Highly Porous Amorphous Calcium Phosphate for Drug Delivery and Bio-Medical Applications


 , ,
, ,  and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of ACPs and CaPs
2.3. Characterization
3. Results and Discussion
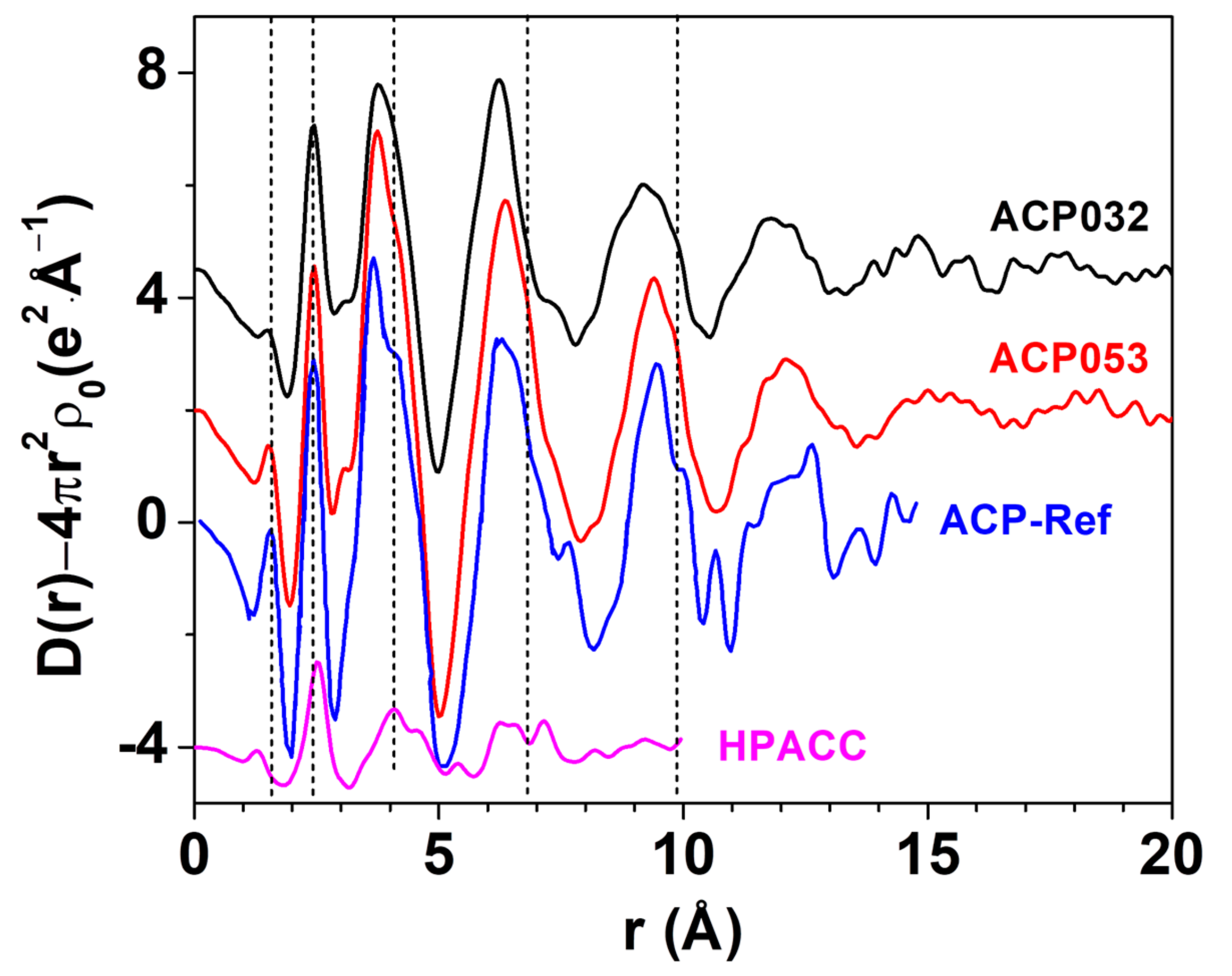
3.1. Synthesis and Characterization of ACPs and CaPs
3.2. Stability of ACPs
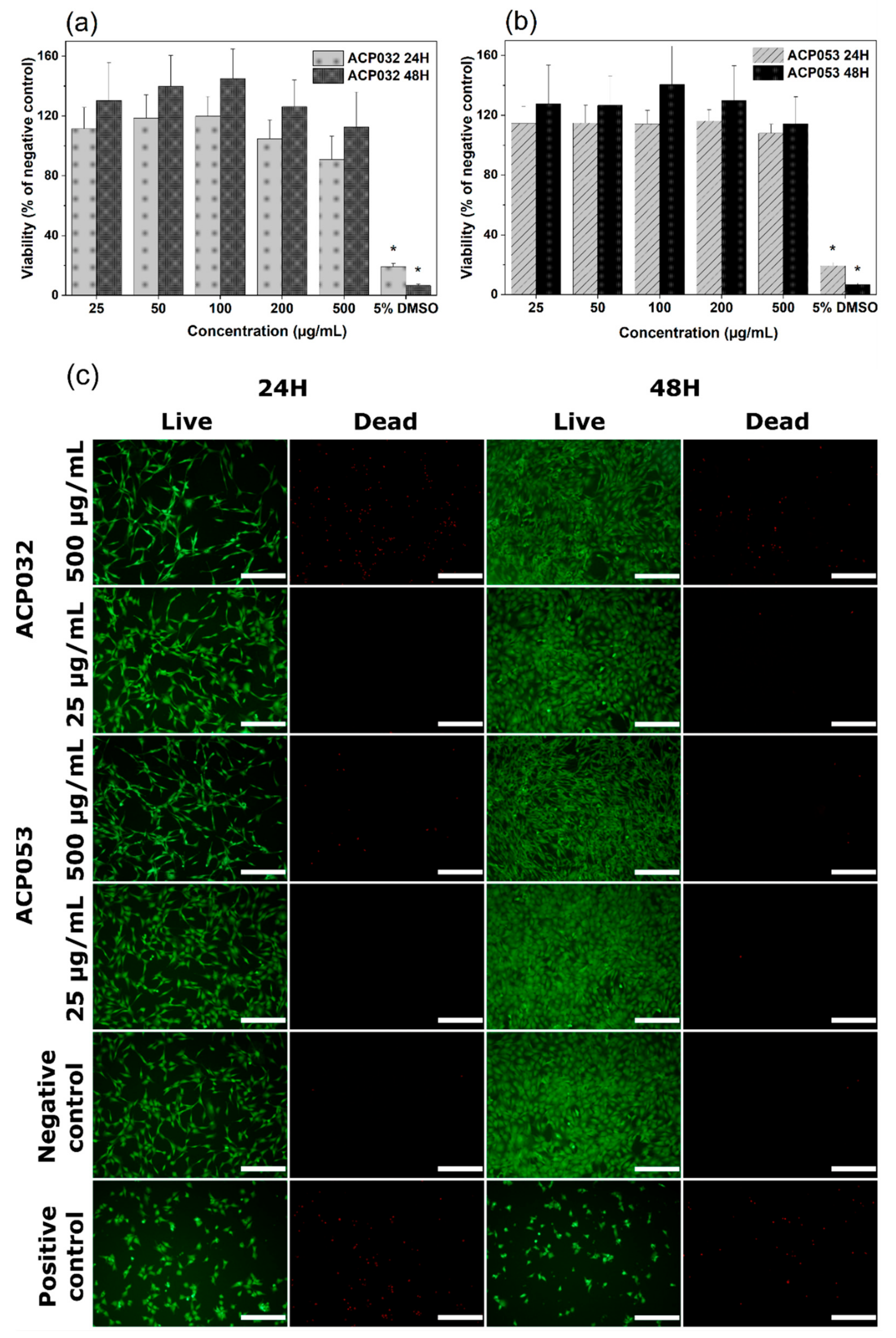
3.3. Cytocompatibility Study of ACPs
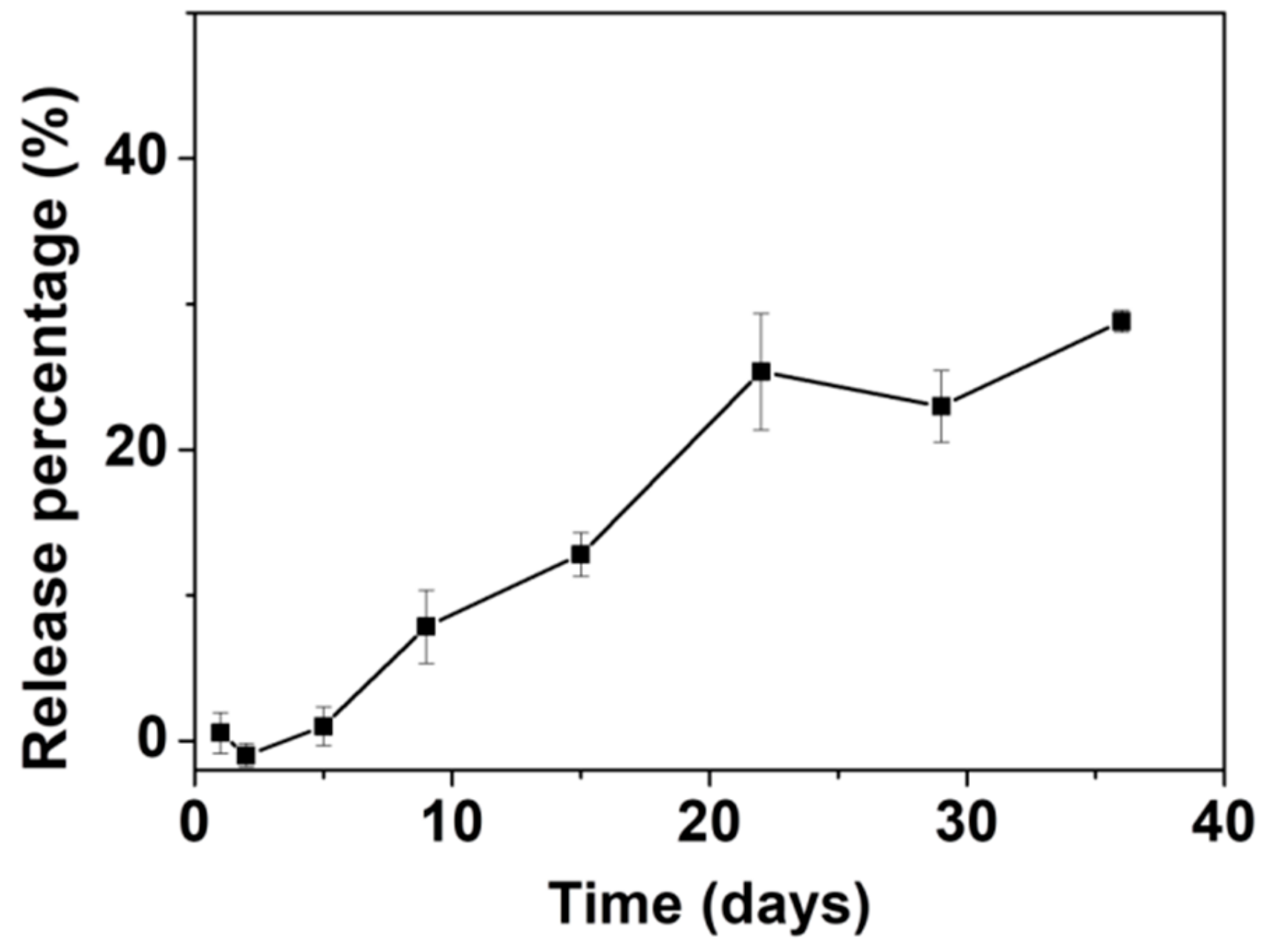
3.4. Drug Loading and In Vitro Release of AL with ACP053 as Carrier
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dorozhkin, S.V. Nanosized and nanocrystalline calcium orthophosphates. Acta Biomater. 2010, 6, 715–734. [Google Scholar] [CrossRef] [PubMed]
- Kumta, P.N.; Sfeir, C.; Lee, D.-H.; Olton, D.; Choi, D. Nanostructured calcium phosphates for biomedical applications: Novel synthesis and characterization. Acta Biomater. 2005, 1, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Zhou, H.; Lee, J. Various preparation methods of highly porous hydroxyapatite/polymer nanoscale biocomposites for bone regeneration. Acta Biomater. 2011, 7, 3813–3828. [Google Scholar] [CrossRef] [PubMed]
- Mahamid, J.; Sharir, A.; Addadi, L.; Weiner, S. Amorphous calcium phosphate is a major component of the forming fin bones of zebrafish: Indications for an amorphous precursor phase. Proc. Natl. Acad. Sci. USA 2008, 105, 12748–12753. [Google Scholar] [CrossRef]
- Lotsari, A.; Rajasekharan, A.K.; Halvarsson, M.; Andersson, M. Transformation of amorphous calcium phosphate to bone-like apatite. Nat. Commun. 2018, 9, 4170. [Google Scholar] [CrossRef]
- Akiva, A.; Kerschnitzki, M.; Pinkas, I.; Wagermaier, W.; Yaniv, K.; Fratzl, P.; Addadi, L.; Weiner, S. Mineral formation in the larval zebrafish tail bone occurs via an acidic disordered calcium phosphate phase. J. Am. Chem. Soc. 2016, 138, 14481–14487. [Google Scholar] [CrossRef]
- Chen, F.; Huang, P.; Qi, C.; Lu, B.-Q.; Zhao, X.-Y.; Li, C.; Wu, J.; Cui, D.-X.; Zhu, Y.-J. Multifunctional biodegradable mesoporous microspheres of Eu3+-doped amorphous calcium phosphate: Microwave-assisted preparation, pH-sensitive drug release, and bioimaging application. J. Mater. Chem. B 2014, 2, 7132–7140. [Google Scholar] [CrossRef]
- Qi, C.; Zhu, Y.-J.; Zhang, Y.-G.; Jiang, Y.-Y.; Wu, J.; Chen, F. Vesicle-like nanospheres of amorphous calcium phosphate: Sonochemical synthesis using the adenosine 5′-triphosphate disodium salt and their application in pH-responsive drug delivery. J. Mater. Chem. B 2015, 3, 7347–7354. [Google Scholar] [CrossRef]
- Yu, W.; Sun, T.-W.; Qi, C.; Ding, Z.; Zhao, H.; Chen, F.; Chen, D.; Zhu, Y.-J.; Shi, Z.; He, Y. Strontium-doped amorphous calcium phosphate porous microspheres synthesized through a microwave-hydrothermal method using fructose 1, 6-bisphosphate as an organic phosphorus source: Application in drug delivery and enhanced bone regeneration. ACS Appl. Mater. Interfaces 2017, 9, 3306–3317. [Google Scholar] [CrossRef]
- Balasundaram, G.; Sato, M.; Webster, T.J. Using hydroxyapatite nanoparticles and decreased crystallinity to promote osteoblast adhesion similar to functionalizing with RGD. Biomaterials 2006, 27, 2798–2805. [Google Scholar] [CrossRef]
- Nagano, M.; Nakamura, T.; Kokubo, T.; Tanahashi, M.; Ogawa, M. Differences of bone bonding ability and degradation behaviour in vivo between amorphous calcium phosphate and highly crystalline. Biomaterials 1996, 17, 1771–1777. [Google Scholar] [CrossRef]
- Yao, S.; Xu, Y.; Zhou, Y.; Shao, C.; Liu, Z.; Jin, B.; Zhao, R.; Cao, H.; Pan, H.; Tang, R. Calcium phosphate nanocluster-loaded injectable hydrogel for bone regeneration. ACS Appl. Bio Mater. 2019, 2, 4408–4417. [Google Scholar] [CrossRef]
- Tovani, C.B.; Gloter, A.; Azaïs, T.; Selmane, M.; Ramos, A.P.; Nassif, N. Formation of stable strontium-rich amorphous calcium phosphate: Possible effects on bone mineral. Acta Biomater. 2019, 92, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Iafisco, M.; Degli Esposti, L.; Ramírez-Rodríguez, G.B.; Carella, F.; Gómez-Morales, J.; Ionescu, A.C.; Brambilla, E.; Tampieri, A.; Delgado-López, J.M. Fluoride-doped amorphous calcium phosphate nanoparticles as a promising biomimetic material for dental remineralization. Sci. Rep. 2018, 8, 17016. [Google Scholar] [CrossRef] [PubMed]
- Van den Vreken, N.M.; Pieters, I.Y.; Declercq, H.A.; Cornelissen, M.J.; Verbeeck, R.M. Characterization of calcium phosphate cements modified by addition of amorphous calcium phosphate. Acta Biomater. 2010, 6, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, Y.; Sun, W.-B.; Zhang, H. Amorphous calcium phosphate and its application in dentistry. Chem. Cent. J. 2011, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Ortali, C.; Julien, I.; Vandenhende, M.; Drouet, C.; Champion, E. Consolidation of bone-like apatite bioceramics by spark plasma sintering of amorphous carbonated calcium phosphate at very low temperature. J. Eur. Ceram. Soc. 2018, 38, 2098–2109. [Google Scholar] [CrossRef]
- Armstrong, W.; Singer, L. Composition and constitution of the mineral phase of bone. Clin. Orthop. Relat. Res. 1965, 38, 179–192. [Google Scholar] [CrossRef]
- Olszta, M.J.; Cheng, X.; Jee, S.S.; Kumar, R.; Kim, Y.-Y.; Kaufman, M.J.; Douglas, E.P.; Gower, L.B. Bone structure and formation: A new perspective. Mater. Sci. Eng. Rep. 2007, 58, 77–116. [Google Scholar] [CrossRef]
- Boanini, E.; Gazzano, M.; Bigi, A. Ionic substitutions in calcium phosphates synthesized at low temperature. Acta Biomater. 2010, 6, 1882–1894. [Google Scholar] [CrossRef]
- Jin, W.; Liu, Z.; Wu, Y.; Jin, B.; Shao, C.; Xu, X.; Tang, R.; Pan, H. Synergic Effect of Sr2+ and Mg2+ on the Stabilization of Amorphous Calcium Phosphate. Cryst. Growth Des. 2018, 18, 6054–6060. [Google Scholar] [CrossRef]
- Tadic, D.; Peters, F.; Epple, M. Continuous synthesis of amorphous carbonated apatites. Biomaterials 2002, 23, 2553–2559. [Google Scholar] [CrossRef]
- Nakamura, M.; Hiratai, R.; Hentunen, T.; Salonen, J.; Yamashita, K. Hydroxyapatite with high carbonate substitutions promotes osteoclast resorption through osteocyte-like cells. ACS Biomater. Sci. Eng. 2016, 2, 259–267. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Amorphous calcium (ortho) phosphates. Acta Biomater. 2010, 6, 4457–4475. [Google Scholar] [CrossRef] [PubMed]
- Combes, C.; Rey, C. Amorphous calcium phosphates: Synthesis, properties and uses in biomaterials. Acta Biomater. 2010, 6, 3362–3378. [Google Scholar] [CrossRef] [PubMed]
- Ibsen, C.J.S.; Leemreize, H.; Mikladal, B.F.; Skovgaard, J.; Bremholm, M.; Eltzholtz, J.R.; Iversen, B.B.; Birkedal, H. Alkali counter ions impact crystallization kinetics of apatite nanocrystals from amorphous calcium phosphate in water at high pH. Cryst. Growth Des. 2018, 18, 6723–6728. [Google Scholar] [CrossRef]
- Meić, I.B.; Kontrec, J.; Jurašin, D.D.; Selmani, A.; Džakula, B.N.; Maltar-Strmečki, N.; Lyons, D.M.; Plodinec, M.; Čeh, M.; Gajović, A. How similar are amorphous calcium carbonate and calcium phosphate? A comparative study of amorphous phase formation conditions. CrystEngComm 2018, 20, 35–50. [Google Scholar] [CrossRef]
- Layrolle, P.; Lebugle, A. Characterization and reactivity of nanosized calcium phosphates prepared in anhydrous ethanol. Chem. Mater. 1994, 6, 1996–2004. [Google Scholar] [CrossRef]
- Ching Lau, C.; Reardon, P.J.T.; Campbell Knowles, J.; Tang, J. Phase-tunable calcium phosphate biomaterials synthesis and application in protein delivery. ACS Biomater. Sci. Eng. 2015, 1, 947–954. [Google Scholar] [CrossRef]
- Mohsen-Nia, M.; Amiri, H.; Jazi, B. Dielectric constants of water, methanol, ethanol, butanol and acetone: Measurement and computational study. J. Solut. Chem. 2010, 39, 701–708. [Google Scholar] [CrossRef]
- Rodrigues, A.; Lebugle, A. Influence of ethanol in the precipitation medium on the composition, structure and reactivity of tricalcium phosphate. Colloids Surf. A Physicochem. Eng. Asp. 1998, 145, 191–204. [Google Scholar] [CrossRef]
- Li, Y.; Weng, W.; Cheng, K.; Du, P.; Shen, G.; Wang, J.; Han, G. Preparation of amorphous calcium phosphate in the presence of poly (ethylene glycol). J. Mater. Sci. Lett. 2003, 22, 1015–1016. [Google Scholar] [CrossRef]
- Sun, R.; Zhang, P.; Bajnóczi, É.G.; Neagu, A.; Tai, C.-W.; Persson, I.; Strømme, M.; Cheung, O. Amorphous Calcium Carbonate Constructed from Nanoparticle Aggregates with Unprecedented Surface Area and Mesoporosity. ACS Appl. Mater. Interfaces 2018, 10, 21556–21564. [Google Scholar] [CrossRef] [PubMed]
- Powell, K.J.; Brown, P.L.; Byrne, R.H.; Gajda, T.; Hefter, G.; Sjöberg, S.; Wanner, H. Chemical speciation of environmentally significant heavy metals with inorganic ligands. Part 1: The Hg2+–Cl−, OH−, CO32−, SO42−, and PO43−aqueous systems (IUPAC Technical Report). Pure Appl. Chem. 2005, 77, 739–800. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- De Aza, P.; Santos, C.; Pazo, A.; De Aza, S.; Cusco, R.; Artus, L. Vibrational properties of calcium phosphate compounds. 1. Raman spectrum of β-tricalcium phosphate. Chem. Mater. 1997, 9, 912–915. [Google Scholar] [CrossRef]
- Chatzipanagis, K.; Iafisco, M.; Roncal-Herrero, T.; Bilton, M.; Tampieri, A.; Kröger, R.; Delgado-López, J.M. Crystallization of citrate-stabilized amorphous calcium phosphate to nanocrystalline apatite: A surface-mediated transformation. CrystEngComm 2016, 18, 3170–3173. [Google Scholar] [CrossRef]
- Casciani, F.; Condrate, R., Sr. The Raman spectrum of monetite, CaHPO4. J. Solid State Chem. 1980, 34, 385–388. [Google Scholar] [CrossRef]
- Zyman, Z.; Epple, M.; Goncharenko, A.; Rokhmistrov, D.; Prymak, O.; Loza, K. Thermally induced crystallization and phase evolution in powders derived from amorphous calcium phosphate precipitates with a Ca/P ratio of 1:1. J. Cryst. Growth 2016, 450, 190–196. [Google Scholar] [CrossRef]
- Berzina-Cimdina, L.; Borodajenko, N. Research of calcium phosphates using Fourier transform infrared spectroscopy. In Infrared Spectroscopy-Materials Science, Engineering and Technology; IntechOpen: London, UK, 2012. [Google Scholar]
- Layrolle, P.; Lebugle, A. Synthesis in pure ethanol and characterization of nanosized calcium phosphate fluoroapatite. Chem. Mater. 1996, 8, 134–144. [Google Scholar] [CrossRef]
- Brangule, A.; Gross, K.A. Importance of FTIR Spectra Deconvolution for the Analysis of Amorphous Calcium Phosphates. In Proceedings of the IOP Conference Series: Materials Science and Engineering, Riga, Latvia, 29 September–2 October 2014; p. 012027. [Google Scholar]
- Yang, L.; Perez-Amodio, S.; Barrère-de Groot, F.Y.; Everts, V.; van Blitterswijk, C.A.; Habibovic, P. The effects of inorganic additives to calcium phosphate on in vitro behavior of osteoblasts and osteoclasts. Biomaterials 2010, 31, 2976–2989. [Google Scholar] [CrossRef]
- Singh, S.S.; Roy, A.; Lee, B.E.; Banerjee, I.; Kumta, P.N. MC3T3-E1 proliferation and differentiation on biphasic mixtures of Mg substituted β-tricalcium phosphate and amorphous calcium phosphate. Mater. Sci. Eng. C 2014, 45, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Uskoković, V.; Marković, S.; Veselinović, L.; Škapin, S.; Ignjatović, N.; Uskoković, D.P. Insights into the kinetics of thermally induced crystallization of amorphous calcium phosphate. PCCP 2018, 20, 29221–29235. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.-J.; Zhu, Y.-J.; Qi, C.; Lu, B.-Q.; Chen, F.; Wu, J. Porous hollow microspheres of amorphous calcium phosphate: Soybean lecithin templated microwave-assisted hydrothermal synthesis and application in drug delivery. J. Mater. Chem. B 2015, 3, 1823–1830. [Google Scholar] [CrossRef]
- Hwang, E.T.; Tatavarty, R.; Chung, J.; Gu, M.B. New functional amorphous calcium phosphate nanocomposites by enzyme-assisted biomineralization. ACS Appl. Mater. Interfaces 2013, 5, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Zhu, Y.J.; Qi, C.; Chen, F.; Lu, B.Q.; Zhao, J.; Wu, J. Hierarchical Hollow Hydroxyapatite Microspheres: Microwave-Assisted Rapid Synthesis by Using Pyridoxal-5′-Phosphate as a Phosphorus Source and Application in Drug Delivery. Chem. Asian J. 2013, 8, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Ranz, X. Développement et Caractérisation de Dépôts D’apatite Obtenus par Projection Plasma sur Prothèses Orthopédiques; INPT: Toulouse, France, 1996. [Google Scholar]
- Betts, F.; Posner, A. An X-ray radial distribution study of amorphous calcium phosphate. Mater. Res. Bull. 1974, 9, 353–360. [Google Scholar] [CrossRef]
- Persson, I.; Trublet, M.; Klysubun, W. Structure Determination of Phosphoric Acid and Phosphate Ions in Aqueous Solution Using EXAFS Spectroscopy and Large Angle X-ray Scattering. J. Phys. Chem. A 2018, 122, 7413–7420. [Google Scholar] [CrossRef] [PubMed]
- Demichelis, R.; Raiteri, P.; Gale, J.D.; Dovesi, R. A new structural model for disorder in vaterite from first-principles calculations. CrystEngComm 2012, 14, 44–47. [Google Scholar] [CrossRef]
- Cheung, O.; Zhang, P.; Frykstrand, S.; Zheng, H.; Yang, T.; Sommariva, M.; Zou, X.; Strømme, M. Nanostructure and pore size control of template-free synthesised mesoporous magnesium carbonate. RSC Adv. 2016, 6, 74241–74249. [Google Scholar] [CrossRef]
- Christoffersen, J.; Christoffersen, M.R.; Kibalczyc, W.; Andersen, F.A. A contribution to the understanding of the formation of calcium phosphates. J. Cryst. Growth 1989, 94, 767–777. [Google Scholar] [CrossRef]
- Palazzo, B.; Iafisco, M.; Laforgia, M.; Margiotta, N.; Natile, G.; Bianchi, C.L.; Walsh, D.; Mann, S.; Roveri, N. Biomimetic hydroxyapatite–drug nanocrystals as potential bone substitutes with antitumor drug delivery properties. Adv. Funct. Mater. 2007, 17, 2180–2188. [Google Scholar] [CrossRef]
- Murugan, R.; Ramakrishna, S. Aqueous mediated synthesis of bioresorbable nanocrystalline hydroxyapatite. J. Cryst. Growth 2005, 274, 209–213. [Google Scholar] [CrossRef]
- Murugan, R.; Ramakrishna, S. Bioresorbable composite bone paste using polysaccharide based nano hydroxyapatite. Biomaterials 2004, 25, 3829–3835. [Google Scholar] [CrossRef] [PubMed]
- Shahrezaee, M.; Raz, M.; Shishehbor, S.; Moztarzadeh, F.; Baghbani, F.; Sadeghi, A.; Bajelani, K.; Tondnevis, F. Synthesis of Magnesium Doped Amorphous Calcium Phosphate as a Bioceramic for Biomedical Application: In Vitro Study. Silicon 2018, 10, 1171–1179. [Google Scholar] [CrossRef]
- Kababya, S.; Gal, A.; Kahil, K.; Weiner, S.; Addadi, L.; Schmidt, A. Phosphate–water interplay tunes amorphous calcium carbonate metastability: Spontaneous phase separation and crystallization vs stabilization viewed by solid state NMR. J. Am. Chem. Soc. 2015, 137, 990–998. [Google Scholar] [CrossRef]
- Blumenthal, N.; Betts, F.; Posner, A. Effect of carbonate and biological macromolecules on formation and properties of hydroxyapatite. Calcif. Tissue Res. 1975, 18, 81–90. [Google Scholar] [CrossRef]
- Sharma, S.; Verma, A.; Teja, B.V.; Pandey, G.; Mittapelly, N.; Trivedi, R.; Mishra, P. An insight into functionalized calcium based inorganic nanomaterials in biomedicine: Trends and transitions. Colloids Surf. B Biointerfaces 2015, 133, 120–139. [Google Scholar] [CrossRef]
- Dizaj, S.M.; Barzegar-Jalali, M.; Zarrintan, M.H.; Adibkia, K.; Lotfipour, F. Calcium carbonate nanoparticles; potential in bone and tooth disorders. Pharm. Sci. 2015, 20, 175–182. [Google Scholar]
- Combes, C.; Bareille, R.; Rey, C. Calcium carbonate–calcium phosphate mixed cement compositions for bone reconstruction. J. Biomed. Mater. Res. Part A 2006, 79, 318–328. [Google Scholar] [CrossRef]
- Tolba, E.; Müller, W.E.; El-Hady, B.M.A.; Neufurth, M.; Wurm, F.; Wang, S.; Schröder, H.C.; Wang, X. High biocompatibility and improved osteogenic potential of amorphous calcium carbonate/vaterite. J. Mater. Chem. B 2016, 4, 376–386. [Google Scholar] [CrossRef]
- Combes, C.; Miao, B.; Bareille, R.; Rey, C. Preparation, physical–chemical characterisation and cytocompatibility of calcium carbonate cements. Biomaterials 2006, 27, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Uskoković, V.; Batarni, S.S.; Schweicher, J.; King, A.; Desai, T.A. Effect of calcium phosphate particle shape and size on their antibacterial and osteogenic activity in the delivery of antibiotics in vitro. ACS Appl. Mater. Interfaces 2013, 5, 2422–2431. [Google Scholar] [CrossRef] [PubMed]
- Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity. 2009. Available online: https://www.iso.org/obp/ui/#iso:std:iso:10993:-10995:ed-10993:v10991:en (accessed on 1 September 2019).
- Termine, J.; Eanes, E.; Conn, K. Phosphoprotein modulation of apatite crystallization. Calcif. Tissue Int. 1980, 31, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Iafisco, M.; Palazzo, B.; Falini, G.; Di Foggia, M.; Bonora, S.; Nicolis, S.; Casella, L.; Roveri, N. Adsorption and conformational change of myoglobin on biomimetic hydroxyapatite nanocrystals functionalized with alendronate. Langmuir 2008, 24, 4924–4930. [Google Scholar] [CrossRef]
- Balas, F.; Manzano, M.; Horcajada, P.; Vallet-Regí, M. Confinement and controlled release of bisphosphonates on ordered mesoporous silica-based materials. J. Am. Chem. Soc. 2006, 128, 8116–8117. [Google Scholar] [CrossRef]
- Huang, W.; Liu, W.; She, Z.; Wu, H.; Shi, X. Alendronate decorated nano hydroxyapatite in mesoporous silica: Cytotoxicity and osteogenic properties. Appl. Surf. Sci. 2011, 257, 9757–9761. [Google Scholar] [CrossRef]
- Pascaud, P.; Errassifi, F.; Brouillet, F.; Sarda, S.; Barroug, A.; Legrouri, A.; Rey, C. Adsorption on apatitic calcium phosphates for drug delivery: Interaction with bisphosphonate molecules. J. Mater. Sci. Mater. Med. 2014, 25, 2373–2381. [Google Scholar] [CrossRef]
- Kim, C.W.; Yun, Y.-P.; Lee, H.J.; Hwang, Y.-S.; Kwon, I.K.; Lee, S.C. In situ fabrication of alendronate-loaded calcium phosphate microspheres: Controlled release for inhibition of osteoclastogenesis. J. Control. Release 2010, 147, 45–53. [Google Scholar] [CrossRef]
- Åberg, J.; Brohede, U.; Mihranyan, A.; Strømme, M.; Engqvist, H. Bisphosphonate incorporation in surgical implant coatings by fast loading and co-precipitation at low drug concentrations. J. Mater. Sci. Mater. Med. 2009, 20, 2053–2061. [Google Scholar] [CrossRef]
- Nancollas, G.; Tang, R.; Phipps, R.; Henneman, Z.; Gulde, S.; Wu, W.; Mangood, A.; Russell, R.; Ebetino, F. Novel insights into actions of bisphosphonates on bone: Differences in interactions with hydroxyapatite. Bone 2006, 38, 617–627. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | H | Ca | P | Ca/P | Estimated Carbonate Content (wt.%) | |
|---|---|---|---|---|---|---|
| ACP032 | 0.26 | 1.56 | 0.84 | 0.38 | 2.21 | ~16% |
| ACP053 | 0.07 | 1.73 | 0.86 | 0.56 | 1.53 | ~4% |
| Samples | Brunauer–Emmett–Teller (BET) Surface Area (m2/g) | Peak Pore-Size a (nm) | Pore Volume b (cm3/g) | Density (g/cm3) c | Calculated Particle Size d (nm) |
|---|---|---|---|---|---|
| ACP032 | 423 | 5.4 | 0.57 | 2.49 | 5.7 |
| ACP053 | 418 | 9.3 | 1.05 | 2.59 | 5.6 |
| CaP058 | 294 | 18.6 | 1.39 | 2.61 | 7.8 |
| CaP068 | 133 | 18.6 | 0.62 | 2.70 | 16.7 |
| CaP078 | 52 | 6.8 | 0.11 | - | - |
| CaP088 | 6 | 18.6 | 0.10 | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, R.; Åhlén, M.; Tai, C.-W.; Bajnóczi, É.G.; de Kleijne, F.; Ferraz, N.; Persson, I.; Strømme, M.; Cheung, O. Highly Porous Amorphous Calcium Phosphate for Drug Delivery and Bio-Medical Applications. Nanomaterials 2020, 10, 20. https://doi.org/10.3390/nano10010020
Sun R, Åhlén M, Tai C-W, Bajnóczi ÉG, de Kleijne F, Ferraz N, Persson I, Strømme M, Cheung O. Highly Porous Amorphous Calcium Phosphate for Drug Delivery and Bio-Medical Applications. Nanomaterials. 2020; 10(1):20. https://doi.org/10.3390/nano10010020
Chicago/Turabian StyleSun, Rui, Michelle Åhlén, Cheuk-Wai Tai, Éva G. Bajnóczi, Fenne de Kleijne, Natalia Ferraz, Ingmar Persson, Maria Strømme, and Ocean Cheung. 2020. "Highly Porous Amorphous Calcium Phosphate for Drug Delivery and Bio-Medical Applications" Nanomaterials 10, no. 1: 20. https://doi.org/10.3390/nano10010020
APA StyleSun, R., Åhlén, M., Tai, C.-W., Bajnóczi, É. G., de Kleijne, F., Ferraz, N., Persson, I., Strømme, M., & Cheung, O. (2020). Highly Porous Amorphous Calcium Phosphate for Drug Delivery and Bio-Medical Applications. Nanomaterials, 10(1), 20. https://doi.org/10.3390/nano10010020