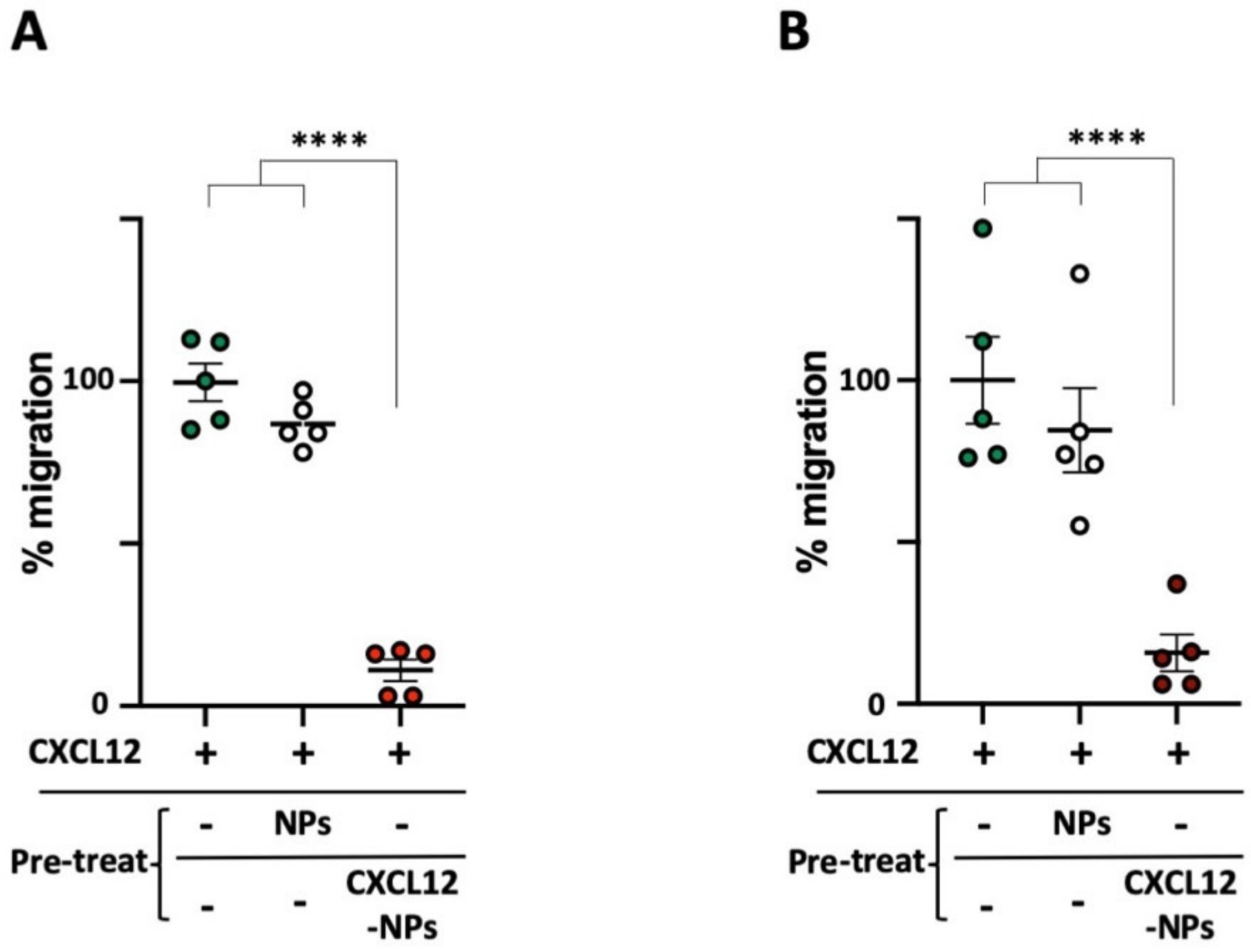
2.6. Cell Migration Assay
2.7. Statistical Analysis
3. Results
3.1. Nanoparticle Sysnthesis and Physicochemical Characterization
3.1.1. Formulation and Characterization of Biotinylated PLGA/Pluronic Nanoparticles
3.1.2. Streptavidin-Mediated Decoration of Biotinylated PLGA/Pluronic Nanoparticles
3.2. Detection of CXCL12 on the Nanoparticle and CXCL12-NP Characterization
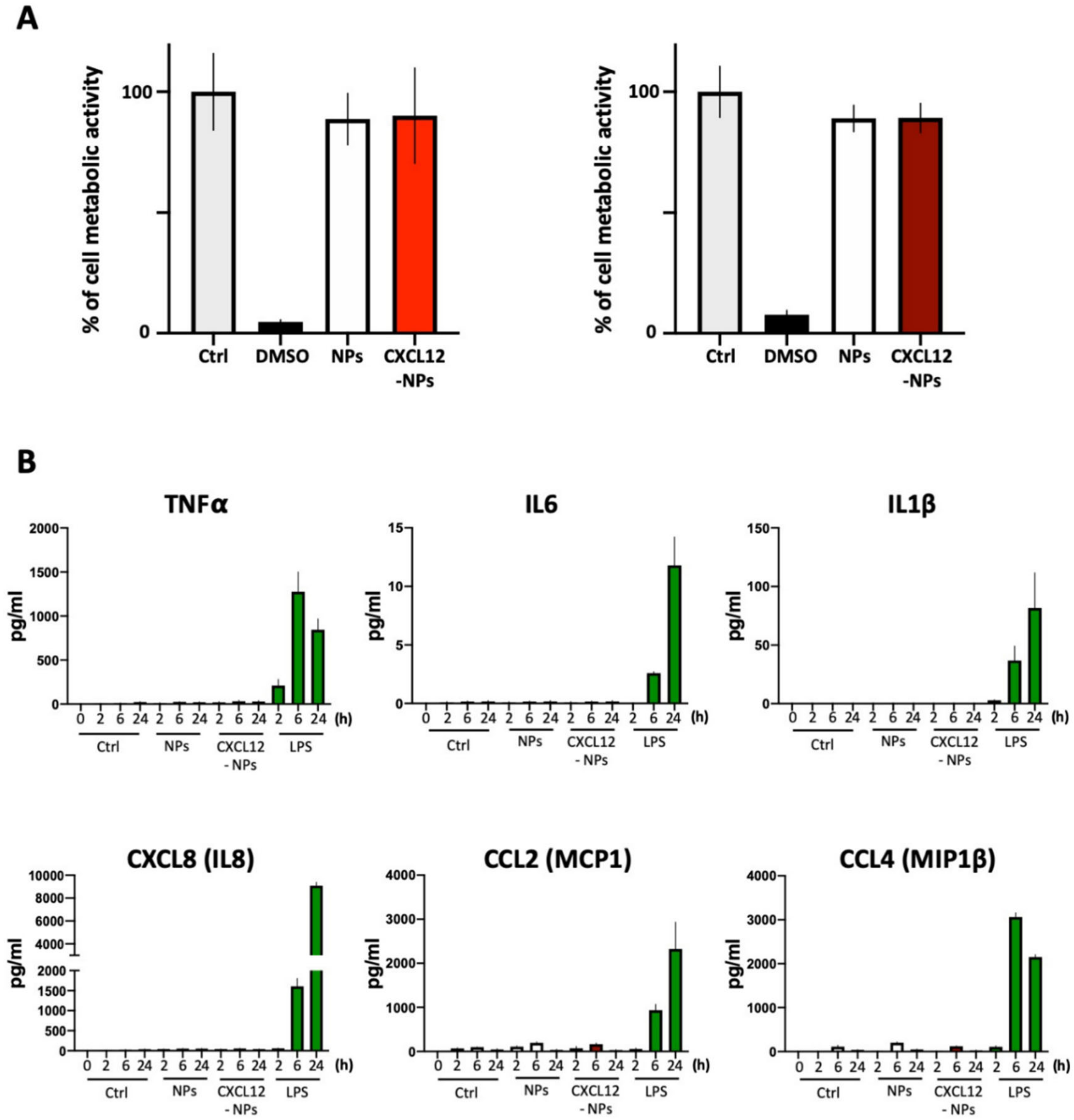
3.3. Biocompatibility of CXCL12-NPs
3.3.1. Toxicity
3.3.2. Inflammatory Cytokine Release
3.4. CXCL12-NP Internalization in THP-1 Cells
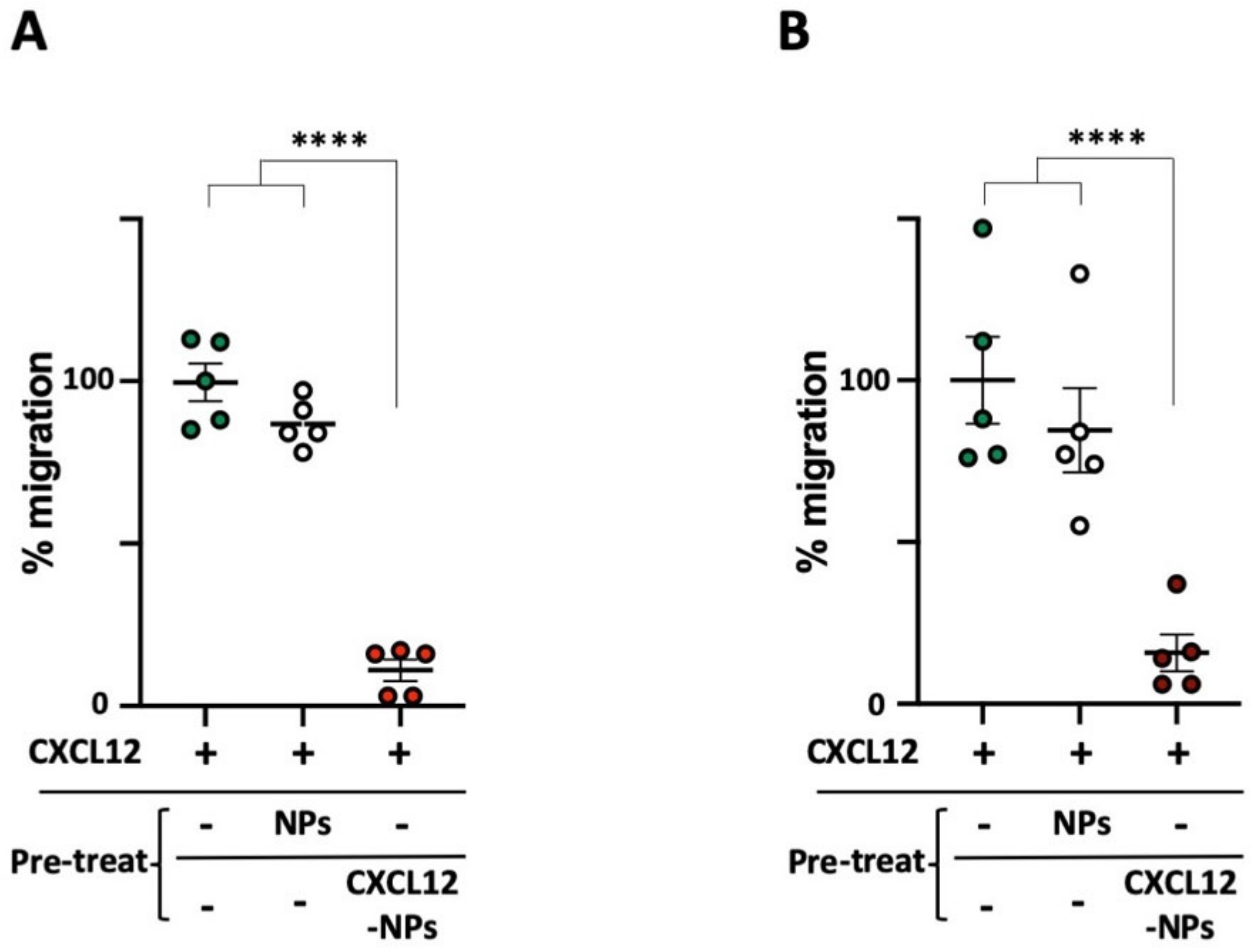
3.5. CXCL12-NPs Do Not Induce THP-1 Migration
3.6. CXCL12-NPs Prevent CXCR4 Mediated Cell Migration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [Green Version]
- Zlotnik, A.; Yoshie, O. The Chemokine Superfamily Revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.R. The chemokine system and cancer. J. Pathol. 2012, 226, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, Y.; Oupický, D. Potential of CXCR4/CXCL12 Chemokine Axis in Cancer Drug Delivery. Curr. Pharmacol. Reports 2016, 2, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, W.; Xue, S.; Chen, Y. The role of CXCL12 in tumor microenvironment. Gene 2018, 641, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Gao, L.; Luan, J.; Zhang, H.; Xiao, T. C-X-C Chemokine Receptor 4 in Diffuse Large B Cell Lymphoma: Achievements and Challenges. Acta Haematol. 2019, 142, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, J.; Feng, Y.; Tian, X.; Wang, B.; Zhou, Y. Oncogenic roles and drug target of CXCR4/CXCL12 axis in lung cancer and cancer stem cell. Tumor Biol. 2016, 37, 8515–8528. [Google Scholar] [CrossRef]
- Xue, L.J.; Mao, X.B.; Ren, L.L.; Chu, X.Y. Inhibition of CXCL12/CXCR4 axis as a potential targeted therapy of advanced gastric carcinoma. Cancer Med. 2017, 6, 1424–1436. [Google Scholar] [CrossRef]
- Mitchell, B.; Mahalingam, M. The CXCR4/CXCL12 axis in cutaneous malignancies with an emphasis on melanoma. Histol. Histopathol. 2014, 29, 1539–1546. [Google Scholar] [CrossRef]
- Xu, C.; Zhao, H.; Chen, H.; Yao, Q. CXCR4 in breast cancer: Oncogenic role and therapeutic targeting. Drug Des. Devel. Ther. 2015, 9, 4953–4964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Wei, Y.; Xia, J.; Wang, S.; Wu, J.; Chen, F.; Huang, G.; Chen, J. CXCL12-CXCR4 contributes to the implication of bone marrow in cancer metastasis. Future Oncol. 2014, 10, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.P.; Gong, J.H.; Loetscher, P.; Rajarathnam, K.; Amara, A.; Arenzana-Seisdedos, F.; Virelizier, J.L.; Baggiolini, M.; Sykes, B.D.; Clark-Lewis, I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997, 16, 6996–7007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajczak, M.Z.; Zuba-Surma, E.; Kucia, M.; Reca, R.; Wojakowski, W.; Ratajczak, J. The pleiotropic effects of the SDF-1-CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia 2006, 20, 1915–1924. [Google Scholar] [CrossRef] [Green Version]
- Noda, M.; Omatsu, Y.; Sugiyama, T.; Oishi, S.; Fujii, N.; Nagasawa, T. CXCL12-CXCR4 chemokine signaling is essential for NK-cell development in adult mice. Blood 2011, 117, 451–458. [Google Scholar] [CrossRef] [Green Version]
- Mandal, M.; Okoreeh, M.K.; Kennedy, D.E.; Maienschein-Cline, M.; Ai, J.; McLean, K.C.; Kaverina, N.; Veselits, M.; Aifantis, I.; Gounari, F.; et al. CXCR4 signaling directs Igk recombination and the molecular mechanisms of late B lymphopoiesis. Nat. Immunol. 2019, 20, 1393–1403. [Google Scholar] [CrossRef]
- Qing, M.; Jones, D.; Springer, T.A. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity 1999, 10, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Mithal, D.S.; Banisadr, G.; Miller, R.J. CXCL12 signaling in the development of the nervous system. J. Neuroimmune Pharmacol. 2012, 7, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Wang, X.; Xia, Z.; Chung, S.K.; Cheung, C.W. CXCL12/CXCR4 axis: An emerging neuromodulator in pathological pain. Rev. Neurosci. 2016, 27, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huang, J.; Li, Y.; Yang, G.-Y. Roles of Chemokine CXCL12 and its Receptors in Ischemic Stroke. Curr. Drug Targets 2012, 13, 166–172. [Google Scholar] [CrossRef]
- Rogers, T.J. Bidirectional Regulation of Opioid and Chemokine Function. Front. Immunol. 2020, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaik, M.M.; Peng, H.; Lu, J.; Rits-Volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 2019, 565, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Baribaud, F.; Romano, J.; Doms, R.W.; Hoxie, J.A. Identification of gp120 Binding Sites on CXCR4 by Using CD4-Independent Human Immunodeficiency Virus Type 2 Env Proteins. J. Virol. 2003, 77, 931–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Silva, E.; Stumpf, M.P.H. HIV and the CCR5-Δ32 resistance allele. FEMS Microbiol. Lett. 2004, 241, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, K.; Harriague, J.; Décrion, C.; Lagane, B.; Shorte, S.; Baleux, F.; Virelizier, J.-L.; Arenzana-Seisdedos, F.; Chakrabarti, L.A. CXCR4-Tropic HIV-1 Envelope Glycoprotein Functions as a Viral Chemokine in Unstimulated Primary CD4 + T Lymphocytes. J. Immunol. 2004, 173, 7150–7160. [Google Scholar] [CrossRef] [Green Version]
- Bardi, G.; Sengupta, R.; Khan, M.Z.; Patel, J.P.; Meucci, O. Human immunodeficiency virus gp120-induced apoptosis of human neuroblastoma cells in the absence of CXCR4 internalization. J. Neurovirol. 2006, 12, 211–218. [Google Scholar] [CrossRef]
- Murakami, T.; Zhang, T.-Y.; Koyanagi, Y.; Tanaka, Y.; Kim, J.; Suzuki, Y.; Minoguchi, S.; Tamamura, H.; Waki, M.; Matsumoto, A.; et al. Inhibitory Mechanism of the CXCR4 Antagonist T22 against Human Immunodeficiency Virus Type 1 Infection. J. Virol. 1999, 73, 7489–7496. [Google Scholar] [CrossRef] [Green Version]
- Unzueta, U.; Céspedes, M.V.; Ferrer-Miralles, N.; Casanova, I.; Cedano, J.; Corchero, J.L.; Domingo-Espín, J.; Villaverde, A.; Mangues, R.; Vázquez, E. Intracellular CXCR4+ cell targeting with T22-empowered protein-only nanoparticles. Int. J. Nanomed. 2012, 7, 4533–4544. [Google Scholar] [CrossRef] [Green Version]
- Sala, R.; Sánchez-García, L.; Serna, N.; Céspedes, M.V.; Casanova, I.; Roldán, M.; Sánchez-Chardi, A.; Unzueta, U.; Vázquez, E.; Mangues, R.; et al. Collaborative membrane activity and receptor-dependent tumor cell targeting for precise nanoparticle delivery in CXCR4+ colorectal cancer. Acta Biomater. 2019, 99, 426–432. [Google Scholar] [CrossRef]
- Díaz, R.; Pallarès, V.; Cano-Garrido, O.; Serna, N.; Sánchez-García, L.; Falgàs, A.; Pesarrodona, M.; Unzueta, U.; Sánchez-Chardi, A.; Sánchez, J.M.; et al. Selective CXCR4+ Cancer Cell Targeting and Potent Antineoplastic Effect by a Nanostructured Version of Recombinant Ricin. Small 2018, 14, 1800665. [Google Scholar] [CrossRef]
- Falgàs, A.; Pallarès, V.; Unzueta, U.; Céspedes, M.V.; Arroyo-Solera, I.; Moreno, M.J.; Gallardo, A.; Mangues, M.A.; Sierra, J.; Villaverde, A.; et al. A CXCR4-targeted nanocarrier achieves highly selective tumor uptake in diffuse large B-cell lymphoma mouse models. Haematologica 2020, 105, 741–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Céspedes, M.V.; Unzueta, U.; Álamo, P.; Gallardo, A.; Sala, R.; Casanova, I.; Pavón, M.A.; Mangues, M.A.; Trías, M.; López-Pousa, A.; et al. Cancer-specific uptake of a liganded protein nanocarrier targeting aggressive CXCR4+ colorectal cancer models. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Pallarès, V.; Unzueta, U.; Falgàs, A.; Sánchez-Garciá, L.; Serna, N.; Gallardo, A.; Morris, G.A.; Alba-Castellón, L.; Álamo, P.; Sierra, J.; et al. An Auristatin nanoconjugate targeting CXCR4+ leukemic cells blocks acute myeloid leukemia dissemination. J. Hematol. Oncol. 2020, 13, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falgàs, A.; Pallarès, V.; Serna, N.; Sánchez-García, L.; Sierra, J.; Gallardo, A.; Alba-Castellón, L.; Álamo, P.; Unzueta, U.; Villaverde, A.; et al. Selective delivery of T22-PE24-H6 to CXCR4+ diffuse large B-cell lymphoma cells leads to wide therapeutic index in a disseminated mouse model. Theranostics 2020, 10, 5169–5180. [Google Scholar] [CrossRef]
- Serna, N.; Sánchez, J.M.; Unzueta, U.; Sánchez-Garcia, L.; Sánchez-Chardi, A.; Mangues, R.; Vázquez, E.; Villaverde, A. Recruiting potent membrane penetrability in tumor cell-targeted protein-only nanoparticles. Nanotechnology 2019, 30, 115101. [Google Scholar] [CrossRef]
- Céspedes, M.V.; Unzueta, U.; Aviñó, A.; Gallardo, A.; Álamo, P.; Sala, R.; Sánchez-Chardi, A.; Casanova, I.; Mangues, M.A.; Lopez-Pousa, A.; et al. Selective depletion of metastatic stem cells as therapy for human colorectal cancer. EMBO Mol. Med. 2018, 10, e8772. [Google Scholar] [CrossRef]
- De la Torre, C.; Casanova, I.; Acosta, G.; Coll, C.; Moreno, M.J.; Albericio, F.; Aznar, E.; Mangues, R.; Royo, M.; Sancenón, F.; et al. Gated Mesoporous Silica Nanoparticles Using a Double-Role Circular Peptide for the Controlled and Target-Preferential Release of Doxorubicin in CXCR4-Expresing Lymphoma Cells. Adv. Funct. Mater. 2015, 25, 687–695. [Google Scholar] [CrossRef]
- Wang, R.T.; Zhi, X.Y.; Yao, S.Y.; Zhang, Y. LFC131 peptide-conjugated polymeric nanoparticles for the effective delivery of docetaxel in CXCR4 overexpressed lung cancer cells. Colloids Surf. B Biointerfaces 2015, 133, 43–50. [Google Scholar] [CrossRef]
- Egorova, A.; Kiselev, A.; Hakli, M.; Ruponen, M.; Baranov, V.; Urtti, A. Chemokine-derived peptides as carriers for gene delivery to CXCR4 expressing cells. J. Gene Med. 2009, 11, 772–781. [Google Scholar] [CrossRef]
- Egorova, A.; Shubina, A.; Sokolov, D.; Selkov, S.; Baranov, V.; Kiselev, A. CXCR4-targeted modular peptide carriers for efficient anti-VEGF siRNA delivery. Int. J. Pharm. 2016, 515, 431–440. [Google Scholar] [CrossRef]
- Cagliani, R.; Gatto, F.; Cibecchini, G.; Marotta, R.; Catalano, F.; Sanchez-Moreno, P.; Pompa, P.P.; Bardi, G. CXCL5 Modified Nanoparticle Surface Improves CXCR2+ Cell Selective Internalization. Cells 2019, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios De La Rosa, J.M.; Spadea, A.; Donno, R.; Lallana, E.; Lu, Y.; Puri, S.; Caswell, P.; Lawrence, M.J.; Ashford, M.; Tirelli, N. Microfluidic-assisted preparation of RGD-decorated nanoparticles: Exploring integrin-facilitated uptake in cancer cell lines. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Donno, R.; Gennari, A.; Lallana, E.; De La Rosa, J.M.R.; d’Arcy, R.; Treacher, K.; Hill, K.; Ashford, M.; Tirelli, N. Nanomanufacturing through microfluidic-assisted nanoprecipitation: Advanced analytics and structure-activity relationships. Int. J. Pharm. 2017, 534, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Geven, M.; Luo, H.; Koo, D.; Panambur, G.; Donno, R.; Gennari, A.; Marotta, R.; Grimaldi, B.; Tirelli, N. Disulfide-Mediated Bioconjugation: Disulfide Formation and Restructuring on the Surface of Nanomanufactured (Microfluidics) Nanoparticles. ACS Appl. Mater. Interfaces 2019, 11, 26607–26618. [Google Scholar] [CrossRef]
- Picciocchi, A.; Šiaučiūnaiteė-Gaubard, L.; Petit-Hartlein, I.; Sadir, R.; Revilloud, J.; Caro, L.; Vivaudou, M.; Fieschi, F.; Moreau, C.; Vivès, C. C-Terminal Engineering of CXCL12 and CCL5 Chemokines: Functional Characterization by Electrophysiological Recordings. PLoS ONE 2014, 9, e87394. [Google Scholar] [CrossRef] [Green Version]
- Dalonneau, F.; Liu, X.Q.; Sadir, R.; Almodovar, J.; Mertani, H.C.; Bruckert, F.; Albiges-Rizo, C.; Weidenhaupt, M.; Lortat-Jacob, H.; Picart, C. The effect of delivering the chemokine SDF-1α in a matrix-bound manner on myogenesis. Biomaterials 2014, 35, 4525–4535. [Google Scholar] [CrossRef] [Green Version]
- Bardi, G.; Lipp, M.; Baggiolini, M.; Loetscher, P. The T cell chemokine receptor CCR7 is internalized on stimulation with ELC, but not with SLC. Eur. J. Immunol. 2001, 31, 3291–3297. [Google Scholar] [CrossRef]
- Jørgensen, A.S.; Rosenkilde, M.M.; Hjortø, G.M. Biased signaling of G protein-coupled receptors—From a chemokine receptor CCR7 perspective. Gen. Comp. Endocrinol. 2018, 258, 4–14. [Google Scholar] [CrossRef]
- Hauser, M.A.; Legler, D.F. Common and biased signaling pathways of the chemokine receptor CCR7 elicited by its ligands CCL19 and CCL21 in leukocytes. J. Leukoc. Biol. 2016, 99, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Hauser, M.A.; Kindinger, I.; Laufer, J.M.; Späte, A.-K.; Bucher, D.; Vanes, S.L.; Krueger, W.A.; Wittmann, V.; Legler, D.F. Distinct CCR7 glycosylation pattern shapes receptor signaling and endocytosis to modulate chemotactic responses. J. Leukoc. Biol. 2016, 99, 993–1007. [Google Scholar] [CrossRef] [Green Version]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F. The significance of cancer cell expression of the chemokine receptor CXCR4. Semin. Cancer Biol. 2004, 14, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Burkhardt, A.M.; Homey, B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 2011, 11, 597–606. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Mozobil® (Plerixafor, AMD3100), 10 years after its approval by the US Food and Drug Administration. Antivir. Chem. Chemother. 2019, 27. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Zhan, W.; Zhu, A.; Yoon, Y.; Lin, S.; Sasaki, M.; Klapproth, J.-M.A.; Yang, H.; Grossniklaus, H.E.; Xu, J.; et al. Development of a Unique Small Molecule Modulator of CXCR4. PLoS ONE 2012, 7, e34038. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Z-Average Size (nm) | PDI | Zeta Potential (mV) | |
|---|---|---|---|
| Non-functionalized NPs | 90 ± 5 | 0.12 ± 0.04 | −10 ± 3 |
| CXCL12-NPs | 90 ± 5 | 0.10 ± 0.03 | 20 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisani, A.; Donno, R.; Gennari, A.; Cibecchini, G.; Catalano, F.; Marotta, R.; Pompa, P.P.; Tirelli, N.; Bardi, G. CXCL12-PLGA/Pluronic Nanoparticle Internalization Abrogates CXCR4-Mediated Cell Migration. Nanomaterials 2020, 10, 2304. https://doi.org/10.3390/nano10112304
Pisani A, Donno R, Gennari A, Cibecchini G, Catalano F, Marotta R, Pompa PP, Tirelli N, Bardi G. CXCL12-PLGA/Pluronic Nanoparticle Internalization Abrogates CXCR4-Mediated Cell Migration. Nanomaterials. 2020; 10(11):2304. https://doi.org/10.3390/nano10112304
Chicago/Turabian StylePisani, Anissa, Roberto Donno, Arianna Gennari, Giulia Cibecchini, Federico Catalano, Roberto Marotta, Pier Paolo Pompa, Nicola Tirelli, and Giuseppe Bardi. 2020. "CXCL12-PLGA/Pluronic Nanoparticle Internalization Abrogates CXCR4-Mediated Cell Migration" Nanomaterials 10, no. 11: 2304. https://doi.org/10.3390/nano10112304
APA StylePisani, A., Donno, R., Gennari, A., Cibecchini, G., Catalano, F., Marotta, R., Pompa, P. P., Tirelli, N., & Bardi, G. (2020). CXCL12-PLGA/Pluronic Nanoparticle Internalization Abrogates CXCR4-Mediated Cell Migration. Nanomaterials, 10(11), 2304. https://doi.org/10.3390/nano10112304