Bioinspired Extracellular Vesicles: Lessons Learned From Nature for Biomedicine and Bioengineering

 , , , ,
, , , ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
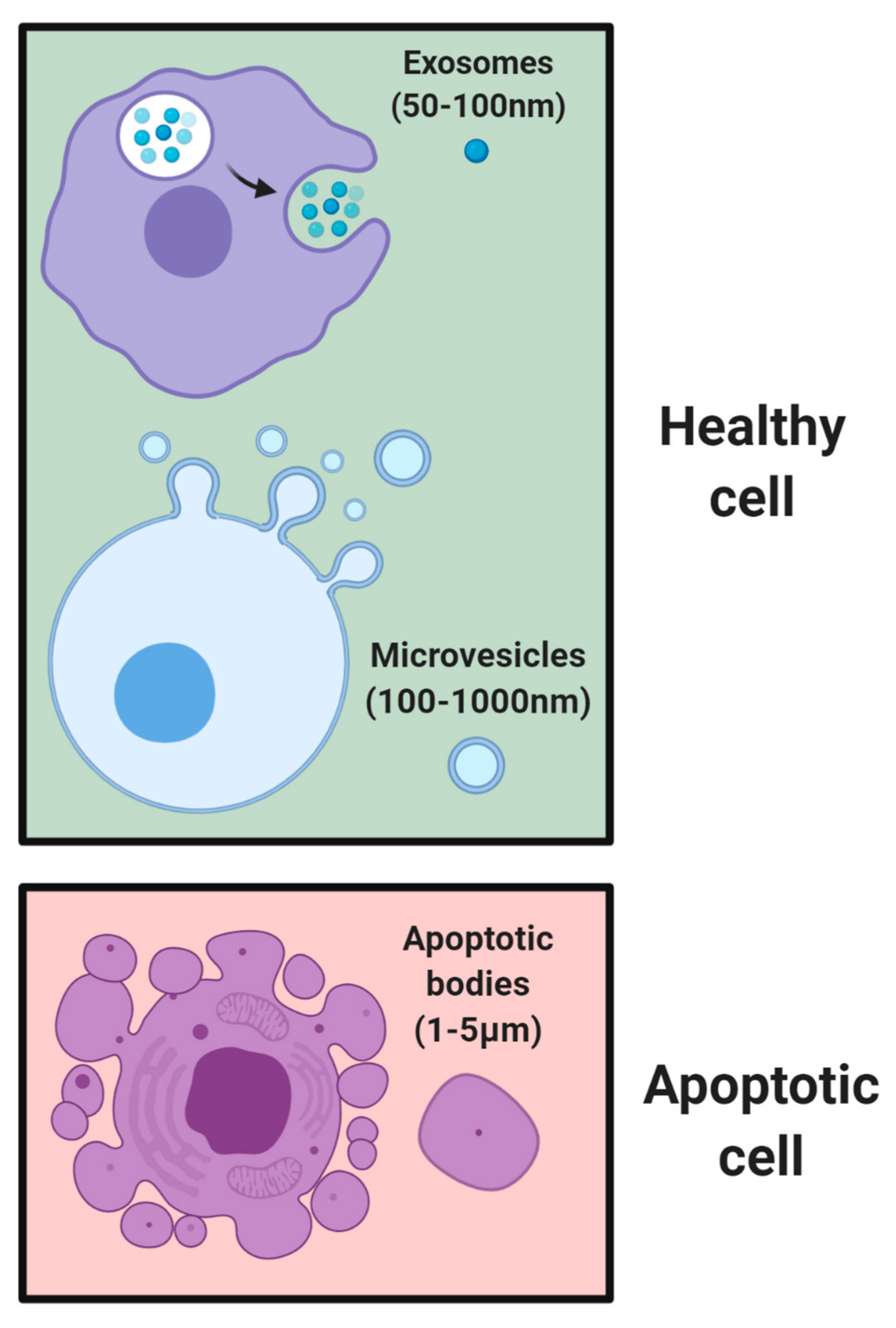
2. Native EV Communication
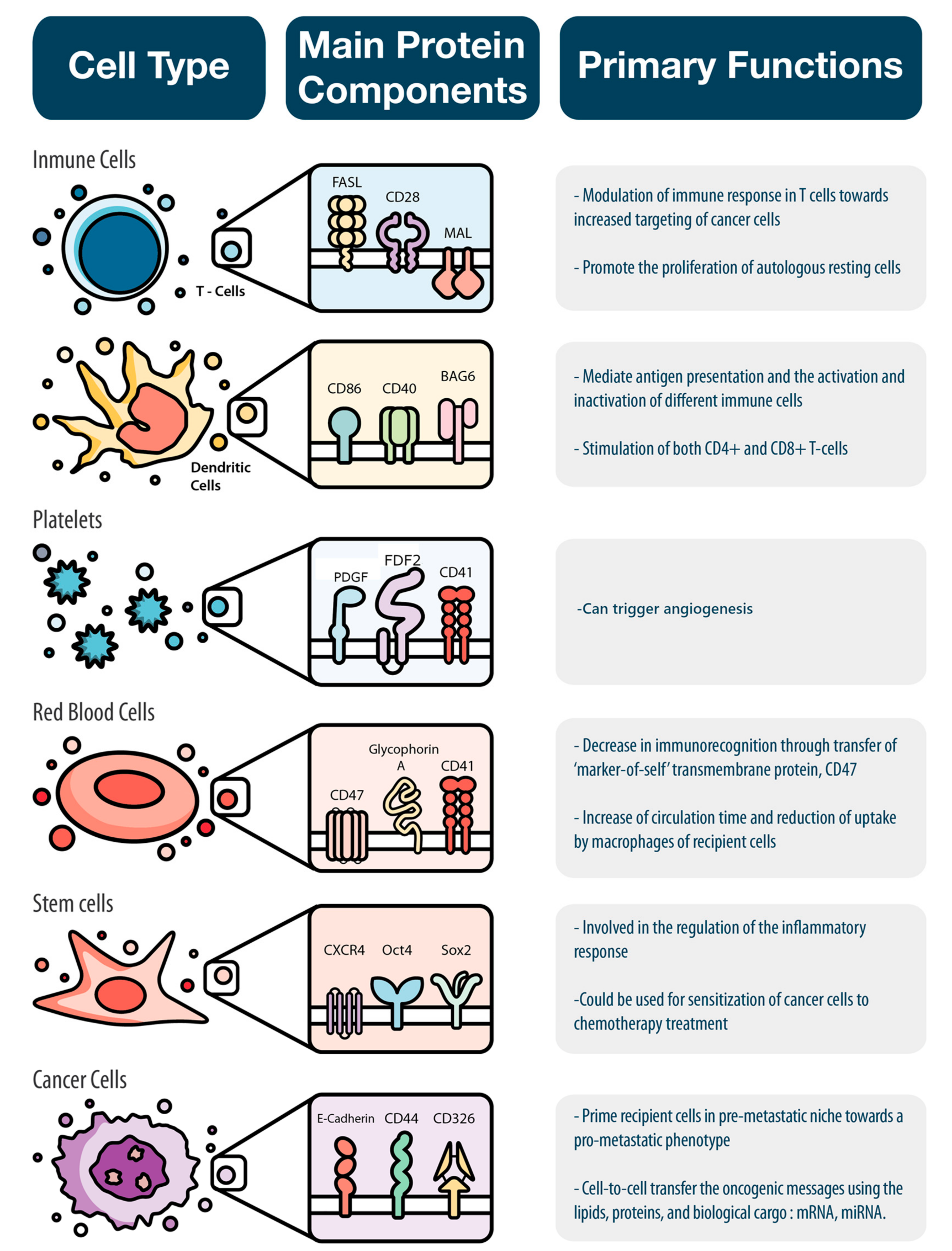
2.1. Immune Cells
2.1.1. Innate Immune System
2.1.2. Adaptive Immune System
2.2. Erythrocytes and Platelets
2.3. Stem Cells
2.3.1. Embryonic Stem Cells
2.3.2. Adult Stem Cells
2.4. Cancer Cells
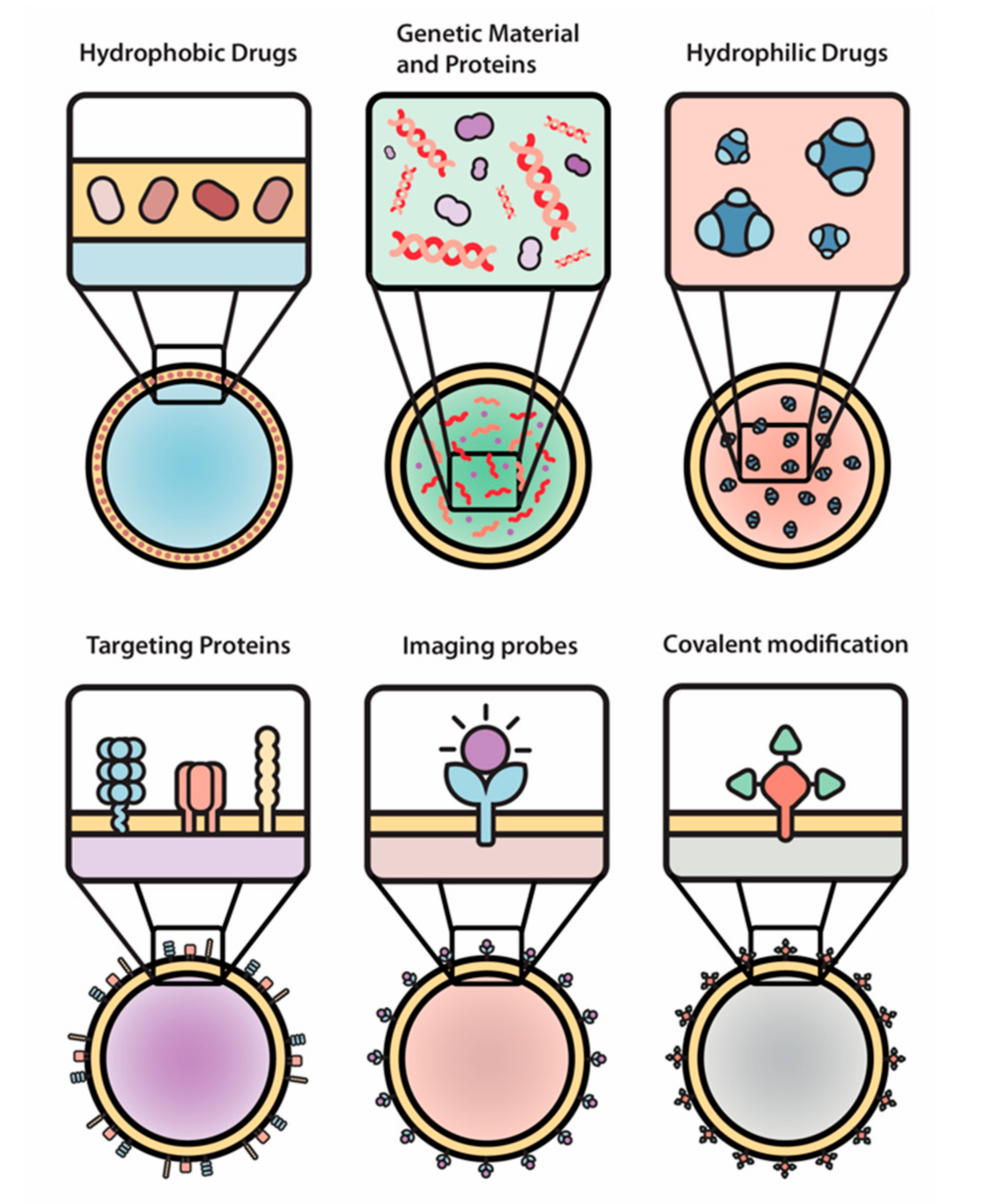
3. Engineered EVs as Drug Delivery Vehicles
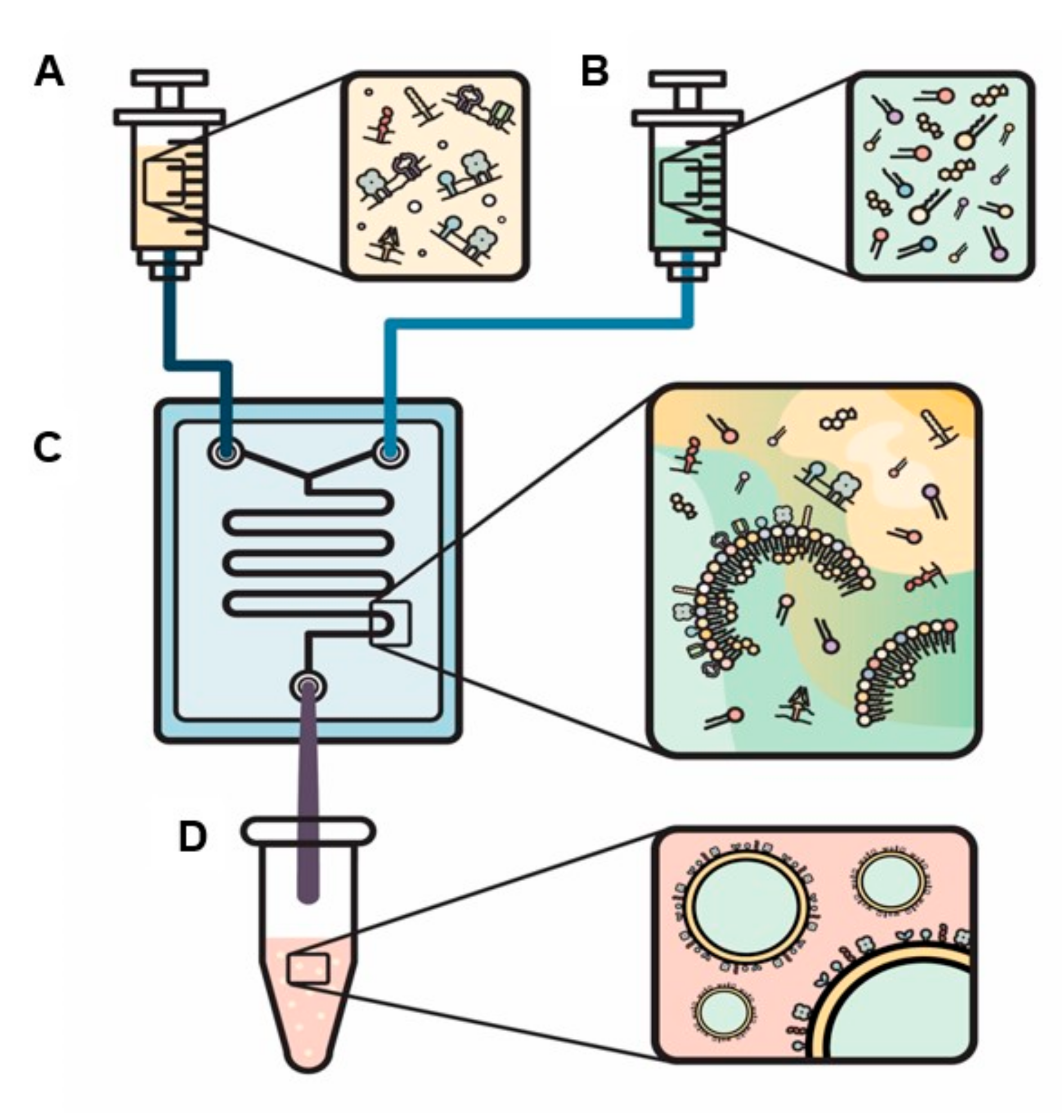
4. New Approaches to Engineer EVs
4.1. Immune Cells
4.2. Erythrocytes and Platelets
4.3. Stem Cells
4.4. Cancer Cells
5. Further Consideration for the Translation of EVs
6. Conclusions
Funding
Conflicts of Interest
References
- Mulcahy, L.; Pink, R.; Carter, D. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.; Aikawa, E.; Alcaraz, M.; Anderson, J.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.; Krishnan, A.; Verma, V.; Bronk, S.; Werneburg, N.; Charlton, M.; Shah, V.; Malhi, H.; Gores, G. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Quesenberry, P.J.; Aliotta, J.; Deregibus, M.C.; Camussi, G. Role of extracellular RNA-carrying vesicles in cell differentiation and reprogramming. Stem Cell Res. 2015, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Hosseini-Beheshti, E.; Choi, W.; Weiswald, L.; Kharmate, G.; Ghaffari, M.; Roshan-Moniri, M.; Hassona, M.; Chan, L.; Chin, M.; Tai, I. Exosomes confer pro-survival signals to alter the phenotype of prostate cells in their surrounding environment. Oncotarget 2016, 7, 14639. [Google Scholar] [CrossRef]
- De Jong, O.; Van Balkom, B.; Schiffelers, R.; Bouten, C.; Verhaar, M. Extracellular vesicles: Potential roles in regenerative medicine. Front. Immunol. 2014, 5, 608. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.; Morelli, A. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.; Weiss, J.; Kim, H.; Peinado, H.; Lyden, D. Extracellular vesicles in cancer: Cell-to-cell mediators of metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.; Sultana, Y.; Aqil, M. Liposomal drug delivery systems: An update review. Curr. Drug Deliv. 2007, 4, 297–305. [Google Scholar] [CrossRef]
- Silva, M.; Melo, S.A. Non-coding RNAs in exosomes: New players in cancer biology. Curr. Genom. 2015, 16, 295–303. [Google Scholar] [CrossRef]
- Lee, Y.; El Andaloussi, S.; Wood, M. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, R125–R134. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yu, J.; Wang, J.; Li, H.; Che, J.; Cao, B. Isolation and Identification of miRNAs in exosomes derived from serum of colon cancer patients. J. Cancer 2017, 8, 1145. [Google Scholar] [CrossRef]
- Kanada, M.; Bachmann, M.; Hardy, J.; Frimannson, D.; Bronsart, L.; Wang, A.; Sylvester, M.; Schmidt, T.; Kaspar, R.; Butte, M. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Mathivanan, S.; Ji, H.; Simpson, R. Exosomes: Extracellular organelles important in intercellular communication. J. Proteom. 2010, 73, 1907–1920. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Gyorgy, B.; Szabo, T.; Pasztoi, M.; Pal, Z.; Misjak, P.; Aradi, B.; Laszlo, V.; Pallinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. CMLS 2011, 68, 2667–2688. [Google Scholar] [CrossRef]
- Maas, S.; Breakefield, X.; Weaver, A. Extracellular vesicles: Unique intercellular delivery vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef]
- Birgegård, G. Advances and challenges in the management of essential thrombocythemia. Ther. Adv. Hematol. 2015, 6, 142–156. [Google Scholar] [CrossRef]
- Willms, E.; Johansson, H.; Mäger, I.; Lee, Y.; Blomberg, K.; Sadik, M.; Alaarg, A.; Smith, C.; Lehtiö, J.; Andaloussi, S. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Zhang, Q.; Higginbotham, J.; Jeppesen, D.; Yang, Y.; Li, W.; McKinley, E.; Graves-Deal, R.; Ping, J.; Britain, C.; Dorsett, K. Transfer of functional cargo in exomeres. Cell Rep. 2019, 27, 940–954. [Google Scholar] [CrossRef]
- Armstrong, J.; Stevens, M. Strategic design of extracellular vesicle drug delivery systems. Adv. Drug Deliv. Rev. 2018, 130, 12–16. [Google Scholar] [CrossRef]
- Parodi, A.; Molinaro, R.; Sushnitha, M.; Evangelopoulos, M.; Martinez, J.; Arrighetti, N.; Corbo, C.; Tasciotti, E. Bio-inspired engineering of cell-and virus-like nanoparticles for drug delivery. Biomaterials 2017, 147, 155–168. [Google Scholar] [CrossRef]
- von Maltzahn, G.; Park, J.; Lin, K.; Singh, N.; Schwoppe, C.; Mesters, R.; Berdel, W.; Ruoslahti, E.; Sailor, M.; Bhatia, S. Nanoparticles that communicate in vivo to amplify tumour targeting. Nat. Mater. 2011, 10, 545–552. [Google Scholar] [CrossRef]
- McKee, A.S.; MacLeod, M.K.; Kappler, J.W.; Marrack, P. Immune mechanisms of protection: Can adjuvants rise to the challenge? BMC Biol. 2010, 8, 37. [Google Scholar] [CrossRef]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Lindenbergh, M.F.S.; Koerhuis, D.G.J.; Borg, E.G.F.; van ‘t Veld, E.M.; Driedonks, T.A.P.; Wubbolts, R.; Stoorvogel, W.; Boes, M. Bystander T-Cells Support Clonal T-Cell Activation by Controlling the Release of Dendritic Cell-Derived Immune-Stimulatory Extracellular Vesicles. Front. Immunol. 2019, 10, 448. [Google Scholar] [CrossRef]
- Maacha, S.; Bhat, A.A.; Jimenez, L.; Raza, A.; Haris, M.; Uddin, S.; Grivel, J.-C. Extracellular vesicles-mediated intercellular communication: Roles in the tumor microenvironment and anti-cancer drug resistance. Mol. Cancer 2019, 18, 55. [Google Scholar] [CrossRef]
- Burrello, J.; Monticone, S.; Gai, C.; Gomez, Y.; Kholia, S.; Camussi, G. Stem cell-derived extracellular vesicles and immune-modulation. Front. Cell Dev. Biol. 2016, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Veerman, R.E.; Akpinar, G.G.; Eldh, M.; Gabrielsson, S. Immune cell-derived extracellular vesicles–Functions and therapeutic applications. Trends Mol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Biemmi, V.; Milano, G.; Ciullo, A.; Cervio, E.; Burrello, J.; Dei Cas, M.; Paroni, R.; Tallone, T.; Moccetti, T.; Pedrazzini, G.; et al. Inflammatory extracellular vesicles prompt heart dysfunction via TRL4-dependent NF-κB activation. Theranostics 2020, 10, 2773–2790. [Google Scholar] [CrossRef]
- Worbs, T.; Hammerschmidt, S.I.; Förster, R. Dendritic cell migration in health and disease. Nat. Rev. Imunnol. 2017, 17, 30. [Google Scholar] [CrossRef]
- Wang, Z.; Ding, L.; Zheng, X.-L.; Wang, H.-X.; Yan, H.-M. DC-derived exosomes induce osteogenic differentiation of mesenchymal stem cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2014, 22, 600–604. [Google Scholar]
- Zhang, B.; Yin, Y.; Lai, R.C.; Lim, S.K. Immunotherapeutic potential of extracellular vesicles. Front. Immunol. 2014, 5, 518. [Google Scholar] [CrossRef]
- Valenzuela-Vazquez, L.; Núñez-Enríquez, J.; Sánchez-Herrera, J.; Jiménez-Hernández, E.; Martín-Trejo, J.; Espinoza-Hernández, L.; Medina-Sanson, A.; Flores-Villegas, L.; Peñaloza-González, J.; Refugio Torres-Nava, J. Functional characterization of NK cells in Mexican pediatric patients with acute lymphoblastic leukemia: Report from the Mexican Interinstitutional Group for the Identification of the Causes of Childhood Leukemia. PLoS ONE 2020, 15, e0227314. [Google Scholar] [CrossRef]
- Jong, A.; Wu, C.; Li, J.; Sun, J.; Fabbri, M.; Wayne, A.; Seeger, R. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J. Extracell. Vesicles 2017, 6, 1294368. [Google Scholar] [CrossRef]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.; Lee, H.; Baek, S.; Jeong, S.; Lee, S.; Lee, J.; Ahn, B. Exosomes derived from natural killer cells exert therapeutic effect in melanoma. Theranostics 2017, 7, 2732. [Google Scholar] [CrossRef]
- Neviani, P.; Wise, P.; Murtadha, M.; Liu, C.; Wu, C.; Jong, A.; Seeger, R.; Fabbri, M. Natural killer–derived exosomal miR-186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res. 2019, 79, 1151–1164. [Google Scholar] [CrossRef]
- Seo, N.; Shirakura, Y.; Tahara, Y.; Momose, F.; Harada, N.; Ikeda, H.; Akiyoshi, K.; Shiku, H. Activated CD8+ T cell extracellular vesicles prevent tumour progression by targeting of lesional mesenchymal cells. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Admyre, C.; Bohle, B.; Johansson, S.; Focke-Tejkl, M.; Valenta, R.; Scheynius, A.; Gabrielsson, S. B cell–derived exosomes can present allergen peptides and activate allergen-specific T cells to proliferate and produce TH2-like cytokines. J. Allergy Clin. Immunol. 2007, 120, 1418–1424. [Google Scholar] [CrossRef]
- Eadie, G.; Brown, I. Analytical review: Red blood cell survival studies. Blood 1953, 8, 1110–1136. [Google Scholar] [CrossRef]
- Merkel, T.; Jones, S.; Herlihy, K.; Kersey, F.; Shields, A.; Napier, M.; Luft, J.; Wu, H.; Zamboni, W.; Wang, A. Using mechanobiological mimicry of red blood cells to extend circulation times of hydrogel microparticles. Proc. Natl. Acad. Sci. USA 2011, 108, 586–591. [Google Scholar] [CrossRef]
- Mause, S.; Ritzel, E.; Liehn, E.; Hristov, M.; Bidzhekov, K.; Muller-Newen, G.; Soehnlein, O.; Weber, C. Platelet microparticles enhance the vasoregenerative potential of angiogenic early outgrowth cells after vascular injury. Circulation 2010, 122, 495–506. [Google Scholar] [CrossRef]
- Hayon, Y.; Dashevsky, O.; Shai, E.; Brill, A.; Varon, D.; Leker, R. Platelet microparticles induce angiogenesis and neurogenesis after cerebral ischemia. Curr. Neurovasc. Res. 2012, 9, 185–192. [Google Scholar] [CrossRef]
- Jeong, H.; Yim, H.; Park, H.; Cho, Y.; Hong, H.; Kim, N.; Oh, I. Mesenchymal Stem Cell Therapy for Ischemic Heart Disease: Systematic Review and Meta-analysis. Int. J. Stem Cells 2018, 11, 1. [Google Scholar] [CrossRef]
- Burdon, T.; Paul, A.; Noiseux, N.; Prakash, S.; Shum-Tim, D. Bone marrow stem cell derived paracrine factors for regenerative medicine: Current perspectives and therapeutic potential. Bone Marrow Res. 2011, 2011, 207326. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Deregibus, M.; Cantaluppi, V. Role of stem-cell-derived microvesicles in the paracrine action of stem cells. Biochem. Soc. Trans. 2013, 41, 283–287. [Google Scholar] [CrossRef]
- Tsuji, K.; Kitamura, S. Trophic Factors from Tissue Stem Cells for Renal Regeneration. Stem Cells Int. 2015, 2015, 537204. [Google Scholar] [CrossRef]
- Ratajczak, J.; Miekus, K.; Kucia, M.; Zhang, J.; Reca, R.; Dvorak, P.; Ratajczak, M. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia 2006, 20, 847–856. [Google Scholar] [CrossRef]
- Deregibus, M.; Cantaluppi, V.; Calogero, R.; Lo Iacono, M.; Tetta, C.; Biancone, L.; Bruno, S.; Bussolati, B.; Camussi, G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 2007, 110, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Quesenberry, P.; Aliotta, J. Cellular phenotype switching and microvesicles. Adv. Drug Deliv. Rev. 2010, 62, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Kamdje, A.; Kamga, P.; Simo, R.; Vecchio, L.; Etet, P.; Muller, J.; Bassi, G.; Lukong, E.; Goel, R.; Amvene, J. Mesenchymal stromal cells’ role in tumor microenvironment: Involvement of signaling pathways. Cancer Biol. Med. 2017, 14, 129. [Google Scholar]
- Balakrishnan, K.; Burger, J.; Quiroga, M.; Henneberg, M.; Ayres, M.; Wierda, W.; Gandhi, V. Influence of bone marrow stromal microenvironment on forodesine-induced responses in CLL primary cells. Blood 2010, 116, 1083–1091. [Google Scholar] [CrossRef]
- Huang, W.; Chang, M.; Tsai, K.; Hung, M.; Chen, H.; Hung, S. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene 2013, 32, 4343–4354. [Google Scholar] [CrossRef]
- Morad, S.; Cabot, M. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Ju, G.; Wu, S.; Cheng, Z.; Cheng, J.; Zou, X.; Zhang, G.; Miao, S.; Liu, G.; Zhu, Y. Microvesicles derived from human Wharton’s jelly mesenchymal stem cells promote human renal cancer cell growth and aggressiveness through induction of hepatocyte growth factor. PLoS ONE 2014, 9, e96836. [Google Scholar] [CrossRef]
- Wu, S.; Ju, G.; Du, T.; Zhu, Y.; Liu, G. Microvesicles derived from human umbilical cord Wharton’s jelly mesenchymal stem cells attenuate bladder tumor cell growth in vitro and in vivo. PLoS ONE 2013, 8, e61366. [Google Scholar] [CrossRef]
- Del Fattore, A.; Luciano, R.; Saracino, R.; Battafarano, G.; Rizzo, C.; Pascucci, L.; Alessandri, G.; Pessina, A.; Perrotta, A.; Fierabracci, A.; et al. Differential effects of extracellular vesicles secreted by mesenchymal stem cells from different sources on glioblastoma cells. Expert Opin. Biol. 2015, 15, 495–504. [Google Scholar] [CrossRef]
- Otsuru, S.; Desbourdes, L.; Guess, A.J.; Hofmann, T.J.; Relation, T.; Kaito, T.; Dominici, M.; Iwamoto, M.; Horwitz, E.M. Extracellular vesicles released from mesenchymal stromal cells stimulate bone growth in osteogenesis imperfecta. Cytotherapy 2018, 20, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G.; Prockop, D.J. Concise review: Mesenchymal stem/multipotent stromal cells: The state of transdifferentiation and modes of tissue repair—Current views. Stem Cells 2007, 25, 2896–2902. [Google Scholar] [CrossRef]
- Zhang, J.; Guan, J.; Niu, X.; Hu, G.; Guo, S.; Li, Q.; Xie, Z.; Zhang, C.; Wang, Y. Exosomes released from human induced pluripotent stem cells-derived MSCs facilitate cutaneous wound healing by promoting collagen synthesis and angiogenesis. J. Transl. Med. 2015, 13, 49. [Google Scholar] [CrossRef]
- Lin, L.; Du, L.; Cao, K.; Huang, Y.; Yu, P.; Zhang, L.; Li, F.; Wang, Y.; Shi, Y. Tumour cell-derived exosomes endow mesenchymal stromal cells with tumour-promotion capabilities. Oncogene 2016, 35, 6038–6042. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Ohyashiki, K.; Kuroda, M.; Ohyashiki, J. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 2013, 32, 2747–2755. [Google Scholar] [CrossRef]
- Yu, D.; Wu, Y.; Shen, H.; Lv, M.M.; Chen, W.; Zhang, X.; Zhong, S.; Tang, J.; Zhao, J. Exosomes in development, metastasis and drug resistance of breast cancer. Cancer Sci. 2015, 106, 959–964. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Rana, S.; Yue, S.; Stadel, D.; Zoller, M. Toward tailored exosomes: The exosomal tetraspanin web contributes to target cell selection. Int. J. Biochem. Cell Biol. 2012, 44, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, H. Engineering of extracellular vesicles as drug delivery vehicles. Stem Cell Investig. 2017, 4, 74. [Google Scholar] [CrossRef]
- Tominaga, N.; Yoshioka, Y.; Ochiya, T. A novel platform for cancer therapy using extracellular vesicles. Adv. Drug Deliv. Rev. 2015, 95, 50–55. [Google Scholar] [CrossRef]
- Kooijmans, S.; Schiffelers, R.; Zarovni, N.; Vago, R. Modulation of tissue tropism and biological activity of exosomes and other extracellular vesicles: New nanotools for cancer treatment. Pharm. Res. 2016, 111, 487–500. [Google Scholar] [CrossRef]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef]
- Tang, K.; Zhang, Y.; Zhang, H.; Xu, P.; Liu, J.; Ma, J.; Lv, M.; Li, D.; Katirai, F.; Shen, G.; et al. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat. Commun. 2012, 3, 1282. [Google Scholar] [CrossRef]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Poste, G.; Nicolson, G. Arrest and metastasis of blood-borne tumor cells are modified by fusion of plasma membrane vesicles from highly metastatic cells. Proc. Natl. Acad. Sci. USA 1980, 77, 399–403. [Google Scholar] [CrossRef]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.; Gainche, L.; Sena-Esteves, M.; Curry, W.; Carter, B.; Krichevsky, A.; Breakefield, X. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Andre, F.; Schartz, N.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; Le Chevalier, T.; et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Thery, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Hellwinkel, J.; Redzic, J.; Harland, T.; Gunaydin, D.; Anchordoquy, T.; Graner, M. Glioma-derived extracellular vesicles selectively suppress immune responses. Neuro Oncol. 2016, 18, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.; Holme, M.; Stevens, M. Re-engineering extracellular vesicles as smart nanoscale therapeutics. ACS Nano 2017, 11, 69–83. [Google Scholar] [CrossRef]
- Kim, H.; Kim, D.; Nam, H.; Moon, S.; Kwon, Y.; Lee, J. Engineered extracellular vesicles and their mimetics for clinical translation. Methods 2019. [Google Scholar] [CrossRef]
- Batrakova, E.; Kim, M. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J. Control. Release 2015, 219, 396–405. [Google Scholar] [CrossRef]
- Mentkowski, K.; Snitzer, J.; Rusnak, S.; Lang, J. Therapeutic potential of engineered extracellular vesicles. AAPS J. 2018, 20, 50. [Google Scholar] [CrossRef]
- Zinger, A.; Baudo, G.; Naoi, T.; Giordano, F.; Lenna, S.; Massaro, M.; Ewing, A.; Kim, H.R.; Tasciotti, E.; Yustein, J.T.; et al. Reproducible and Characterized Method for Ponatinib Encapsulation into Biomimetic Lipid Nanoparticles as a Platform for Multi Tyrosine Kinase Targeted Therapy. ACS Appl. Bio Mater. 2020, 3. [Google Scholar] [CrossRef]
- Haney, M.; Klyachko, N.; Zhao, Y.; Gupta, R.; Plotnikova, E.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.; de Jong, O.; Brouwer, M.; Wood, M.; Lavieu, G.; Schiffelers, R.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shtam, T.; Kovalev, R.; Varfolomeeva, E.; Makarov, E.; Kil, Y.; Filatov, M. Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun. Signal. 2013, 11, 88. [Google Scholar] [CrossRef]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacologica Sinica 2017, 38, 754–763. [Google Scholar] [CrossRef]
- O’Neill, C.; Gilligan, K.; Dwyer, R. Role of extracellular vesicles (EVs) in cell stress response and resistance to cancer therapy. Cancers 2019, 11, 136. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Zipkin, M. Exosome redux. Nat. Biotechnol. 2019, 37, 1395–1400. [Google Scholar] [CrossRef]
- Haraszti, R.; Miller, R.; Stoppato, M.; Sere, Y.; Coles, A.; Didiot, M.; Wollacott, R.; Sapp, E.; Dubuke, M.; Li, X. Exosomes produced from 3D cultures of MSCs by tangential flow filtration show higher yield and improved activity. Mol. Ther. 2018, 26, 2838–2847. [Google Scholar] [CrossRef]
- Smyth, T.; Petrova, K.; Payton, N.; Persaud, I.; Redzic, J.; Graner, M.; Smith-Jones, P.; Anchordoquy, T. Surface functionalization of exosomes using click chemistry. Bioconjugate Chem. 2014, 25, 1777–1784. [Google Scholar] [CrossRef]
- Kim, M.; Haney, M.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.; Kabanov, A.; Batrakova, E. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Llorente, A.; Skotland, T.; Sylvänne, T.; Kauhanen, D.; Róg, T.; Orłowski, A.; Vattulainen, I.; Ekroos, K.; Sandvig, K. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochimica Biophysica Acta Mol. Cell Biol. Lipids 2013, 1831, 1302–1309. [Google Scholar] [CrossRef]
- Van Dommelen, S.; Vader, P.; Lakhal, S.; Kooijmans, S.; van Solinge, W.; Wood, M.; Schiffelers, R. Microvesicles and exosomes: Opportunities for cell-derived membrane vesicles in drug delivery. J. Control. Release 2012, 161, 635–644. [Google Scholar] [CrossRef]
- Sato, Y.; Umezaki, K.; Sawada, S.; Mukai, S.; Sasaki, Y.; Harada, N.; Shiku, H.; Akiyoshi, K. Engineering hybrid exosomes by membrane fusion with liposomes. Sci. Rep. 2016, 6, 21933. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.; Zhang, L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, R.; Evangelopoulos, M.; Hoffman, J.; Corbo, C.; Taraballi, F.; Martinez, J.; Hartman, K.; Cosco, D.; Costa, G.; Romeo, I. Design and Development of Biomimetic Nanovesicles Using a Microfluidic Approach. Adv. Mater. 2018, 30, 1702749. [Google Scholar] [CrossRef]
- Zhai, Y.; Su, J.; Ran, W.; Zhang, P.; Yin, Q.; Zhang, Z.; Yu, H.; Li, Y. Preparation and Application of Cell Membrane-Camouflaged Nanoparticles for Cancer Therapy. Theranostics 2017, 7, 2575–2592. [Google Scholar] [CrossRef]
- Corbo, C.; Parodi, A.; Evangelopoulos, M.; Engler, D.; Matsunami, R.; Engler, A.; Molinaro, R.; Scaria, S.; Salvatore, F.; Tasciotti, E. Proteomic Profiling of a Biomimetic Drug Delivery Platform. Curr. Drug Targets 2015, 16, 1540–1547. [Google Scholar] [CrossRef]
- Evangelopoulos, M.; Parodi, A.; Martinez, J.; Yazdi, I.; Cevenini, A.; van de Ven, A.; Quattrocchi, N.; Boada, C.; Taghipour, N.; Corbo, C. Cell source determines the immunological impact of biomimetic nanoparticles. Biomaterials 2016, 82, 168–177. [Google Scholar] [CrossRef]
- Parodi, A.; Quattrocchi, N.; Van De Ven, A.; Chiappini, C.; Evangelopoulos, M.; Martinez, J.; Brown, B.; Khaled, S.; Yazdi, I.; Enzo, M. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2013, 8, 61. [Google Scholar] [CrossRef]
- Molinaro, R.; Corbo, C.; Martinez, J.; Taraballi, F.; Evangelopoulos, M.; Minardi, S.; Yazdi, I.; Zhao, P.; De Rosa, E.; Sherman, M.; et al. Biomimetic proteolipid vesicles for targeting inflamed tissues. Nat. Mater. 2016, 15, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Boada, C.; Zinger, A.; Tsao, C.; Zhao, P.; Martinez, J.O.; Hartman, K.; Naoi, T.; Sukhoveshin, R.; Sushnitha, M.; Molinaro, R. Rapamycin-loaded biomimetic nanoparticles reverse vascular inflammation. Circ. Res. 2020, 126, 25–37. [Google Scholar] [CrossRef]
- Pitchaimani, A.; Nguyen, T.D.T.; Aryal, S. Natural killer cell membrane infused biomimetic liposomes for targeted tumor therapy. Biomaterials 2018, 160, 124–137. [Google Scholar] [CrossRef]
- Kalaydina, R.-V.; Bajwa, K.; Qorri, B.; Decarlo, A.; Szewczuk, M.R. Recent advances in “smart” delivery systems for extended drug release in cancer therapy. Int. J. Nanomed. 2018, 13, 4727. [Google Scholar] [CrossRef]
- Doshi, N.; Zahr, A.; Bhaskar, S.; Lahann, J.; Mitragotri, S. Red blood cell-mimicking synthetic biomaterial particles. Proc. Natl. Acad. Sci. USA 2009, 106, 21495–21499. [Google Scholar] [CrossRef]
- Usman, W.; Pham, T.; Kwok, Y.; Vu, L.; Ma, V.; Peng, B.; San Chan, Y.; Wei, L.; Chin, S.; Azad, A. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef]
- Hu, C.; Fang, R.; Luk, B.; Chen, K.; Carpenter, C.; Gao, W.; Zhang, K.; Zhang, L. ‘Marker-of-self’ functionalization of nanoscale particles through a top-down cellular membrane coating approach. Nanoscale 2013, 5, 2664–2668. [Google Scholar] [CrossRef]
- Anselmo, A.; Modery-Pawlowski, C.; Menegatti, S.; Kumar, S.; Vogus, D.; Tian, L.; Chen, M.; Squires, T.; Sen Gupta, A.; Mitragotri, S. Platelet-like nanoparticles: Mimicking shape, flexibility, and surface biology of platelets to target vascular injuries. ACS Nano 2014, 8, 11243–11253. [Google Scholar] [CrossRef]
- Riazifar, M.; Pone, E.; Lotvall, J.; Zhao, W. Stem Cell Extracellular Vesicles: Extended Messages of Regeneration. Annu. Rev. Pharm. Toxicol. 2017, 57, 125–154. [Google Scholar] [CrossRef]
- Yuan, Z.; Kolluri, K.; Gowers, K.; Janes, S. TRAIL delivery by MSC-derived extracellular vesicles is an effective anticancer therapy. J. Extracell. Vesicles 2017, 6, 1265291. [Google Scholar] [CrossRef]
- Glinsky, V.; Glinsky, G.; Glinskii, O.; Huxley, V.; Turk, J.; Mossine, V.; Deutscher, S.; Pienta, K.; Quinn, T. Intravascular metastatic cancer cell homotypic aggregation at the sites of primary attachment to the endothelium. Cancer Res. 2003, 63, 3805–3811. [Google Scholar] [CrossRef]
- Sun, H.; Su, J.; Meng, Q.; Yin, Q.; Chen, L.; Gu, W.; Zhang, P.; Zhang, Z.; Yu, H.; Wang, S.; et al. Cancer-Cell-Biomimetic Nanoparticles for Targeted Therapy of Homotypic Tumors. Adv. Mater. 2016, 28, 9581–9588. [Google Scholar] [CrossRef]
- Sun, H.; Su, J.; Meng, Q.; Yin, Q.; Chen, L.; Gu, W.; Zhang, Z.; Yu, H.; Zhang, P.; Wang, S.; et al. Cancer Cell Membrane-Coated Gold Nanocages with Hyperthermia-Triggered Drug Release and Homotypic Target Inhibit Growth and Metastasis of Breast Cancer. Adv. Funct. Mater. 2017, 27, 1604300. [Google Scholar] [CrossRef]
- Rodriguez, P.; Harada, T.; Christian, D.; Pantano, D.; Tsai, R.; Discher, D. Minimal “Self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science 2013, 339, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, Y.; Tang, K.; Zhang, H.; Yin, X.; Li, Y.; Xu, P.; Sun, Y.; Ma, R.; Ji, T. Reversing drug resistance of soft tumor-repopulating cells by tumor cell-derived chemotherapeutic microparticles. Cell Res. 2016, 26, 713–727. [Google Scholar] [CrossRef]
- Furi, I.; Momen-Heravi, F.; Szabo, G. Extracellular vesicle isolation: Present and future. Ann. Transl. Med. 2017, 5. [Google Scholar] [CrossRef]
- Hartjes, T.A.; Mytnyk, S.; Jenster, G.W.; van Steijn, V.; van Royen, M.E. Extracellular vesicle quantification and characterization: Common methods and emerging approaches. Bioengineering 2019, 6, 7. [Google Scholar] [CrossRef]
- Jeyaram, A.; Jay, S.M. Preservation and storage stability of extracellular vesicles for therapeutic applications. AAPS J. 2018, 20, 1. [Google Scholar] [CrossRef]
- Gardiner, C.; Vizio, D.D.; Sahoo, S.; Théry, C.; Witwer, K.W.; Wauben, M.; Hill, A.F. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. J. Extracell. Vesicles 2016, 5, 32945. [Google Scholar] [CrossRef]
- Bosch, S.; De Beaurepaire, L.; Allard, M.; Mosser, M.; Heichette, C.; Chrétien, D.; Jegou, D.; Bach, J.-M. Trehalose prevents aggregation of exosomes and cryodamage. Sci. Rep. 2016, 6, 36162. [Google Scholar] [CrossRef]
- Frank, J.; Richter, M.; de Rossi, C.; Lehr, C.; Fuhrmann, K.; Fuhrmann, G. Extracellular vesicles protect glucuronidase model enzymes during freeze-drying. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zinger, A.; Brozovich, A.; Pasto, A.; Sushnitha, M.; Martinez, J.O.; Evangelopoulos, M.; Boada, C.; Tasciotti, E.; Taraballi, F. Bioinspired Extracellular Vesicles: Lessons Learned From Nature for Biomedicine and Bioengineering. Nanomaterials 2020, 10, 2172. https://doi.org/10.3390/nano10112172
Zinger A, Brozovich A, Pasto A, Sushnitha M, Martinez JO, Evangelopoulos M, Boada C, Tasciotti E, Taraballi F. Bioinspired Extracellular Vesicles: Lessons Learned From Nature for Biomedicine and Bioengineering. Nanomaterials. 2020; 10(11):2172. https://doi.org/10.3390/nano10112172
Chicago/Turabian StyleZinger, Assaf, Ava Brozovich, Anna Pasto, Manuela Sushnitha, Jonathan O. Martinez, Michael Evangelopoulos, Christian Boada, Ennio Tasciotti, and Francesca Taraballi. 2020. "Bioinspired Extracellular Vesicles: Lessons Learned From Nature for Biomedicine and Bioengineering" Nanomaterials 10, no. 11: 2172. https://doi.org/10.3390/nano10112172
APA StyleZinger, A., Brozovich, A., Pasto, A., Sushnitha, M., Martinez, J. O., Evangelopoulos, M., Boada, C., Tasciotti, E., & Taraballi, F. (2020). Bioinspired Extracellular Vesicles: Lessons Learned From Nature for Biomedicine and Bioengineering. Nanomaterials, 10(11), 2172. https://doi.org/10.3390/nano10112172