Comprehensive Density Functional Theory Studies of Vibrational Spectra of Carbonates


Abstract
1. Introduction
2. Calculation Method
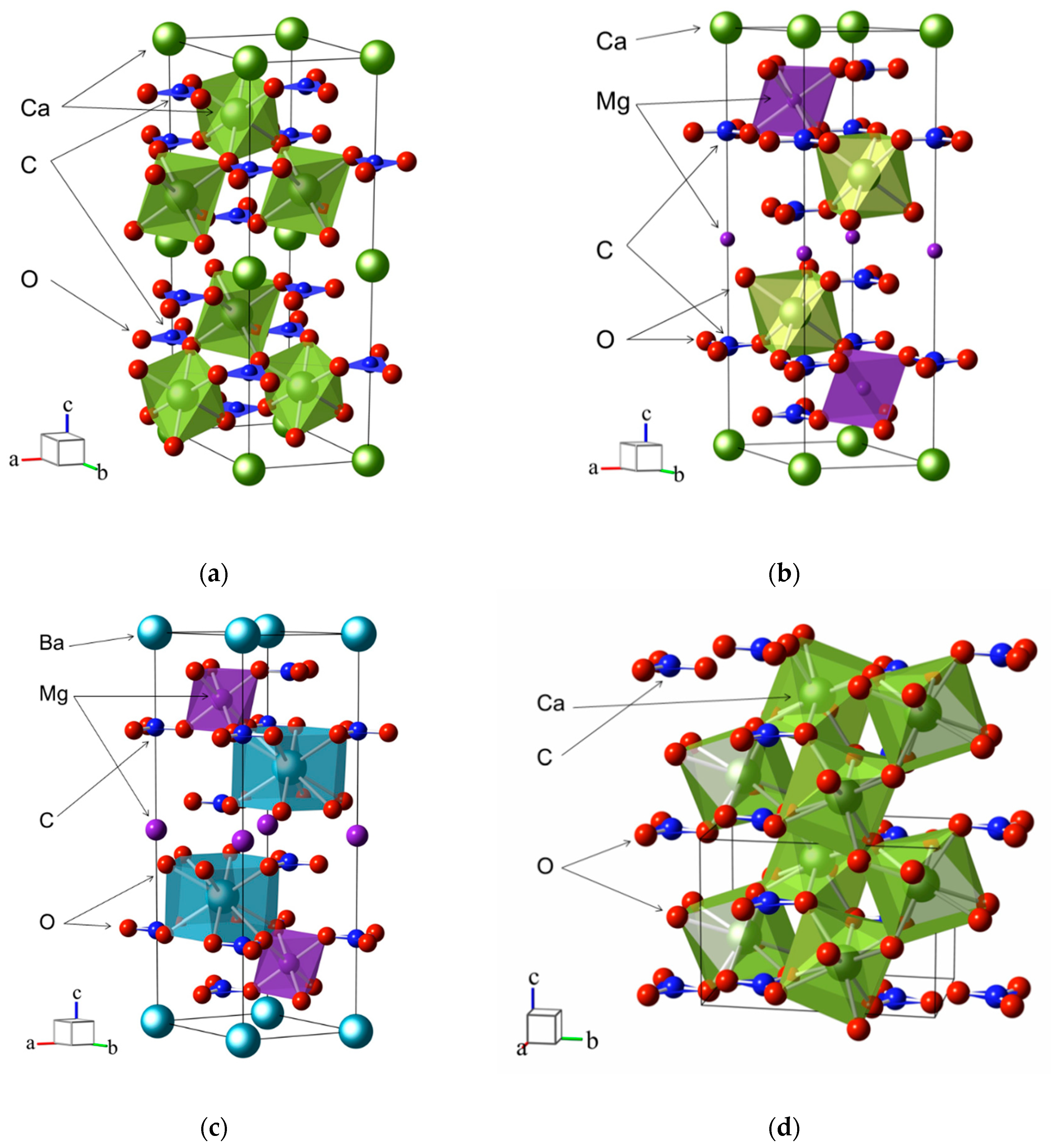
3. Crystal Structure
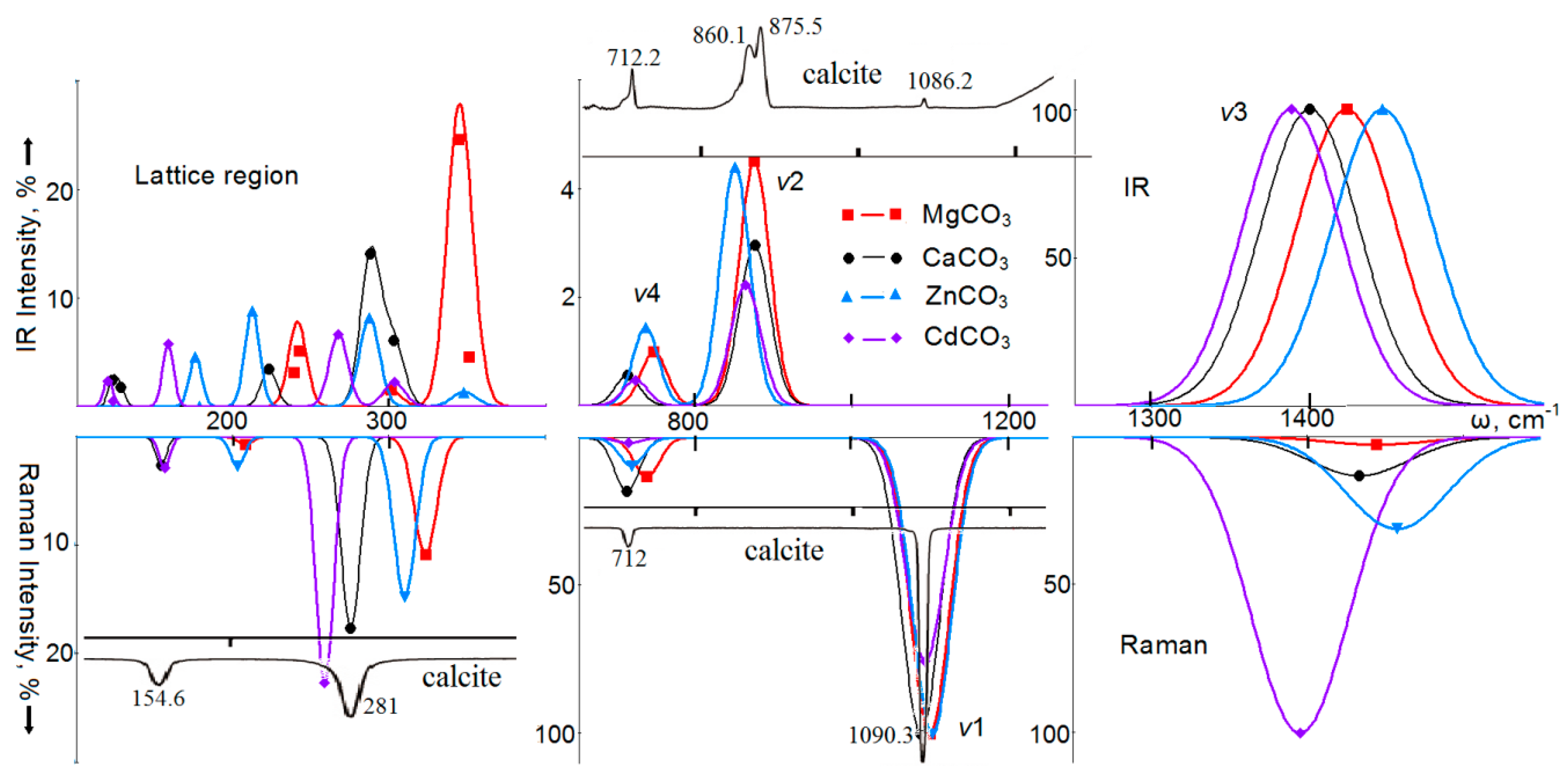
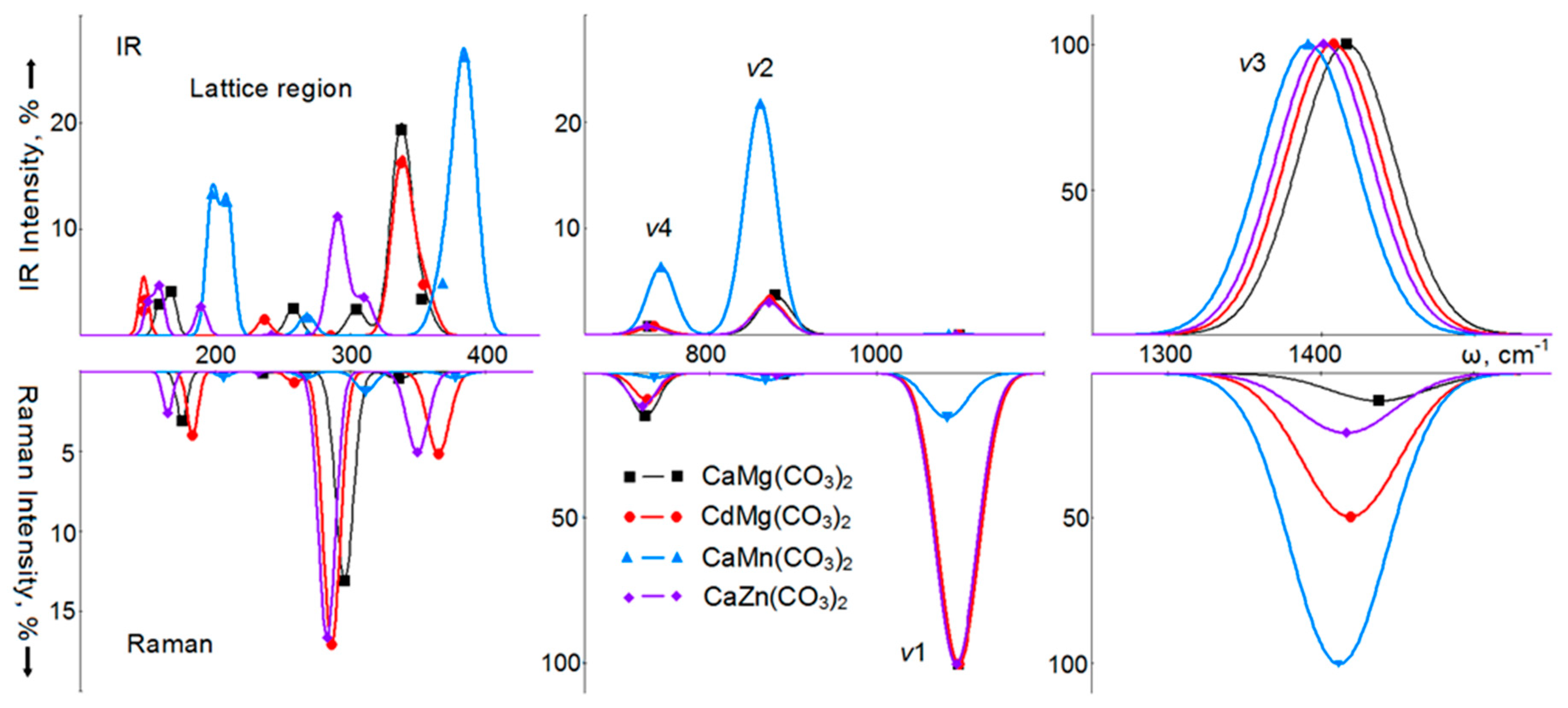
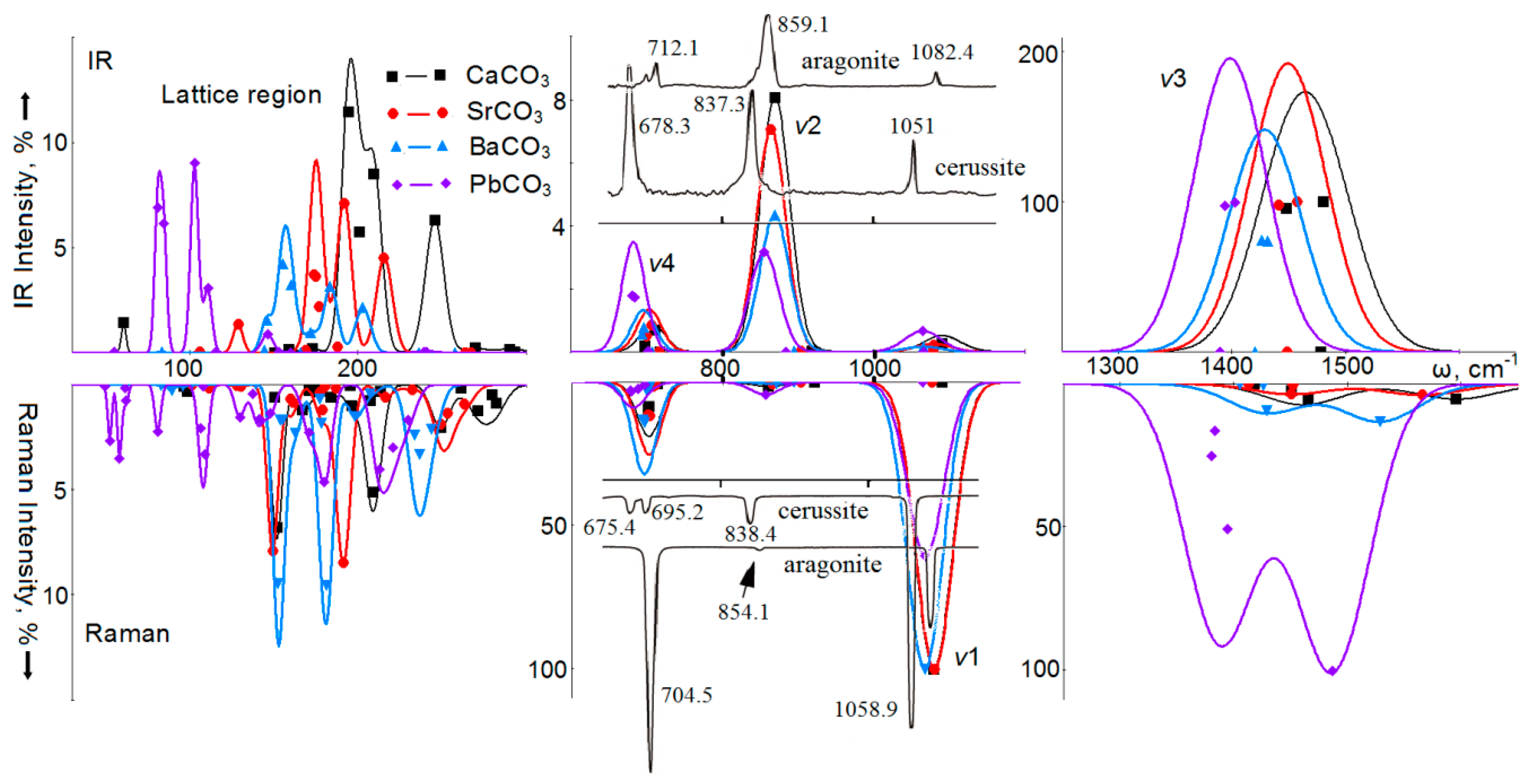
4. Vibrational Spectra
5. Optical Spectra of Crystals with a Calcite Structure
6. Vibrational Spectra of Crystals with a Dolomite Structure
7. Vibrational Spectra of Crystals with Aragonite Structure
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Liu, Y.; Shen, Y.; Zhao, S.; Luo, J. Structure-property relationship in nonlinear optical materials with p-conjugated CO3 triangles. Coord. Chem. Rev. 2020, 407, 213152. [Google Scholar] [CrossRef]
- Hazen, R.M.; Downs, R.T.; Jones, A.P.; Kah, L. Carbon Mineralogy and Crystal Chemistry. Rev. Mineral. Geochem. 2013, 75, 7–46. [Google Scholar] [CrossRef]
- Miller, K.G.; Kominz, M.A.; Browning, J.V.; Wright, J.D.; Mountain, G.S.; Katz, M.E.; Sugarman, P.J.; Cramer, B.S.; Christie-Blick, N.; Pekar, S.F. The Phanerozoic Record of Global Sea-Level Change. Science 2005, 310, 1293–1298. [Google Scholar] [CrossRef]
- Dupraz, C.; Reid, R.P.; Braissant, O.; Decho, A.W.; Norman, R.S.; Visscher, P.T. Processes of carbonate precipitation in modern microbial mats. Earth-Sci. Rev. 2009, 96, 141–162. [Google Scholar] [CrossRef]
- Litasov, K.D.; Goncharov, A.F.; Hemley, R.J. Crossover from melting to dissociation of CO2 under pressure: Implications for the lower mantle. Earth Planet. Sci. Lett. 2011, 309, 318–323. [Google Scholar] [CrossRef]
- Litasov, K.D.; Shatskiy, A.; Ohtani, E.; Yaxley, G.M. Solidus of alkaline carbonatite in the deep mantie. Geology 2013, 41, 79–82. [Google Scholar] [CrossRef]
- Montañez, I.P.; McElwain, J.C.; Poulsen, C.J.; White, J.D.; DiMichele, W.A.; Wilson, J.P.; Griggs, G.; Hren, M.T. Climate, pCO2 and terrestrial carbon cycle linkages during late Palaeozoic glacial–interglacial cycles. Nat. Geosci. 2016, 9, 824–828. [Google Scholar] [CrossRef]
- Sieber, M.J.; Wilke, F.; Koch-Müller, M. Partition coefficients of trace elements between carbonates and melt and suprasolidus phase relation of Ca-Mg-carbonates at 6 GPa. Am. Mineral. 2020, 105, 922–931. [Google Scholar] [CrossRef]
- Xie, M.; Yang, L.; Ji, Y.; Wang, Z.; Ren, X.; Liu, Z.; Asiri, A.M.; Xiong, X.; Sun, X. An amorphous Co-carbonate-hydroxide nanowire array for efficient and durable oxygen evolution reaction in carbonate electrolytes. Nanoscale 2017, 9, 16612–16615. [Google Scholar] [CrossRef]
- Sobhani-Nasab, A.; Pourmohamadian, H.; Rahimi-Nasrabadi, M.; Sheikhzadeh, G.A.; Tabrizi, H.B. Evaluation of the thermal properties of SrCO3-microencapsulated palmitic acid composites as thermal energy storage materials. J. Therm. Anal. Calorim. 2020, 140, 2123–2130. [Google Scholar] [CrossRef]
- De Beauvoir, T.H.; Sangregorio, A.; Cornu, I.; Josse, M. Synthesis, sintering by Cool-SPS and characterization of A2Cu(CO3)2 (A = K, Na): Evidence for multiferroic and magnetoelectric cupricarbonates. Dalton Trans. 2020, 49, 7820–7828. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ryu, K.H.; Ha, H.Y.; Jung, K.-D.; Lee, J.H. Techno-economic and environmental evaluation of nano calcium carbonate production utilizing the steel slag. J. CO2 Util. 2020, 37, 113–121. [Google Scholar] [CrossRef]
- Mazhar, S.F.B.; Meyer, H.J.; Samuels, T.; Sharonov, M.; Shi, L.; Alfano, R.R. Exploration of the competition between O- and E-wave induced stimulated Raman and supercontinuum in calcite under ultrafast laser excitation. Appl. Opt. 2020, 59, 5252–5257. [Google Scholar] [CrossRef] [PubMed]
- Karuppaiah, M.; Akilan, R.; Sakthivel, P.; Asaithambi, S.; Shankar, R.; Yuvakkumar, R.; Hayakawa, Y.; Ravi, G. Synthesis of self-essembled micro/nano structured manganese carbonate for high performance, long lifespan asymmetric supercapacitors and investigation of atomic-level intercalation properties of OH− ions via first principle calculation. J. Energy Storage 2020, 27, 101138. [Google Scholar] [CrossRef]
- Assaedi, H.; Alomayri, T.; Kaze, C.R.; Jindal, B.B.; Subaer, S.; Shaikh, F.; Alraddadi, S. Characterization and properties of geopolymer nanocomposites with different contents of nano-CaCO3. Constr. Build. Mater. 2020, 252, 119137. [Google Scholar] [CrossRef]
- Atuchin, V.V.; Kesler, V.G.; Kokh, A.E.; Pokrovsky, L.D. X-ray photoelectron spectroscopy study of b-BaB2O4 optical surface. Appl. Surf. Sci. 2004, 223, 352–360. [Google Scholar] [CrossRef]
- Ramana, C.V.; Vemuri, R.S.; Kaichev, V.V.; Kochubey, V.A.; Saraev, A.A.; Atuchin, V.V. X-ray photoelectron spectroscopy depth profiling of La2O3/Si thin films deposited by reactive magnetron sputtering. ACS Appl. Mater. Interfaces 2011, 3, 4370–4373. [Google Scholar] [CrossRef]
- Atuchin, V.V.; Molokeev, M.S.; Yurkin, G.Y.; Gavrilova, T.A.; Kesler, V.G.; Laptash, N.M.; Flerov, I.N.; Patrin, G.S. Synthesis, Structural, Magnetic, and Electronic Properties of Cubic CsMnMoO3F3 Oxyfluoride. J. Phys. Chem. C 2012, 116, 10162–10170. [Google Scholar] [CrossRef]
- Rubio, E.J.; Atuchin, V.V.; Kruchinin, V.N.; Pokrovsky, L.D.; Prosvirin, I.P.; Ramana, C.V. Electronic Structure and Optical Quality of Nanocrystalline Y2O3 Film Surfaces and Interfaces on Silicon. J. Phys. Chem. C 2014, 118, 13644–13651. [Google Scholar] [CrossRef]
- Atuchin, V.V.; Vinnik, D.; Gavrilova, T.A.; Gudkova, S.; Isaenko, L.I.; Jiang, X.; Pokrovsky, L.D.; Prosvirin, I.P.; Mashkovtseva, L.S.; Lin, Z. Flux Crystal Growth and the Electronic Structure of BaFe12O19 Hexaferrite. J. Phys. Chem. C 2016, 120, 5114–5123. [Google Scholar] [CrossRef]
- Kang, L.; Lin, Z.; Qin, J.; Chen, C. Two novel nonlinear optical carbonates in the dee-ultraviolet region: KBeCO3F and RbAlCO3F2. Sci. Rep. 2013, 3, 1366. [Google Scholar] [CrossRef] [PubMed]
- Than, T.T.; Young, J.; Rondinelli, J.M.; Halasyamani, P.S. Mixed-metal carbonate fluorides as deep-ultraviolet non-linear optical materials. J. Am. Chem. Soc. 2017, 139, 1285–1295. [Google Scholar]
- Zhang, X.; Wu, H.; Cheng, S.; Han, G.; Yang, Z.; Pan, S. K9[B4O5(OH)4]3(CO3)X×7H2O (X = Cl, Br): Synthesis, characterizations, and theoretical studies of noncentrosymmetric halogen borate-carbonates with short UV cutoff edges. Inorg. Chem. 2019, 58, 6974–6982. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Lin, C.S.; Yang, Y.; Zhao, D.; Lin, Z.; Ye, N.; Huang, J.S. Y2(CO3)3×H2O and (NH4)2Ca2Y4(CO3)9×H2O: Partial aviovalent cation substitution enabling evolution from centrosymmetry to noncentrosymmetry for nonlinear optical response. Chem. Mater. 2019, 31, 52–56. [Google Scholar] [CrossRef]
- Chen, K.; Peng, G.; Lin, C.; Luo, M.; Fan, H.; Yang, S.; Ye, N. NaPb2(CO3)2F2(OH)1−x(0 < x ≤ 1): A new member of alkali-lead carbonate fluoride system with large birefringence. J. Solid State Chem. 2020, 288, 121407. [Google Scholar] [CrossRef]
- Zou, G.; Ok, K.M. Novel ultraviolet (UV) nonlinear optical (NLO) materials discovered by chemical substitution-oriented design. Chem. Sci. 2020, 11, 5404–5409. [Google Scholar] [CrossRef]
- Gong, P.; Liu, X.; Kang, L.; Lin, Z. Inorganic planar p-conjugated groups in nonlinear optical crystals: Review and outlook. Inorg. Chem. Front. 2020, 7, 839–952. [Google Scholar] [CrossRef]
- Dorfman, S.M.; Badro, J.; Nabiei, F.; Prakapenka, V.B.; Cantoni, M.; Gillet, P. Carbonate stability in the reduced lower mantle. Earth Planet. Sci. Lett. 2018, 489, 84–91. [Google Scholar] [CrossRef]
- Zolotoyabko, E.; Caspi, E.N.; Fieramosca, J.S.; Von Dreele, R.B.; Marin, F.; Mor, G.; Addadi, L.; Weiner, S.; Politi, Y. Differences between Bond Lengths in Biogenic and Geological Calcite. Cryst. Growth Des. 2010, 10, 1207–1214. [Google Scholar] [CrossRef]
- Liang, W.; Li, Z.; Yin, Y.; Li, R.; Chen, L.; He, Y.; Dong, H.; Dai, L.; Li, H. Single crystal growth, characterization and high-pressure Raman spectroscopy of impurity-free magnesite (MgCO3). Phys. Chem. Miner. 2018, 45, 423–434. [Google Scholar] [CrossRef]
- Liang, W.; Yin, Y.; Li, Z.; Li, R.; Li, L.; He, Y.; Dong, H.; Li, Z.; Yan, S.; Zhai, S.; et al. Single crystal growth, crystalline structure investigation and high-pressure behavior of impurity-free siderite (FeCO3). Phys. Chem. Miner. 2018, 45, 831–842. [Google Scholar] [CrossRef]
- Gao, J.; Zhu, F.; Lai, X.-J.; Huang, R.; Qin, S.; Chen, D.; Liu, J.; Zheng, L.-R.; Wu, X. Compressibility of a natural smithsonite ZnCO3 up to 50 GPa. High Press. Res. 2014, 34, 89–99. [Google Scholar] [CrossRef]
- Liang, W.; Li, L.; Li, R.; Yin, Y.; Li, Z.; Liu, X.; Shan, S.; He, Y.; Meng, Y.; Li, Z.; et al. Crystal structure of impurity-free rhodochrosite (MnCO3) and thermal expansion properties. Phys. Chem. Miner. 2020, 47, 1–11. [Google Scholar] [CrossRef]
- Chariton, S.; Cerantola, V.; Ismailova, L.; Bykova, E.; Bykov, M.; Kupenko, I.; McCammon, C.; Dubrovinsky, L.S. The high-pressure behavior of spherocobaltite (CoCO3): A single crystal Raman spectroscopy and XRD study. Phys. Chem. Miner. 2018, 45, 59–68. [Google Scholar] [CrossRef]
- Reddy, B.J.; Frost, R.L. Electronic and vibrational spectra of gaspeite. Neues Jahrb. Mineral. Mon. 2004, 525–536. [Google Scholar] [CrossRef]
- Liu, L.G.; Lin, C.C. A Calcite → aragonite-type phase transition in CdCO3. Am. Mineral. 1997, 82, 643–646. [Google Scholar] [CrossRef]
- Liu, Z.T.Y.; Burton, B.P.; Khare, S.V.; Sarin, P. First-principles phase diagram calculations for the carbonate quasibinary systems CaCO3-ZnCO3, CdCO3-ZnCO3, CaCO3-CdCO3 and MgCO3-ZnCO3. Chem. Geol. 2016, 443, 137–145. [Google Scholar] [CrossRef]
- Zucchini, A.; Comodi, P.; Nazzareni, S.; Hanfland, M. The effect of cation ordering and temperature on the high-pressure behaviour of dolomite. Phys. Chem. Miner. 2014, 41, 783–793. [Google Scholar] [CrossRef]
- Garavelli, C.G.; Vurro, F.; Fioravanti, G.C. Minrecordite, a new mineral from Tsumeb. Mineral. Rec. 1982, 13, 131–136. [Google Scholar]
- Chai, L.; Navrotsky, A. Synthesis, characterization, and energetics of solid solution along the dolomite-ankerite join, and implications for the stability of ordered CaFe(CO3)2. Am. Mineral. 1996, 81, 1141–1147. [Google Scholar] [CrossRef]
- Frondel, C.; Bauer, C.L.H. Kutnahorite: A manganese dolomite, CaMn(CO3)2. Am. Mineral. 1955, 40, 748–760. [Google Scholar]
- Tareen, J.A.K.; Fazeli, A.R.; Basavalingu, B.; Bhandige, G.T. Decarbonation curves and associated thermodynamic data for synthetic Cd-dolomites CdMg(CO3)2, CdMn(CO3)2 and CdZn(CO3)2. J. Therm. Anal. 1995, 44, 937–954. [Google Scholar] [CrossRef]
- Bromiley, F.A.; Ballaran, T.B.; Langenhorst, F.; Seifert, F. Order and miscibility in the otavite-magnesite solid solution. Am. Mineral. 2007, 92, 829–836. [Google Scholar] [CrossRef]
- Effenberger, H.; Zemann, J. Single crystal X-ray investigation of norsethite, BaMg(CO3)2: One more mineral with an aplanar carbonate group. Z. Krist. 1985, 171, 275–280. [Google Scholar] [CrossRef]
- Liang, W.; Li, L.; Yin, Y.; Li, R.; Li, Z.; Liu, X.; Zhao, C.; Yang, S.; Meng, Y.; Li, Z.; et al. Crystal structure of norsethite-type BaMn(CO3)2 and its pressure-induced transition investigated by Raman spectroscopy. Phys. Chem. Miner. 2019, 46, 771–781. [Google Scholar] [CrossRef]
- Pippinger, T.; Miletich, R.; Effenberger, H.; Hofer, G.; Lotti, P.; Merlini, M. High-pressure polymorphism and structural transitions of norsethite, BaMg(CO3)2. Phys. Chem. Miner. 2014, 41, 737–755. [Google Scholar] [CrossRef]
- Antao, S.M.; Hassan, I. The orthorhombic structure of CaCO3, SrCO3, PbCO3 and BaCO3: Linear structure trends. Can. Mineral. 2009, 47, 1245–1255. [Google Scholar] [CrossRef]
- Ye, Y.; Smyth, J.R.; Boni, P. Crystal structure and thermal expansion of aragonite-group carbonates by single-crystal X-ray diffraction. Am. Mineral. 2012, 97, 707–712. [Google Scholar] [CrossRef]
- Kaabar, W.; Bott, S.; Devonshire, R. Raman spectroscopic study of mixed carbonate materials. Spectrochim. Acta A 2011, 78, 136–141. [Google Scholar] [CrossRef]
- Litasov, K.; Shatskiy, A.; Gavryushkin, P.N.; Bekhtenova, A.; Dorogokupets, P.I.; Danilov, B.S.; Higo, Y.; Akilbekov, A.T.; Inerbaev, T.M. P-V-T equation of state of CaCO3 aragonite to 29 GPa and 1673 K: In situ X-ray diffraction study. Phys. Earth Planet. Inter. 2017, 265, 82–91. [Google Scholar] [CrossRef]
- Lobanov, S.S.; Dong, X.; Martirosyan, N.S.; Samtsevich, A.I.; Stevanovic, V.; Gavryushkin, P.N.; Litasov, K.D.; Greenberg, E.; Prakapenka, V.B.; Oganov, A.R.; et al. Raman spectroscopy and x-ray diffraction of sp3CaCO3 at lower mantle pressures. Phys. Rev. B 2017, 96, 104101. [Google Scholar] [CrossRef]
- Smith, D.; Lawler, K.V.; Martinez-Canales, M.; Daykin, A.W.; Fussell, Z.; Smith, G.A.; Childs, C.; Smith, J.S.; Pickard, C.J.; Salamat, A. Postaragonite phases of CaCO3 at lower mantle pressures. Phys. Rev. Mater. 2018, 2, 013605. [Google Scholar] [CrossRef]
- Zhang, Z.; Mao, Z.; Liu, X.; Zhang, Y.; Brodholt, J.P. Stability and Reactions of CaCO3 polymorphs in the Earth’s Deep Mantle. J. Geophys. Res. Solid Earth 2018, 123, 6491–6500. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Y.; Ding, H.; Lu, A.; Li, Y.; Wang, C. Infrared emission properties of a kind of natural carbonate: Interpretation from mineralogical analysis. Phys. Chem. Miner. 2020, 47, 1–15. [Google Scholar] [CrossRef]
- Weir, C.E.; Lippincott, E.R. Infrared studies of aragonite, calcite, and vaterite type structures in the borates, carbonates, and nitrates. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1961, 65, 173–180. [Google Scholar] [CrossRef]
- Bischoff, W.D.; Sharma, S.K.; Mackenzie, F.T. Carbonate ion disorder in synthetic and biogenic magnesian calcites—A Raman spectral study. Am. Mineral. 1985, 70, 581–589. [Google Scholar]
- Lin, C.-C.; Liu, L.-G. Post-aragonite phase transitions in strontianite and cerussite—A high pressure Raman spectroscopic study. J. Phys. Chem. Solids 1997, 58, 977–987. [Google Scholar] [CrossRef]
- Valenzano, L.; Noël, Y.; Orlando, R.; Zicovich-Wilson, C.M.; Ferrero, M.; Dovesi, R. Ab initio vibrational spectra and dielectric properties of carbonates: Magnesite, calcite and dolomite. Theor. Chem. Acc. 2007, 117, 991–1000. [Google Scholar] [CrossRef]
- Kalinin, N.V.; Saleev, V.A. Ab initio modeling of Raman and infrared spectra of calcite. Comput. Opt. 2018, 42, 263–266. [Google Scholar] [CrossRef]
- Sun, J.; Wu, Z.; Cheng, H.; Zhang, Z.; Frost, R.L. A Raman spectroscopic comparison of calcite and dolomite. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 117, 158–162. [Google Scholar] [CrossRef]
- Perrin, J.; Vielzeuf, D.; Laporte, D.; Ricolleau, A.; Rossman, G.R.; Floquet, N. Raman characterization of synthetic magnesian calcites. Am. Mineral. 2016, 101, 2525–2538. [Google Scholar] [CrossRef]
- Liu, J.; Caracas, R.; Fan, D.; Bobocioiu, E.; Zhang, D.; Mao, W.L. High-pressure compressibility and vibrational properties of (Ca,Mn)CO3. Am. Mineral. 2016, 101, 2723–2730. [Google Scholar] [CrossRef]
- Minch, R.; Seoung, D.-H.; Ehm, L.; Winkler, B.; Knorr, K.; Peters, L.; Borkowski, L.; Parise, J.B.; Lee, Y.; Dubrovinsky, L.S.; et al. High-pressure behavior of otavite (CdCO3). J. Alloy Compd. 2010, 508, 251–257. [Google Scholar] [CrossRef]
- Dufresne, W.J.; Rufledt, C.J.; Marshall, C.P. Raman spectroscopy of the eight natural carbonate minerals of calcite structure. J. Raman Spectrosc. 2018, 49, 1999–2007. [Google Scholar] [CrossRef]
- Efthimiopoulos, I.; Germer, M.; Jahn, S.; Harms, M.; Reichmann, H.J.; Speziale, S.; Schade, U.; Sieber, M.; Koch-Müller, M. Effects of hydrostaticity on the structural stability of carbonates at lower mantle pressures: The case study of dolomite. High Press. Res. 2018, 39, 36–49. [Google Scholar] [CrossRef]
- Efthimiopoulos, I.; Jahn, S.; Kuras, A.; Schade, U.; Koch-Müller, M. Combined high-pressure and high-temperature vibrational studies of dolomite: Phase diagram and evidence of a new distorted modification. Phys. Chem. Miner. 2017, 44, 465–476. [Google Scholar] [CrossRef]
- Martens, W.N.; Rintoul, L.; Kloprogge, J.T.; Frost, R.L. Single crystal raman spectroscopy of cerussite. Am. Mineral. 2004, 89, 352–358. [Google Scholar] [CrossRef]
- Wang, X.; Ye, Y.; Wu, X.; Smyth, J.R.; Yang, Y.; Zhang, Z.; Wang, Z. High-temperature Raman and FTIR study of aragonite-group carbonates. Phys. Chem. Miner. 2018, 46, 51–62. [Google Scholar] [CrossRef]
- Efthimiopoulos, I.; Müller, J.; Winkler, B.; Otzen, C.; Harms, M.; Schade, U.; Koch-Müller, M. Vibrational response of strontianite at high pressures and high temperatures and construction of P–T phase diagram. Phys. Chem. Miner. 2019, 46, 27–35. [Google Scholar] [CrossRef]
- Lane, M.D.; Christensen, P.R. Thermal infrared emission spectroscopy of anhydrous carbonates. J. Geophys. Res. 1997, 102, 25581–25592. [Google Scholar] [CrossRef]
- Deines, P. Carbon isotope effects in carbonate systems. Geochim. Cosmochim. Acta 2004, 68, 2659–2679. [Google Scholar] [CrossRef]
- Brusentsova, T.N.; Peale, R.E.; Maukonen, D.; Harlow, G.E.; Boesenberg, J.S.; Ebel, D.; Prêt, D.; Sammartino, S.; Beaufort, D.; Fialin, M.; et al. Far infrared spectroscopy of carbonate minerals. Am. Mineral. 2010, 95, 1515–1522. [Google Scholar] [CrossRef]
- Farsang, S.; Facq, S.; Redfern, S.A.T. Raman modes of carbonate minerals as pressure and temperature gauges up to 6 GPa and 500 °C. Am. Mineral. 2018, 103, 1988–1998. [Google Scholar] [CrossRef]
- Erba, A.; Baima, J.; Bush, I.; Orlando, R.; Dovesi, R. Large-Scale Condensed Matter DFT Simulations: Performance and Capabilities of the CRYSTAL Code. J. Chem. Theory Comput. 2017, 13, 5019–5027. [Google Scholar] [CrossRef] [PubMed]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Valenzano, L.; Torres, F.J.; Doll, K.; Pascale, F.; Zicovich-Wilson, C.M.; Dovesi, R. Ab InitioStudy of the Vibrational Spectrum and Related Properties of Crystalline Compounds; the Case of CaCO3Calcite. Z. Phys. Chem. 2006, 220, 893–912. [Google Scholar] [CrossRef]
- Peintinger, M.F.; Oliveira, D.V.; Bredow, T. Consistent Gaussian basis sets of triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2013, 34, 451–459. [Google Scholar] [CrossRef]
- Laun, J.; Oliveira, D.V.; Bredow, T. Consistent gaussian basis sets of double- and triple-zeta valence with polarization quality of the fifth period for solid-state calculations. J. Comput. Chem. 2018, 39, 1285–1290. [Google Scholar] [CrossRef]
- Piskunov, S.; Heifets, E.; Eglitis, R.; Borstel, G. Bulk properties and electronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: An ab initio HF/DFT study. Comput. Mater. Sci. 2004, 29, 165–178. [Google Scholar] [CrossRef]
- Evarestov, R.; Kotomin, E.A.; Mastrikov, Y.; Gryaznov, D.; Heifets, E.; Maier, J. Comparative density-functional LCAO and plane-wave calculations ofLaMnO3surfaces. Phys. Rev. B 2005, 72, 214411. [Google Scholar] [CrossRef]
- Sophia, G.; Baranek, P.; Sarrazin, C.; Rérat, M.; Dovesi, R. First-principles study of the mechanisms of the pressure-induced dielectric anomalies in ferroelectric perovskites. Phase Transit. 2013, 86, 1069–1084. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Pascale, F.; Zicovich-Wilson, C.M.; Gejo, F.L.; Civalleri, B.; Orlando, R.; Dovesi, R. The calculation of the vibrational frequencies of crystalline compounds and its implementation in the CRYSTAL code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Zicovich-Wilson, C.M.; Pascale, F.; Roetti, C.; Saunders, V.R.; Orlando, R.; Dovesi, R. Calculation of the vibration frequencies of alpha-quartz: The effect of Hamiltonian and basis set. J. Comput. Chem. 2004, 25, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Carteret, C.; De La Pierre, M.; Dossot, M.; Pascale, F.; Erba, A.; Dovesi, R. The vibrational spectrum of CaCO3 aragonite: A combined experimental and quantum-mechanical investigation. J. Chem. Phys. 2013, 138, 014201. [Google Scholar] [CrossRef]
- Baima, J.; Ferrabone, M.; Orlando, R.; Erba, A.; Dovesi, R. Thermodynamics and phonon dispersion of pyrope and grossular silicate garnets from ab initio simulations. Phys. Chem. Miner. 2016, 43, 137–149. [Google Scholar] [CrossRef][Green Version]
- Maschio, L.; Kirtman, B.; Rérat, M.; Orlando, R.; Dovesi, R. Ab initio analytical Raman intensities for periodic systems through a coupled perturbed Hartree-Fock/Kohn-Sham method in an atomic orbital basis. II. Validation and comparison with experiments. J. Chem. Phys. 2013, 139, 164102. [Google Scholar] [CrossRef]
- Korabel’Nikov, D.; Zhuravlev, Y.N. Structural, elastic, electronic and vibrational properties of a series of sulfates from first principles calculations. J. Phys. Chem. Solids 2018, 119, 114–121. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Grzechnik, A.; Simon, P.; Gillet, P.; McMillan, P. An infrared study of MgCO3 at high pressure. Phys. B Condens. Matter 1999, 262, 67–73. [Google Scholar] [CrossRef]
- Clark, S.J.; Jouanna, P.; Haines, J.; Mainprice, D. Calculation of infrared and Raman vibration modes of magnesite at high pressure by density-functional perturbation theory and comparison with experiments. Phys. Chem. Miner. 2010, 38, 193–202. [Google Scholar] [CrossRef]
- Yao, C.; Wu, Z.; Zou, F.; Sun, W. Thermodynamic and Elastic Properties of Magnesite at Mantle Conditions: First-Principles Calculations. Geochem. Geophys. Geosyst. 2018, 19, 2719–2731. [Google Scholar] [CrossRef]
- Gillet, P.; Biellmann, C.; Reynard, B.; McMillan, P. Raman spectroscopic studies of carbonates part I: High-pressure and high-temperature behaviour of calcite, magnesite, dolomite and aragonite. Phys. Chem. Miner. 1993, 20, 1–18. [Google Scholar] [CrossRef]
- Schauble, E.A.; Ghosh, P.; Eiler, J.M. Preferential formation of 13C–18O bonds in carbonate minerals, estimated using first-principles lattice dynamics. Geochim. Cosmochim. Acta 2006, 70, 2510–2529. [Google Scholar] [CrossRef]
- De La Pierre, M.; Carteret, C.; Maschio, L.; André, E.; Orlando, R.; Dovesi, R. The Raman spectrum of CaCO3polymorphs calcite and aragonite: A combined experimental and computational study. J. Chem. Phys. 2014, 140, 164509. [Google Scholar] [CrossRef]
- Spivak, A.V.; Solopova, N.; Cerantola, V.; Bykova, E.; Zakharchenko, E.; Dubrovinsky, L.S.; Litvin, Y. Raman study of MgCO3–FeCO3 carbonate solid solution at high pressures up to 55 GPa. Phys. Chem. Miner. 2014, 41, 633–638. [Google Scholar] [CrossRef]
- Siva, T.; Muralidharan, S.; Sathiyanarayanan, S.; Manikandan, E.; Jayachandran, M. Enhanced Polymer Induced Precipitation of Polymorphous in Calcium Carbonate: Calcite Aragonite Vaterite Phases. J. Inorg. Organomet. Polym. Mater. 2017, 27, 770–778. [Google Scholar] [CrossRef]
- Farfan, G.A.; Boulard, E.; Wang, S.; Mao, W.L. Bonding and electronic changes in rhodochrosite at high pressure. Am. Mineral. 2013, 98, 1817–1823. [Google Scholar] [CrossRef]
- Zhao, C.; Li, H.; Jiangb, J.; He, Y.; Liang, W. Phase Transition and vibration properties of MnCO3 at high pressure and high-temperature by Raman spectroscopy. High Press. Res. 2018, 38, 212–223. [Google Scholar] [CrossRef]
- Böttcher, M.E.; Gehlken, P.-L.; Skogby, H.; Reutel, C. The vibrational spectra of BaMg(CO3)2 (norsethite). Miner. Mag. 1997, 61, 249–256. [Google Scholar] [CrossRef]
- Chaney, J.; Santillán, J.D.; Knittle, E.; Williams, Q. A high-pressure infrared and Raman spectroscopic study of BaCO3: The aragonite, trigonal and Pmmn structures. Phys. Chem. Miner. 2015, 42, 83–93. [Google Scholar] [CrossRef]
- Brooker, M.H.; Sunder, S.; Taylor, P.; Lopata, V.J. Infrared and Raman spectra and X-ray diffraction studies of solid lead(II) carbonates. Can. J. Chem. 1982, 61, 494–502. [Google Scholar] [CrossRef]
- Catalli, K.; Santillán, J.; Williams, Q. A high pressure infrared spectroscopic study of PbCO3-cerussite: Constraints on the structure of the post-aragonite phase. Phys. Chem. Miner. 2005, 32, 412–417. [Google Scholar] [CrossRef]
- Frech, R.; Wang, E.C.; Bates, J.B. The i.r. and Raman spectra of CaCO3 (aragonite). Spectrochim. Acta Part A Mol. Spectrosc. 1980, 36, 915–919. [Google Scholar] [CrossRef]
- Bayarjargal, L.; Fruhner, C.-J.; Schrodt, N.; Winkler, B. CaCO3 phase diagram studied with Raman spectroscopy at pressures up to 50 GPa and high temperatures and DFT modeling. Phys. Earth Planet. Inter. 2018, 281, 31–45. [Google Scholar] [CrossRef]
- Biedermann, N.; Speziale, S.; Winkler, B.; Reichmann, H.J.; Koch-Müller, M.; Heide, G. High-pressure phase behavior of SrCO3: An experimental and computational Raman scattering study. Phys. Chem. Miner. 2017, 44, 335–343. [Google Scholar] [CrossRef]
- Minch, R.; Dubrovinsky, L.S.; Kurnosov, A.; Ehm, L.; Knorr, K.; Depmeier, W. Raman spectroscopic study of PbCO3 at high pressures and temperatures. Phys. Chem. Miner. 2010, 37, 45–56. [Google Scholar] [CrossRef]
- Alía, J.M.; de Mera, Y.D.; Edwards, H.G.M.; Martín, P.G.; Andres, S.L. FT-Raman and infrared spectroscopic study of aragonite-strontianite (CaxSr1−xCO3) solid solution. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1997, 53, 2347–2362. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbonate | a, Å | b, Å | c, Å | V, Å3 | RM–O, Å | RC–O, Å |
|---|---|---|---|---|---|---|
| MgCO3 | 4.6624 | 4.6624 | 15.1891 | 285.9527 | 2.1229 | 1.2857 |
| CaCO3_C | 5.0385 | 5.0385 | 17.3168 | 380.7118 | 2.3905 | 1.2878 |
| ZnCO3 | 4.7094 | 4.7094 | 15.1297 | 290.5952 | 2.1344 | 1.2973 |
| CdCO3 | 4.9819 | 4.9819 | 16.6163 | 357.1529 | 2.3312 | 1.2874 |
| CaMg(CO3)2 | 4.8382 | 4.8382 | 16.2563 | 329.5605 | 2.2571 | 1.2865 |
| CdMg(CO3)2 | 4.8140 | 4.8140 | 15.8629 | 318.3695 | 2.2253 | 1.2862 |
| CaMn(CO3)2 | 4.8295 | 4.8295 | 15.8159 | 319.4686 | 2.2302 | 1.2874 |
| CaZn(CO3)2 | 4.8558 | 4.8558 | 16.2964 | 332.7782 | 2.2648 | 1.2866 |
| BaMg(CO3)2 | 5.0637 | 5.0637 | 17.0662 | 378.9683 | 2.6811 | 1.2846 |
| CaCO3_A | 5.0020 | 8.0175 | 5.8581 | 234.9323 | 2.5604 | 1.2857 |
| SrCO3 | 5.1469 | 8.4418 | 6.1947 | 269.1522 | 2.6838 | 1.2884 |
| BaCO3 | 5.3665 | 8.9327 | 6.6847 | 320.4459 | 2.8567 | 1.2910 |
| PbCO3 | 5.2453 | 8.5723 | 6.3725 | 286.5319 | 2.7451 | 1.2897 |
| Method | ν4 | ν1 | ν3 | v3 |
|---|---|---|---|---|
| Experiment [68] | 713.2 − 0.173·M (0.986) | 1090.1 − 0.17·M (0.919) | 1495.3 − 0.588·M (0.989) | 1597.2 − 0.582·M (0.99) |
| B3LYP | 714.3 − 0.128·M (0.933) | 1087.059 − 0.116·M (0.894) | 1486.0 − 0.426·M (0.992) | 1619.0 − 0.638·M (0.998) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuravlev, Y.N.; Atuchin, V.V. Comprehensive Density Functional Theory Studies of Vibrational Spectra of Carbonates. Nanomaterials 2020, 10, 2275. https://doi.org/10.3390/nano10112275
Zhuravlev YN, Atuchin VV. Comprehensive Density Functional Theory Studies of Vibrational Spectra of Carbonates. Nanomaterials. 2020; 10(11):2275. https://doi.org/10.3390/nano10112275
Chicago/Turabian StyleZhuravlev, Yurii N., and Victor V. Atuchin. 2020. "Comprehensive Density Functional Theory Studies of Vibrational Spectra of Carbonates" Nanomaterials 10, no. 11: 2275. https://doi.org/10.3390/nano10112275
APA StyleZhuravlev, Y. N., & Atuchin, V. V. (2020). Comprehensive Density Functional Theory Studies of Vibrational Spectra of Carbonates. Nanomaterials, 10(11), 2275. https://doi.org/10.3390/nano10112275