Synergy between Ni and Co Nanoparticles Supported on Carbon in Guaiacol Conversion



Abstract
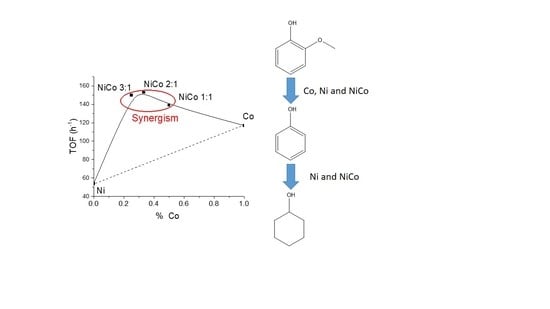
1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Catalyst Characterization
2.3. Catalyst Activity Measurement
3. Results
3.1. Catalyst Characterization
3.2. Catalytic Properties
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Joffres, B.; Lorentz, C.; Vidalie, M.; Laurenti, D.; Quoineaud, A.A.; Charon, N.; Daudin, A.; Quignard, A.; Geantet, C. Catalytic hydroconversion of a wheat straw soda lignin: Characterization of the products and the lignin residue. Appl. Catal. B 2014, 145, 167–176. [Google Scholar] [CrossRef]
- Pandey, M.P.; Kim, C.S. Lignin Depolymerization and Conversion: A Review of Thermochemical Methods. Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin Valorization: Improving Lignin Processing in the Biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef] [PubMed]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.C.; Rahimpour, M.R. Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation. Energy Environ. Sci. 2014, 7, 103–129. [Google Scholar] [CrossRef]
- Boonyasuwat, S.; Omotoso, T.; Resasco, D.E.; Crossley, S.P. Conversion of Guaiacol over Supported Ru Catalysts. Catal. Lett. 2013, 143, 783–791. [Google Scholar] [CrossRef]
- Nimmanwudipong, T.; Aydin, C.; Lu, J.; Runnebaum, R.C.; Brodwater, K.C.; Browning, N.D.; Block, D.E.; Gates, B.C. Selective Hydrodeoxygenation of Guaiacol Catalyzed by Platinum Supported on Magnesium Oxide. Catal. Lett. 2012, 142, 1190–1196. [Google Scholar] [CrossRef]
- Chang, J.; Danuthai, T.; Dewiyanti, S.; Wang, C.; Borgna, A. Hydrodeoxygenation of Guaiacol over Carbon-Supported Metal Catalysts. ChemCatChem 2013, 5, 3041–3049. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Afanasiev, P.; Geantet, C. Hydrodeoxygenation of guaiacol with CoMo catalysts. Part I: Promoting effect of cobalt on HDO selectivity and activity. Appl. Catal. B 2011, 101, 239–245. [Google Scholar] [CrossRef]
- Feitosa, L.F.; Berhault, G.; Laurenti, D.; da Silva, V.T. Effect of the Nature of the Carbon Support on the Guaiacol Hydrodeoxygenation Performance of Nickel Phosphide: Comparison between Carbon Nanotubes and a Mesoporous Carbon Support. Ind. Eng. Chem. Res. 2019, 58, 16164–16181. [Google Scholar] [CrossRef]
- Ghampson, I.T.; Sepulveda, C.; Garcia, R.; Frederick, B.G.; Wheeler, M.C.; Escalona, N.; DeSisto, W.J. Guaiacol transformation over unsupported molybdenum-based nitride catalysts. Appl. Catal. A 2012, 413–414, 78–84. [Google Scholar] [CrossRef]
- Jongerius, A.L.; Gosselink, R.W.; Dijkstra, J.; Bitter, J.H.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Carbon Nanofiber Supported Transition-Metal Carbide Catalysts for the Hydrodeoxygenation of Guaiacol. ChemCatChem 2013, 5, 2964–2972. [Google Scholar] [CrossRef]
- Blanco, E.; Dongil, A.B.; García-Fierro, J.L.; Escalona, N. Insights in supported rhenium carbide catalysts for hydroconversion of lignin-derived compounds. Appl. Catal. A 2020, 599, 117600. [Google Scholar] [CrossRef]
- Leiva, K.; Martinez, N.; Sepulveda, C.; García, R.; Jimenez, C.A.; Laurenti, D.; Vrinat, M.; Geantet, C.; Fierro, J.L.G.; Ghampson, I.T.; et al. Hydrodeoxygenation of 2-methoxyphenol over different Re active phases supported on SiO2 catalysts. Appl. Catal. A 2015, 490, 71–79. [Google Scholar] [CrossRef]
- Blanco, E.; Aguirre-Abarca, D.A.; Díaz de León, J.N.; Escalona, N. Relevant aspects of the conversion of guaiacol as a model compound for bio-oil over supported molybdenum oxycarbide catalysts. New J. Chem. 2020, 44, 12027–12035. [Google Scholar] [CrossRef]
- Alda-Onggar, M.; Mäki-Arvela, P.; Aho, A.; Simakova, I.L.; Murzin, D.Y. Hydrodeoxygenation of phenolic model compounds over zirconia supported Ir and Ni-catalysts. React. Kinet. Mech. Catal. 2019, 126, 737–759. [Google Scholar] [CrossRef]
- Lindfors, C.; Mäki-Arvela, P.; Paturi, P.; Aho, A.; Eränen, K.; Hemming, J.; Peurla, M.; Kubička, D.; Simakova, I.L.; Murzin, D.Y. Hydrodeoxygenation of Isoeugenol over Ni- and Co-Supported Catalysts. ACS Sustain. Chem. Eng. 2019, 7, 14545–14560. [Google Scholar] [CrossRef]
- Barton, R.R.; Carrier, M.; Segura, C.; Fierro, J.L.G.; Park, S.; Lamb, H.H.; Escalona, N.; Peretti, S.W. Ni/HZSM-5 catalyst preparation by deposition-precipitation. Part 2. Catalytic hydrodeoxygenation reactions of lignin model compounds in organic and aqueous systems. Appl. Catal. A 2018, 562, 294–309. [Google Scholar] [CrossRef]
- Ghampson, I.T.; Sepúlveda, C.; Dongil, A.B.; Pecchi, G.; García, R.; Fierro, J.L.G.; Escalona, N. Phenol hydrodeoxygenation: Effect of support and Re promoter on the reactivity of Co catalysts. Catal. Sci. Technol. 2016, 6, 7289–7306. [Google Scholar] [CrossRef]
- Ranaware, V.; Verma, D.; Insyani, R.; Riaz, A.; Kim, S.M.; Kim, J. Highly-efficient and magnetically-separable ZnO/Co@N-CNTs catalyst for hydrodeoxygenation of lignin and its derived species under mild conditions. Green Chem. 2019, 21, 1021–1042. [Google Scholar] [CrossRef]
- Liu, X.; Jia, W.; Xu, G.; Zhang, Y.; Fu, Y. Selective Hydrodeoxygenation of Lignin-Derived Phenols to Cyclohexanols over Co-Based Catalysts. ACS Sustain. Chem. Eng. 2017, 5, 8594–8601. [Google Scholar] [CrossRef]
- Tran, N.T.T.; Uemura, Y.; Ramli, A. Hydrodeoxygenation of Guaiacol over Al-MCM-41 Supported Metal Catalysts: A Comparative Study of Co and Ni. Procedia Eng. 2016, 148, 1252–1258. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Gardini, D.; de Carvalho, H.W.P.; Damsgaard, C.D.; Grunwaldt, J.-D.; Jensen, P.A.; Wagner, J.B.; Jensen, A.D. Stability and resistance of nickel catalysts for hydrodeoxygenation: Carbon deposition and effects of sulfur, potassium, and chlorine in the feed. Catal. Sci. Technol. 2014, 4, 3672–3686. [Google Scholar] [CrossRef]
- Bykova, M.V.; Zavarukhin, S.G.; Trusov, L.I.; Yakovlev, V.A. Guaiacol hydrodeoxygenation kinetics with catalyst deactivation taken into consideration. Kinet. Catal. 2013, 54, 40–48. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, C.S.; Liu, Y.G.; Hou, X.X.; Zhang, R.Q.; Tang, X.Y. Coke Deposition on Ni/HZSM-5 in Bio-oil Hydrodeoxygenation Processing. Energy Fuels 2015, 29, 1722–1728. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, C.S.; Liu, Y.G.; Tang, S.S.; Chen, G.H.; Zhang, R.Q.; Tang, X.Q. Coke formation on the surface of Ni/HZSM-5 and Ni–Cu/HZSM-5 catalysts during bio-oil hydrodeoxygenation. Fuel 2017, 189, 23–31. [Google Scholar] [CrossRef]
- Jahromi, H.; Agblevor, F.A. Hydrodeoxygenation of Aqueous-Phase Catalytic Pyrolysis Oil to Liquid Hydrocarbons Using Multifunctional Nickel Catalyst. Ind. Eng. Chem. Res. 2018, 57, 13257–13268. [Google Scholar] [CrossRef]
- Schmitt, C.C.; Reolon, M.B.G.; Zimmermann, M.; Raffelt, K.; Grunwaldt, J.D.; Dahmen, N. Synthesis and Regeneration of Nickel-Based Catalysts for Hydrodeoxygenation of Beech Wood Fast Pyrolysis Bio-Oil. Catalysts 2018, 8, 449. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Bimetallic catalysts for upgrading of biomass to fuels and chemicals. Chem. Soc. Rev. 2012, 41, 8075–8098. [Google Scholar] [CrossRef]
- Dongil, A.B.; Bachiller-Baeza, B.; Rodríguez-Ramos, I.; Fierro, J.L.G.; Escalona, N. The effect of Cu loading on Ni/carbon nanotubes catalysts for hydrodeoxygenation of guaiacol. RSC Adv. 2016, 6, 26658–26667. [Google Scholar] [CrossRef]
- Liu, X.; An, W.; Wang, Y.; Turner, C.H.; Resasco, D.E. Hydrodeoxygenation of guaiacol over bimetallic Fe-alloyed (Ni, Pt) surfaces: Reaction mechanism, transition-state scaling relations and descriptor for predicting C–O bond scission reactivity. Catal. Sci. Technol. 2018, 8, 2146–2158. [Google Scholar] [CrossRef]
- Han, Q.; Rehman, M.U.; Wang, J.; Rykov, A.; Gutiérrez, O.Y.; Zhao, Y.; Wang, S.; Ma, X.; Lercher, J.A. The synergistic effect between Ni sites and Ni–Fe alloy sites on hydrodeoxygenation of lignin-derived phenols. Appl. Catal. B 2019, 253, 348–358. [Google Scholar] [CrossRef]
- Luo, Z.; Zheng, Z.; Li, L.; Cui, Y.-T.; Zhao, C. Bimetallic Ru–Ni Catalyzed Aqueous-Phase Guaiacol Hydrogenolysis at Low H2 Pressures. ACS Catal. 2017, 7, 8304–8313. [Google Scholar] [CrossRef]
- Huynh, T.M.; Armbruster, U.; Pohl, M.-M.; Schneider, M.; Radnik, J.; Hoang, D.-L.; Phan, B.M.Q.; Nguyen, D.A.; Martin, A. Hydrodeoxygenation of Phenol as a Model Compound for Bio-oil on Non-noble Bimetallic Nickel-based Catalysts. ChemCatChem 2014, 6, 1940–1951. [Google Scholar] [CrossRef]
- Tran, N.T.T.; Uemura, Y.; Chowdhury, S.; Ramli, A. Vapor-phase hydrodeoxygenation of guaiacol on Al-MCM-41 supported Ni and Co catalysts. Appl. Catal. A 2016, 512, 93–100. [Google Scholar] [CrossRef]
- Zhou, M.; Ye, J.; Liu, P.; Xu, J.; Jiang, J. Water-Assisted Selective Hydrodeoxygenation of Guaiacol to Cyclohexanol over Supported Ni and Co Bimetallic Catalysts. ACS Sustain. Chem. Eng. 2017, 5, 8824–8835. [Google Scholar] [CrossRef]
- Zanuttini, M.S.; Dalla Costa, B.O.; Querini, C.A.; Peralta, M.A. Hydrodeoxygenation of m-cresol with Pt supported over mild acid materials. Appl. Catal. A 2014, 482, 352–361. [Google Scholar] [CrossRef]
- Zhu, X.; Lobban, L.L.; Mallinson, R.G.; Resasco, D.E. Bifunctional transalkylation and hydrodeoxygenation of anisole over a Pt/HBeta catalyst. J. Catal. 2011, 281, 21–29. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Ghampson, I.T.; Pecchi, G.; Fierro, J.L.G.; Videla, A.; Escalona, N. Catalytic hydrodeoxygenation of anisole over Re–MoOx/TiO2 and Re–VOx/TiO2 catalysts. Appl. Catal. B 2017, 208, 60–74. [Google Scholar] [CrossRef]
- Huynh, T.; Armbruster, U.; Kreyenschulte, C.; Nguyen, L.; Phan, B.; Nguyen, D.; Martin, A. Understanding the Performance and Stability of Supported Ni–Co-Based Catalysts in Phenol HDO. Catalysts 2016, 6, 176. [Google Scholar] [CrossRef]
- Takanabe, K.; Nagaoka, K.; Nariai, K.; Aika, K.-I. Titania-supported cobalt and nickel bimetallic catalysts for carbon dioxide reforming of methane. J. Catal. 2005, 232, 268–275. [Google Scholar] [CrossRef]
- Reuel, R.C.; Bartholomew, C.H. The stoichiometries of H2 and CO adsorptions on cobalt: Effects of support and preparation. J. Catal. 1984, 85, 63–77. [Google Scholar] [CrossRef]
- Dongil, A.B.; Pastor-Pérez, L.; Escalona, N.; Sepúlveda-Escribano, A. Carbon nanotube-supported Ni–CeO2 catalysts. Effect of the support on the catalytic performance in the low-temperature WGS reaction. Carbon 2016, 101, 296–304. [Google Scholar] [CrossRef]
- Ma, Q.; Wang, D.; Wu, M.; Zhao, T.; Yoneyama, Y.; Tsubaki, N. Effect of catalytic site position: Nickel nanocatalyst selectively loaded inside or outside carbon nanotubes for methane dry reforming. Fuel 2013, 108, 430–438. [Google Scholar] [CrossRef]
- van Deelen, T.W.; Su, H.; Sommerdijk, N.A.J.M.; de Jong, K.P. Assembly and activation of supported cobalt nanocrystal catalysts for the Fischer–Tropsch synthesis. Chem. Commun. 2018, 54, 2530–2533. [Google Scholar] [CrossRef] [PubMed]
- van Deelen, T.W.; Yoshida, H.; Oord, R.; Zečević, J.; Weckhuysen, B.M.; de Jong, K.P. Cobalt nanocrystals on carbon nanotubes in the Fischer-Tropsch synthesis: Impact of support oxidation. Appl. Catal. A 2020, 593, 117441. [Google Scholar] [CrossRef]
- Dongil, A.B.; Ghampson, I.T.; García, R.; Fierro, J.L.G.; Escalona, N. Hydrodeoxygenation of guaiacol over Ni/carbon catalysts: Effect of the support and Ni loading. RSC Adv. 2016, 6, 2611–2623. [Google Scholar] [CrossRef]
- Han, G.-H.; Lee, M.W.; Park, S.; Kim, H.J.; Ahn, J.-P.; Seo, M.-G.; Lee, K.-Y. Revealing the factors determining the selectivity of guaiacol HDO reaction pathways using ZrP-supported Co and Ni catalysts. J. Catal. 2019, 377, 343–357. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, J.; Zheng, L.; Fan, G.; Yang, L.; Li, F. Significant Promotion of Surface Oxygen Vacancies on Bimetallic CoNi Nanocatalysts for Hydrodeoxygenation of Biomass-derived Vanillin to Produce Methylcyclohexanol. ACS Sustain. Chem. Eng. 2020, 8, 6075–6089. [Google Scholar] [CrossRef]
- Mochizuki, T.; Chen, S.-Y.; Toba, M.; Yoshimura, Y. Deoxygenation of guaiacol and woody tar over reduced catalysts. Appl. Catal. B 2014, 146, 237–243. [Google Scholar] [CrossRef]
- Herrera, C.; Ghampson, I.T.; Cruces, K.; Sepúlveda, C.; Barrientos, L.; Laurenti, D.; Geantet, C.; Serpell, R.; Contreras, D.; Melin, V.; et al. Valorization of biomass derivatives through the conversion of phenol over silica-supported Mo-Re oxide catalysts. Fuel 2020, 259, 116245. [Google Scholar] [CrossRef]
- Lu, M.; Sun, Y.; Zhang, P.; Zhu, J.; Li, M.; Shan, Y.; Shen, J.; Song, C. Hydrodeoxygenation of Guaiacol Catalyzed by High-Loading Ni Catalysts Supported on SiO2–TiO2 Binary Oxides. Ind. Eng. Chem. Res. 2019, 58, 1513–1524. [Google Scholar] [CrossRef]
- Dongil, A.B.; Pastor-Pérez, L.; Sepúlveda-Escribano, A.; García, R.; Escalona, N. Hydrodeoxygenation of guaiacol: Tuning the selectivity to cyclohexene by introducing Ni nanoparticles inside carbon nanotubes. Fuel 2016, 172, 65–69. [Google Scholar] [CrossRef]
- Rather, S.U. Preparation, characterization and hydrogen storage studies of carbon nanotubes and their composites: A review. Int. J. Hydrog. Energy 2020, 45, 4653–4672. [Google Scholar] [CrossRef]
- Dee la Peña O’Shea, V.A.; De la Piscina, P.R.; Homs, N.; Aromí, G.; Fierro, J.L.G. Development of Hexagonal Closed-Packed Cobalt Nanoparticles Stable at High Temperature. Chem. Mater. 2009, 21, 5637–5643. [Google Scholar] [CrossRef]
- Tsakoumis, N.E.; Patanou, E.; Lögdberg, S.; Johnsen, R.E.; Myrstad, R.; van Beek, W.; Rytter, E.; Blekkan, E.A. Structure–Performance Relationships on Co-Based Fischer–Tropsch Synthesis Catalysts: The More Defect-Free, the Better. ACS Catal. 2019, 9, 511–520. [Google Scholar] [CrossRef]
- Kitakami, O.; Sato, H.; Shimada, Y.; Sato, F.; Tanaka, M. Size effect on the crystal phase of cobalt fine particles. Phys. Rev. B 1997, 56, 13849–13854. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G | Ni/G | NiCo3:1/G | NiCo2:1/G | NiCo1:1/G | Co/G | ||
|---|---|---|---|---|---|---|---|
| SBET (m2 g−1) | 422 | 272 | 289 | 260 | 254 | 194 | |
| Vp (mL g−1) | 0.62 | 0.46 | 0.51 | 0.50 | 0.50 | 0.33 | |
| CO uptake (µmol g−1) | - | 311 | 81 | 104 | 123 | 96 | |
| Particle size (nm) | CO-chem. | - | 8 | 31 | 24 | 20 | 25 |
| TEM | - | 7 ± 1 | 9 ± 1 | 11 ± 2 | 5 ± 1 | 7 ± 1 | |
| Catalyst | Ni/G | NiCo3:1/G | NiCo2:1/G | NiCo1:1/G | Ni/G + Co/G (1:1) * | Co/G |
|---|---|---|---|---|---|---|
| r0 (10−6 mol g−1 s−1) | 8.0 | 21.6 | 16.9 | 23.5 | 11.5 | 19.9 |
| TOF (h−1) | 54 | 150 | 153 | 139 | 68 | 117 |
| SBET (m2 g−1) | Ni/G | NiCo1:1/G | Co/G |
|---|---|---|---|
| Fresh | 272 | 254 | 194 |
| Spent | 263 | 227 | 207 |
| dTEM (nm) | 7 | 6 | >50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco, E.; Dongil, A.B.; Escalona, N. Synergy between Ni and Co Nanoparticles Supported on Carbon in Guaiacol Conversion. Nanomaterials 2020, 10, 2199. https://doi.org/10.3390/nano10112199
Blanco E, Dongil AB, Escalona N. Synergy between Ni and Co Nanoparticles Supported on Carbon in Guaiacol Conversion. Nanomaterials. 2020; 10(11):2199. https://doi.org/10.3390/nano10112199
Chicago/Turabian StyleBlanco, Elodie, Ana Belen Dongil, and Néstor Escalona. 2020. "Synergy between Ni and Co Nanoparticles Supported on Carbon in Guaiacol Conversion" Nanomaterials 10, no. 11: 2199. https://doi.org/10.3390/nano10112199
APA StyleBlanco, E., Dongil, A. B., & Escalona, N. (2020). Synergy between Ni and Co Nanoparticles Supported on Carbon in Guaiacol Conversion. Nanomaterials, 10(11), 2199. https://doi.org/10.3390/nano10112199