Ultrafast Charge Generation Enhancement in Nanoscale Polymer Solar Cells with DIO Additive

 ,
, 
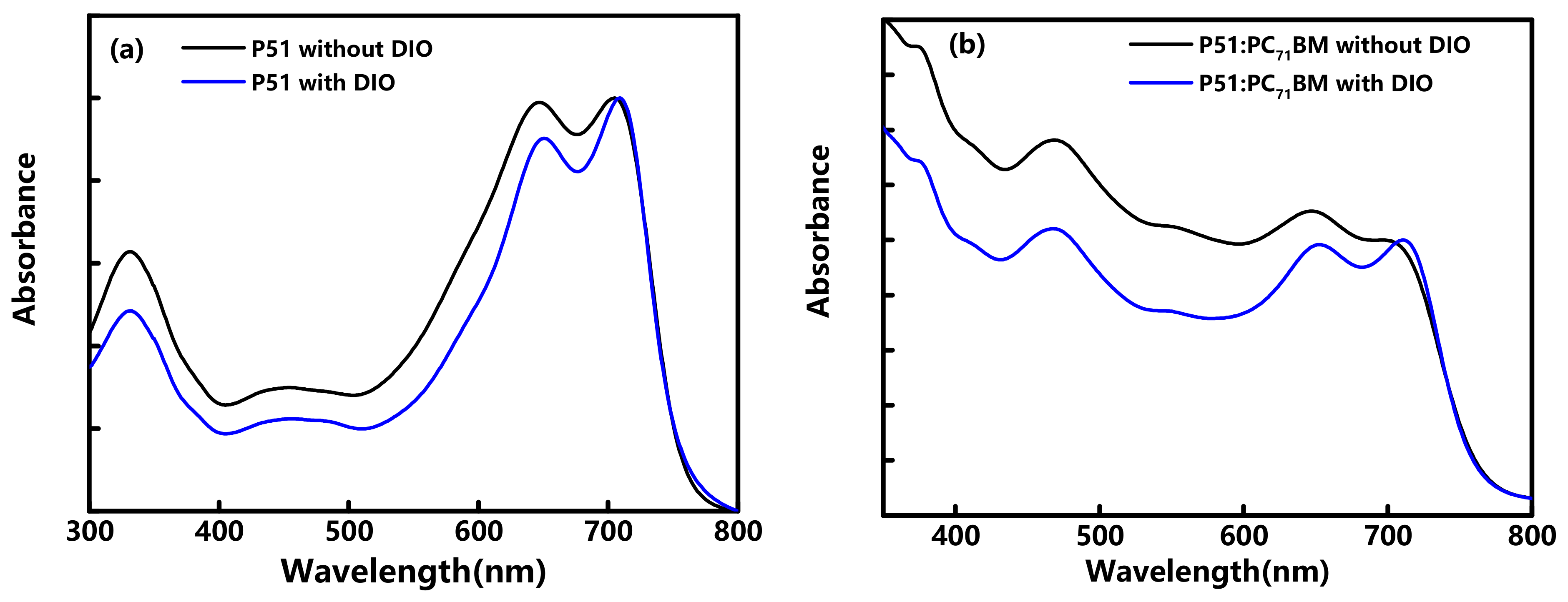{kind=link}
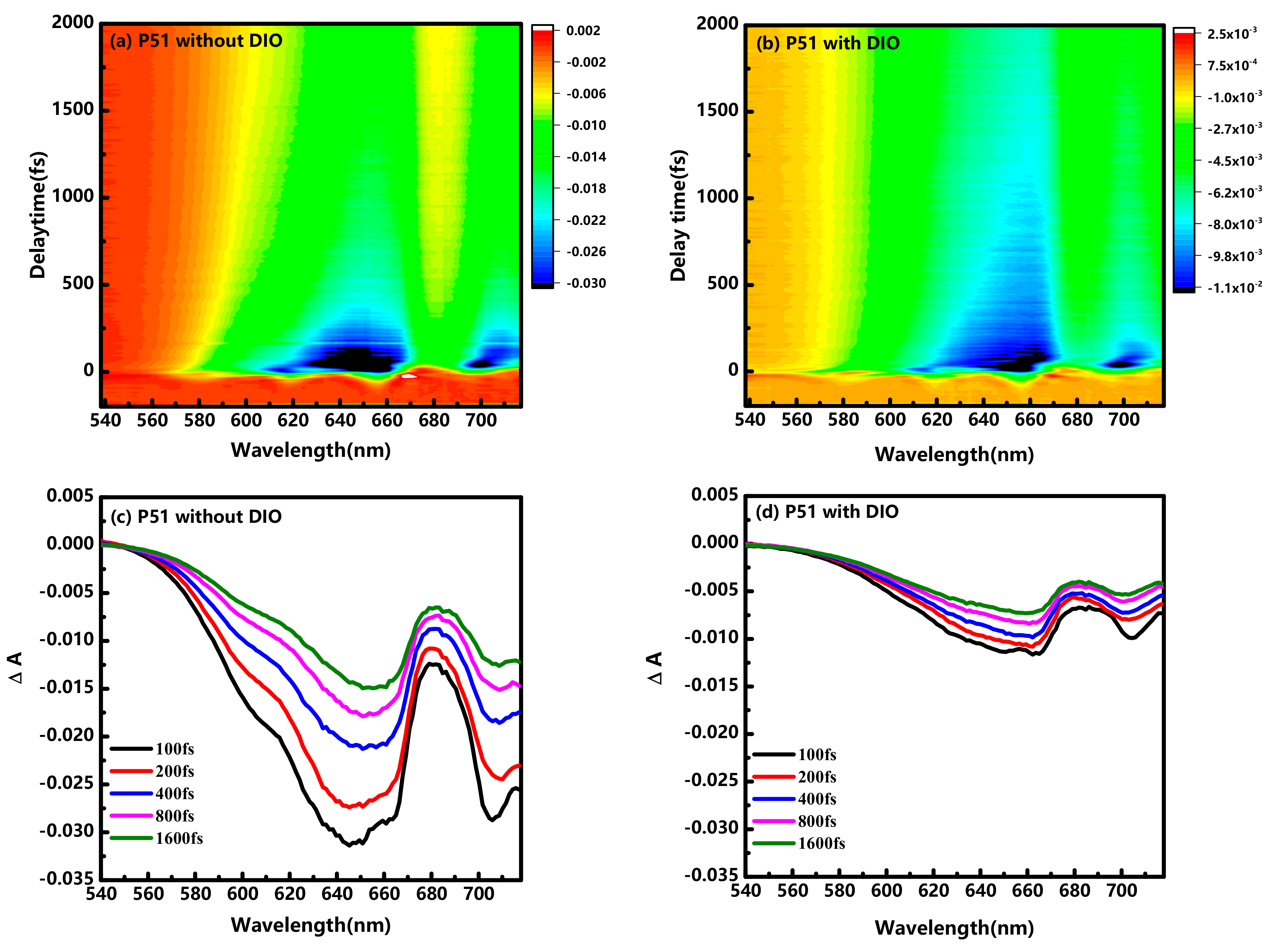{kind=link}
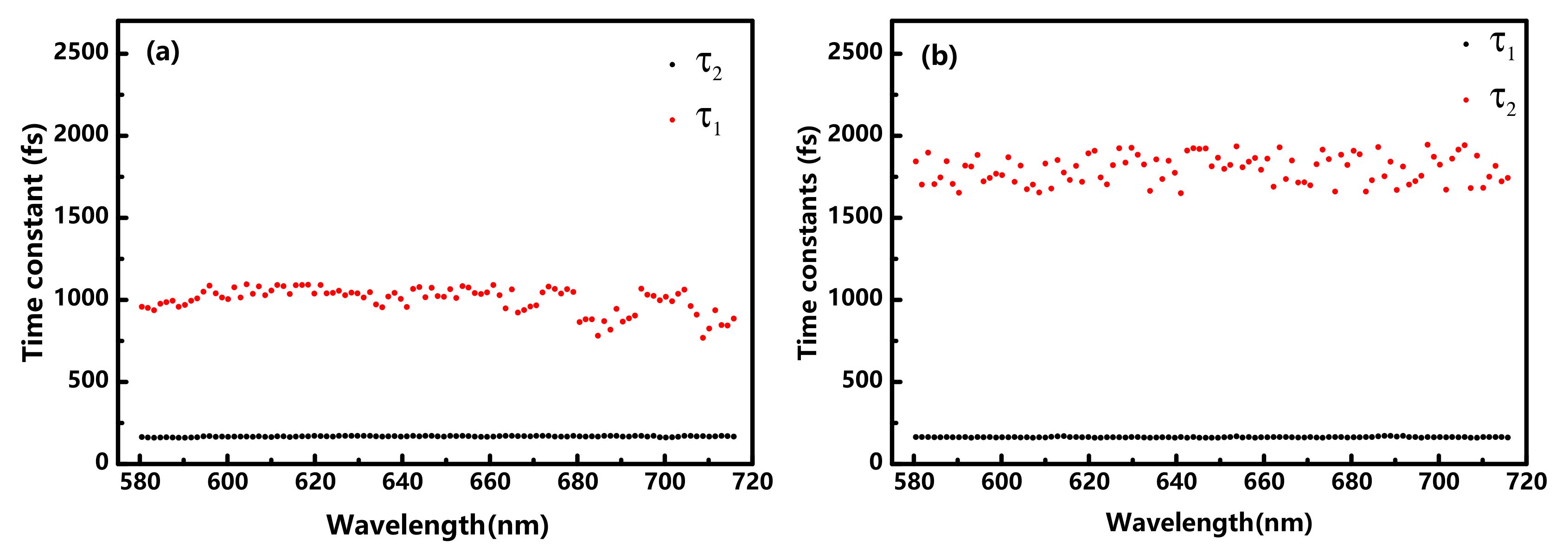{kind=link}
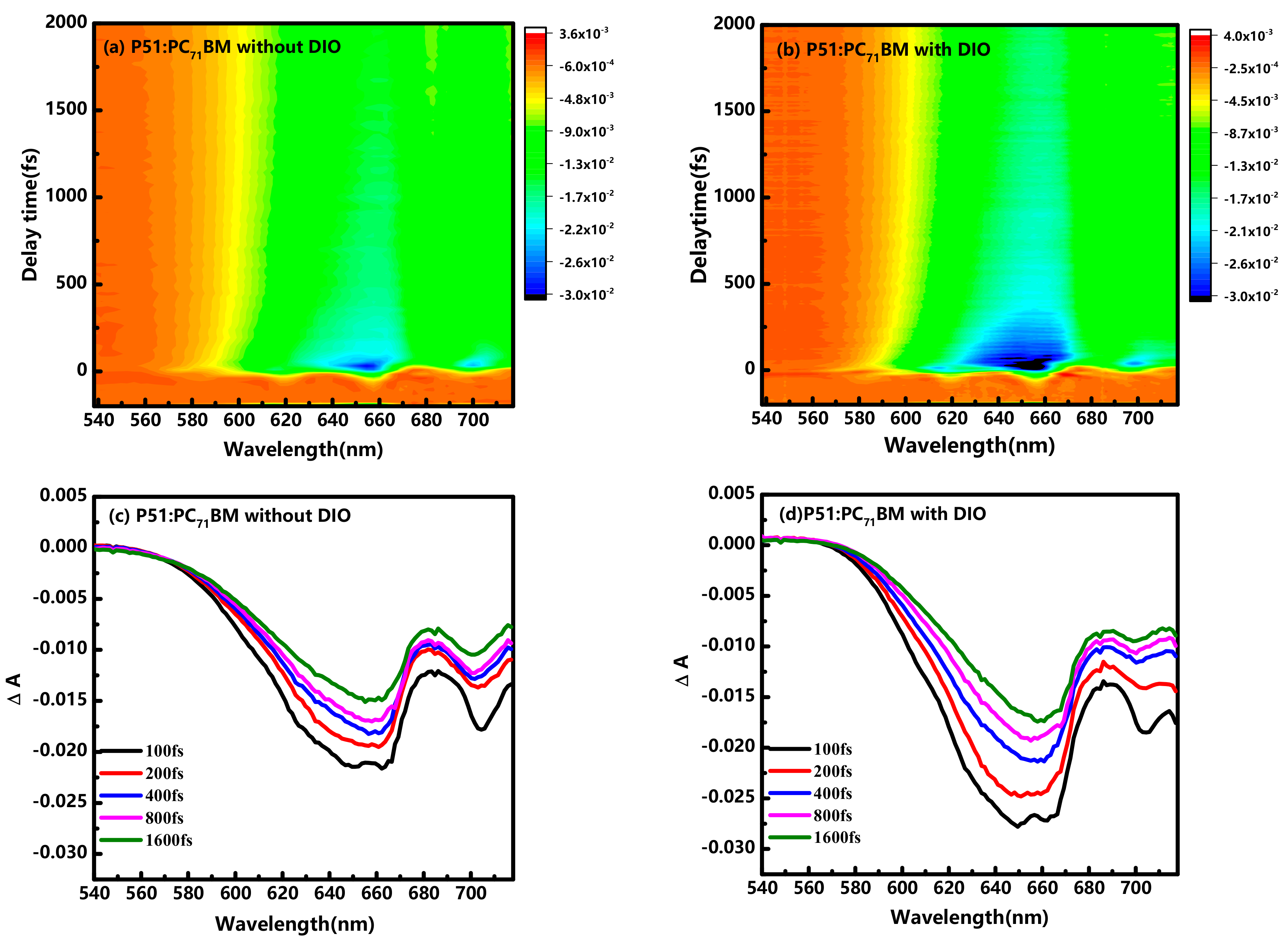{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
2.1. Material Synthesis and Optical Properties
2.2. Ultrafast Transient Absorption System
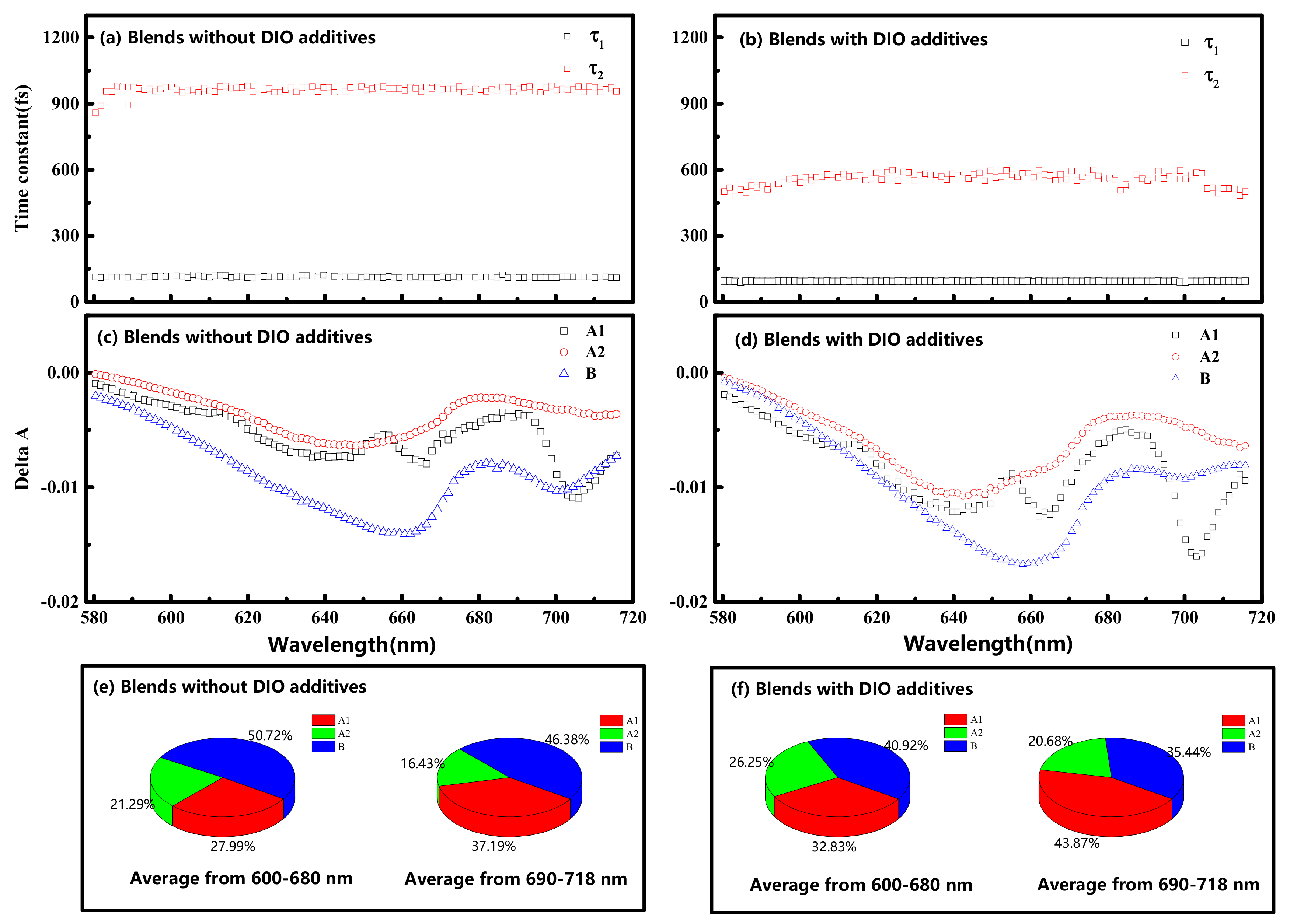
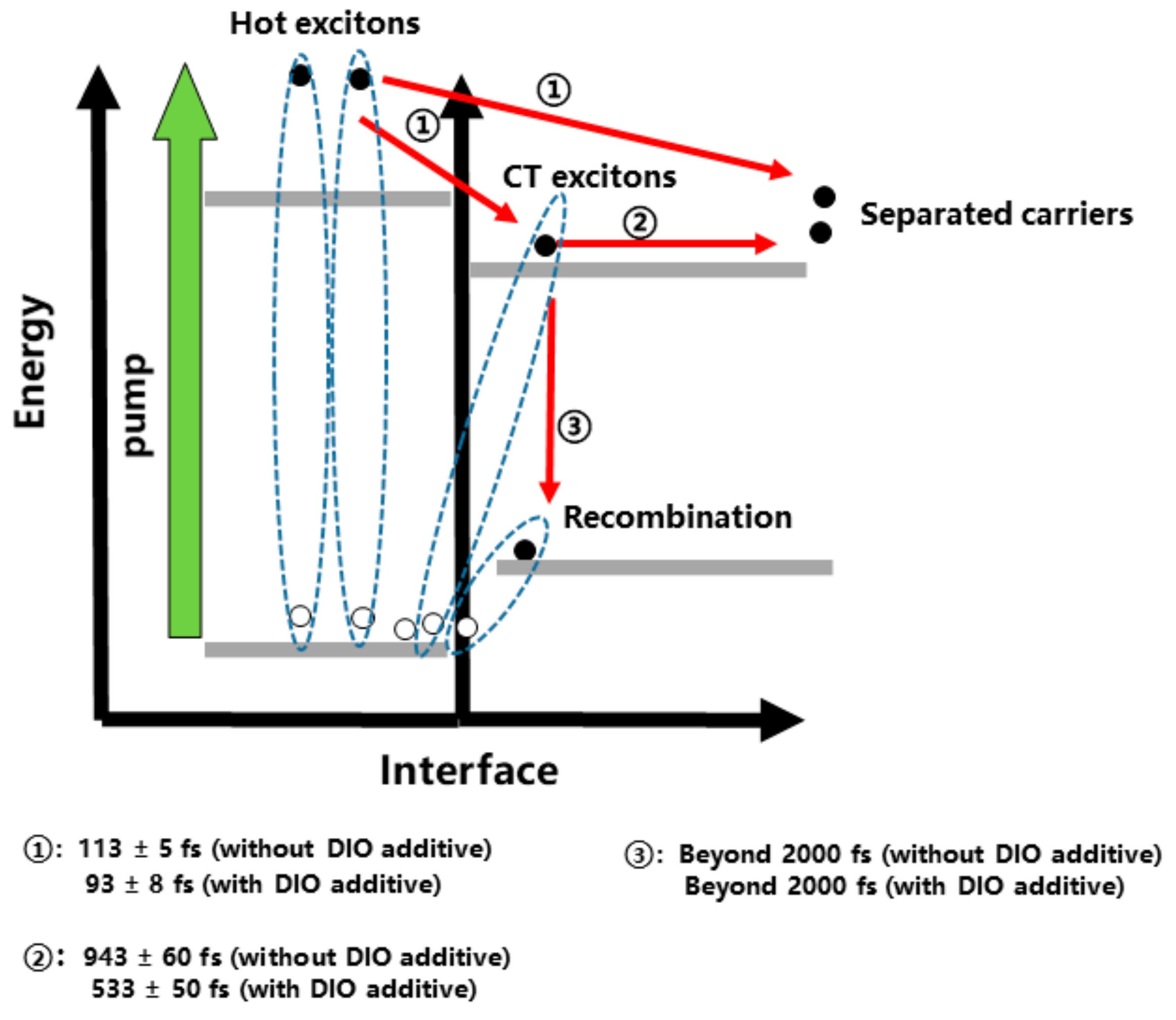
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, S.; Ye, L.; Zhao, W.; Yan, H.; Yang, B.; Liu, D.; Li, W.; Ade, H.; Hou, J. A Wide Band-Gap Polymer with a Deep HOMO Level Enables 14.2% Efficiency in Polymer Solar Cells. J. Am. Chem. Soc. 2018, 23, 7159–7167. [Google Scholar] [CrossRef]
- Hou, J.; Inganas, O.; Friend, R.H.; Gao, F. Organic solar cells based on non-fullerene acceptors. Nat. Mater. 2018, 17, 119–128. [Google Scholar] [CrossRef]
- Cui, Y.; Yao, H.; Hong, L.; Zhang, T.; Xu, Y.; Xian, K.; Gao, B.; Qin, J.; Zhang, J.; Wei, Z.; et al. Achieving Over 15% Efficiency in Organic Photovoltaic Cells via Copolymer Design. Adv. Mater. 2019, 31, 1808356. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhang, Y.; Zhou, L.; Zhang, G.; Yip, H.-L.; Lau, T.-K.; Lu, X.; Zhu, C.; Peng, H.; Johnson, P.A.; et al. Single-Junction Organic Solar Cell with over 15% Efficiency Using Fused-Ring Acceptor with Electron-Deficient Core. Joule 2019, 3, 1140–1151. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, J.; Zhang, Z.G.; Bai, H.; Li, Y.; Zhu, D.; Zhan, X. An electron acceptor challenging fullerenes for efficient polymer solar cells. Adv. Mater. 2015, 27, 1170–1174. [Google Scholar] [CrossRef] [PubMed]
- Howard, I.A.; Mauer, R.; Meister, M.; Laquai, F. Effect of morphology on ultrafast free carrier generation in polythiophene:fullerene organic solar cells. J. Am. Chem. Soc. 2010, 132, 14866. [Google Scholar] [CrossRef]
- Wan, Q.; Guo, X.; Wang, Z.; Li, W.; Guo, B.; Ma, W.; Zhang, M.; Li, Y. 10.8% Efficiency Polymer Solar Cells Based on PTB7-Th and PC71BM via Binary Solvent Additives Treatment. Adv. Funct. Mater. 2016, 26, 6635–6640. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, F.; Li, L.; An, Q.; Wang, J.; Zhang, J. The underlying reason of DIO additive on the improvement polymer solar cells performance. Appl. Surf. Sci. 2014, 305, 221–226. [Google Scholar] [CrossRef]
- Kniepert, J.; Lange, I.; Heidbrink, J.; Kurpiers, J.; Brenner, T.J.K.; Koster, L.J.A.; Neher, D. Effect of Solvent Additive on Generation, Recombination, and Extraction in PTB7:PCBM Solar Cells: A Conclusive Experimental and Numerical Simulation Study. J. Phys. Chem. C 2015, 119, 8310–8320. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, M.; Ma, W.; Ye, L.; Zhang, S.; Liu, S.; Ade, H.; Huang, F.; Hou, J. Enhanced photovoltaic performance by modulating surface composition in bulk heterojunction polymer solar cells based on PBDTTT-C-T/PC71 BM. Adv. Mater. 2014, 26, 4043–4049. [Google Scholar] [CrossRef]
- Kaake, L.G.; Welch, G.C.; Moses, D.; Bazan, G.C.; Heeger, A.J. Influence of Processing Additives on Charge-Transfer Time Scales and Sound Velocity in Organic Bulk Heterojunction Films. J. Phys. Chem. Lett. 2012, 3, 1253–1257. [Google Scholar] [CrossRef]
- Zusan, A.; Gieseking, B.; Zerson, M.; Dyakonov, V.; Magerle, R.; Deibel, C. The effect of diiodooctane on the charge carrier generation in organic solar cells based on the copolymer PBDTTT-C. Sci. Rep. 2015, 5, 8286. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Lorrmann, J.; Rauh, D.; Deibel, C.; Dyakonov, V. A New Approach for Probing the Mobility and Lifetime of Photogenerated Charge Carriers in Organic Solar Cells under Real Operating Conditions. Adv. Mater. 2012, 24, 4381–4386. [Google Scholar] [CrossRef]
- Grancini, G.; Maiuri, M.; Fazzi, D.; Petrozza, A.; Egelhaaf, H.J.; Brida, D.; Cerullo, G.; Lanzani, G. Hot exciton dissociation in polymer solar cells. Nat. Mater. 2013, 12, 29–33. [Google Scholar] [CrossRef]
- Bakulin, A.A.; Rao, A.; Pavelyev, V.G.; van Loosdrecht, P.H.M.; Pshenichnikov, M.S.; Niedzialek, D.; Cornil, J.; Beljonne, D.; Friend, R.H. The Role of Driving Energy and Delocalized States for Charge Separation in Organic Semiconductors. Science 2012, 335, 1340–1344. [Google Scholar] [CrossRef]
- Wang, R.; Yao, Y.; Zhang, C.; Zhang, Y.; Bin, H.; Xue, L.; Zhang, Z.G.; Xie, X.; Ma, H.; Wang, X.; et al. Ultrafast hole transfer mediated by polaron pairs in all-polymer photovoltaic blends. Nat. Commun. 2019, 10, 398. [Google Scholar] [CrossRef] [PubMed]
- Kaake, L.G.; Zhong, C.; Love, J.A.; Nagao, I.; Bazan, G.C.; Nguyen, T.Q.; Huang, F.; Cao, Y.; Moses, D.; Heeger, A.J. Ultrafast Charge Generation in an Organic Bilayer Film. J. Phys. Chem. Lett. 2014, 5, 2000–2006. [Google Scholar] [CrossRef]
- Tamai, Y.; Fan, Y.; Kim, V.O.; Ziabrev, K.; Rao, A.; Barlow, S.; Marder, S.R.; Friend, R.H.; Menke, S.M. Ultrafast Long-Range Charge Separation in Nonfullerene Organic Solar Cells. ACS Nano 2017, 11, 12473–12481. [Google Scholar] [CrossRef]
- Huo, L.; Zhang, S.; Guo, X.; Xu, F.; Li, Y.; Hou, J. Replacing alkoxy groups with alkylthienyl groups: A feasible approach to improve the properties of photovoltaic polymers. Angew. Chem. Int. Ed. Engl. 2011, 50, 9697–9702. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Hou, J.; Zhang, S.; Liang, Y.; Yang, G.; Yang, Y.; Yu, L.; Wu, Y.; Li, G. Polymer solar cells with enhanced open-circuit voltage and efficiency. Nat. Photonics 2009, 3, 649–653. [Google Scholar] [CrossRef]
- Shirakawa, A.; Sakane, I.; Kobayashi, T. Pulse-front-matched optical parametric amplification for sub-10-fs pulse generation tunable in the visible and near infrared. Opt. Lett. 1998, 23, 1292. [Google Scholar] [CrossRef]
- Shi, T.; Liu, Z.; Miyatake, T.; Tamiaki, H.; Kobayashi, T.; Zhang, Z.; Du, J.; Leng, Y. Ultrafast dynamics of multi-exciton state coupled to coherent vibration in zinc chlorin aggregates for artificial photosynthesis. Opt. Express 2017, 25, 29667–29675. [Google Scholar] [CrossRef] [PubMed]
- Arkhipov, V.I.; Emelianova, E.V.; Bässler, H. Hot Exciton Dissociation in a Conjugated Polymer. Phys. Rev. Lett. 1999, 82, 1321–1324. [Google Scholar] [CrossRef]
- Guo, J.; Ohkita, H.; Benten, H.; Ito, S. Near-IR Femtosecond Transient Absorption Spectroscopy of Ultrafast Polaron and Triplet Exciton Formation in Polythiophene Films with Different Regioregularities. J. Am. Chem. Soc. 2009, 131, 16869–16880. [Google Scholar] [CrossRef]
- Gélinas, S.; Paré-Labrosse, O.; Brosseau, C.-N.; Albert-Seifried, S.; McNeill, C.R.; Kirov, K.R.; Howard, I.A.; Leonelli, R.; Friend, R.H.; Silva, C. The Binding Energy of Charge-Transfer Excitons Localized at Polymeric Semiconductor Heterojunctions. J. Phys. Chem. C 2011, 115, 7114–7119. [Google Scholar] [CrossRef]
- Arkhipov, V.I.; Bässler, H. Exciton dissociation and charge photogeneration in pristine and doped conjugated polymers. Phys. Status Solidi A 2004, 201, 1152–1187. [Google Scholar] [CrossRef]
- Ai, X.; Beard, M.C.; Knutsen, K.P.; Shaheen, S.E.; Rumbles, G.; Ellingson, R.J. Photoinduced charge carrier generation in a poly(3-hexylthiophene) and methanofullerene bulk heterojunction investigated by time-resolved terahertz spectroscopy. J. Phys. Chem. B 2006, 110, 25462–25471. [Google Scholar] [CrossRef]
- Lee, Y.H.; Yabushita, A.; Hsu, C.S.; Yang, S.H.; Iwakura, I.; Luo, C.W.; Wu, K.H.; Kobayashi, T. Ultrafast relaxation dynamics of photoexcitations in poly(3-hexylthiophene) for the determination of the defect concentration. Chem. Phys. Lett. 2010, 498, 71–76. [Google Scholar] [CrossRef]
- Rao, A.; Chow, P.C.; Gelinas, S.; Schlenker, C.W.; Li, C.Z.; Yip, H.L.; Jen, A.K.; Ginger, D.S.; Friend, R.H. The role of spin in the kinetic control of recombination in organic photovoltaics. Nature 2013, 500, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Himmelberger, S.; Andersson, M.; Hanifi, D.; Xia, Y.; Zhang, S.; Wang, J.; Hou, J.; Salleo, A.; Inganas, O. The Effect of Processing Additives on Energetic Disorder in Highly Efficient Organic Photovoltaics: A Case Study on PBDTTT-C-T:PC71 BM. Adv. Mater. 2015, 27, 3868–3873. [Google Scholar] [CrossRef]
- Han, D.; Du, J.; Kobayashi, T.; Miyatake, T.; Tamiaki, H.; Li, Y.; Leng, Y. Excitonic Relaxation and Coherent Vibrational Dynamics in Zinc Chlorin Aggregates for Artificial Photosynthetic Systems. J. Phys. Chem. B 2015, 119, 12265–12273. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Nakata, K.; Jiang, Y.; Tokunaga, E.; Kobayashi, T. Spectral modulation observed in Chl-a by ultrafast laser spectroscopy. Opt. Express 2011, 19, 22480–22485. [Google Scholar] [CrossRef]
- Falke, S.M.; Rozzi, C.A.; Brida, D.; Maiuri, M.; Amato, M.; Sommer, E.; De Sio, A.; Rubio, A.; Cerullo, G.; Molinari, E.; et al. Coherent ultrafast charge transfer in an organic photovoltaic blend. Science 2014, 344, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Gélinas, S.; Rao, A.; Kumar, A.; Smith, S.L.; Chin, A.W.; Clark, J.; van der Poll, T.S.; Bazan, G.C.; Friend, R.H. Ultrafast Long-Range Charge Separation in Organic Semiconductor Photovoltaic Diodes. Science 2014, 343, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Jakowetz, A.C.; Bohm, M.L.; Zhang, J.; Sadhanala, A.; Huettner, S.; Bakulin, A.A.; Rao, A.; Friend, R.H. What Controls the Rate of Ultrafast Charge Transfer and Charge Separation Efficiency in Organic Photovoltaic Blends. J. Am. Chem. Soc. 2016, 138, 11672–11679. [Google Scholar] [CrossRef] [PubMed]
- Kaake, L.G.; Moses, D.; Heeger, A.J. Coherence and Uncertainty in Nanostructured Organic Photovoltaics. J. Phys. Chem. Lett. 2013, 4, 2264–2268. [Google Scholar] [CrossRef]
- Fu, G.; Yang, S.; Shi, J.; Zhang, Z.; Liu, B.; Zhao, X.; Li, G.; Li, X. Formation of charge-transfer complexes significantly improves the performance of polymer solar cells based on PBDTTT-C-T: PC71BM. Prog. Photovolt. Res. Appl. 2015, 23, 783–792. [Google Scholar] [CrossRef]
- Etzold, F.; Howard, I.A.; Mauer, R.; Meister, M.; Kim, T.D.; Lee, K.S.; Baek, N.S.; Laquai, F. Ultrafast exciton dissociation followed by nongeminate charge recombination in PCDTBT:PCBM photovoltaic blends. J. Am. Chem. Soc. 2011, 133, 9469–9479. [Google Scholar] [CrossRef]
- Shivanna, R.; Shoaee, S.; Dimitrov, S.; Kandappa, S.K.; Rajaram, S.; Durrant, J.R.; Narayan, K.S. Charge generation and transport in efficient organic bulk heterojunction solar cells with a perylene acceptor. Energ. Environ. Sci. 2014, 7, 435–441. [Google Scholar] [CrossRef]
- Gehrig, D.W.; Howard, I.A.; Laquai, F. Charge Carrier Generation Followed by Triplet State Formation, Annihilation, and Carrier Recreation in PBDTTT-C/PC60BM Photovoltaic Blends. J. Phys. Chem. C 2015, 119, 13509–13515. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, T.; Zhang, Z.; Guo, X.; Liu, Z.; Wang, C.; Huang, S.; Jia, T.; Quan, C.; Xiong, Q.; Zhang, M.; et al. Ultrafast Charge Generation Enhancement in Nanoscale Polymer Solar Cells with DIO Additive. Nanomaterials 2020, 10, 2174. https://doi.org/10.3390/nano10112174
Shi T, Zhang Z, Guo X, Liu Z, Wang C, Huang S, Jia T, Quan C, Xiong Q, Zhang M, et al. Ultrafast Charge Generation Enhancement in Nanoscale Polymer Solar Cells with DIO Additive. Nanomaterials. 2020; 10(11):2174. https://doi.org/10.3390/nano10112174
Chicago/Turabian StyleShi, Tongchao, Zeyu Zhang, Xia Guo, Zhengzheng Liu, Chunwei Wang, Sihao Huang, Tingyuan Jia, Chenjing Quan, Qian Xiong, Maojie Zhang, and et al. 2020. "Ultrafast Charge Generation Enhancement in Nanoscale Polymer Solar Cells with DIO Additive" Nanomaterials 10, no. 11: 2174. https://doi.org/10.3390/nano10112174
APA StyleShi, T., Zhang, Z., Guo, X., Liu, Z., Wang, C., Huang, S., Jia, T., Quan, C., Xiong, Q., Zhang, M., Du, J., & Leng, Y. (2020). Ultrafast Charge Generation Enhancement in Nanoscale Polymer Solar Cells with DIO Additive. Nanomaterials, 10(11), 2174. https://doi.org/10.3390/nano10112174