
Composites Composed of Hydrophilic and Hydrophobic Polymers, and Hydroxyapatite Nanoparticles: Synthesis, Characterization, and Study of Their Biocompatible Properties

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
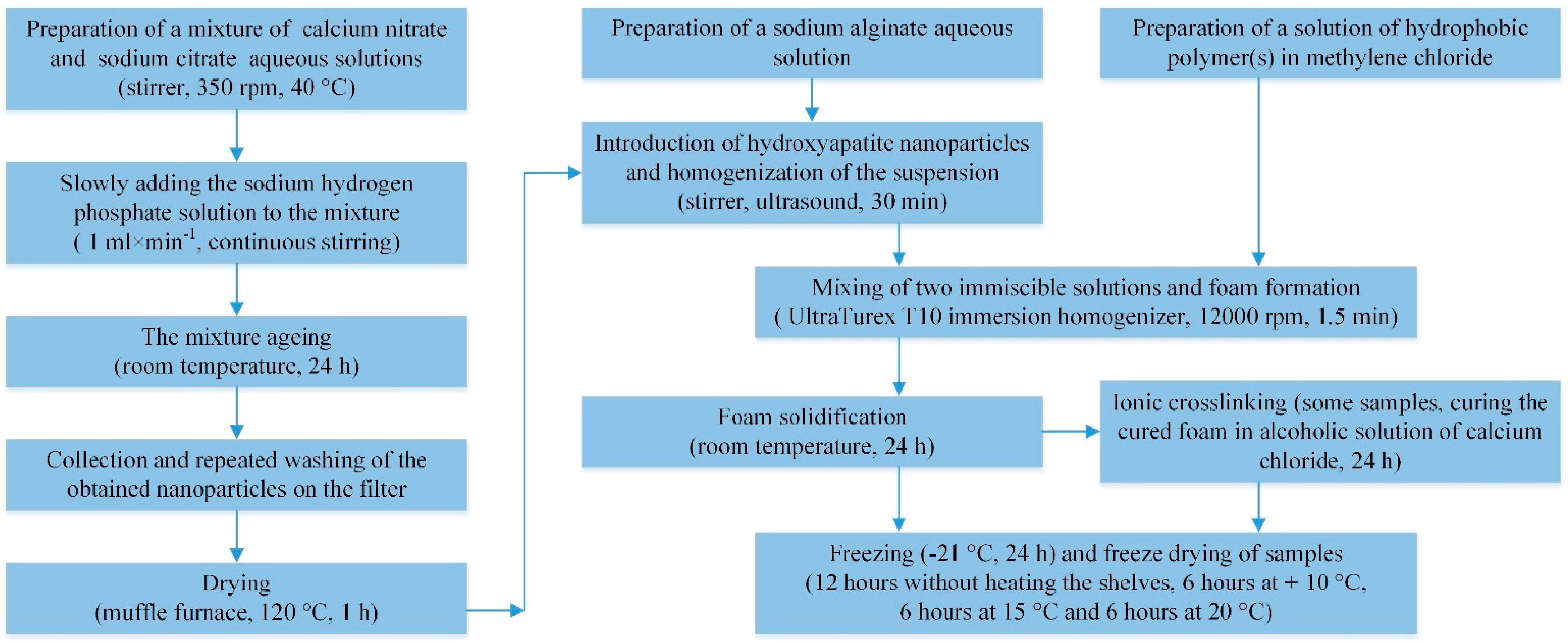
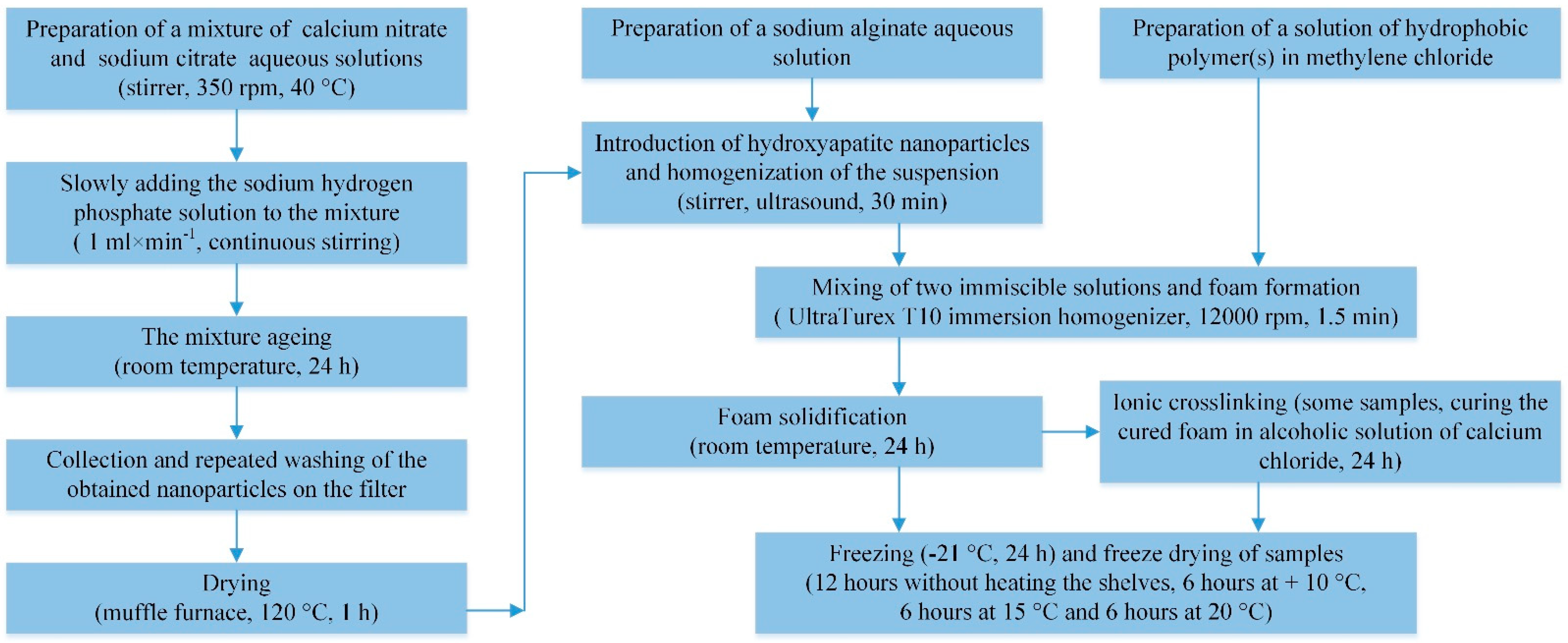
2.2. Synthesis of HAP Nanoparticles
2.3. Synthesis of Porous Hydrophobic Polymer(s)/Alginate/HAP NPs Matrices
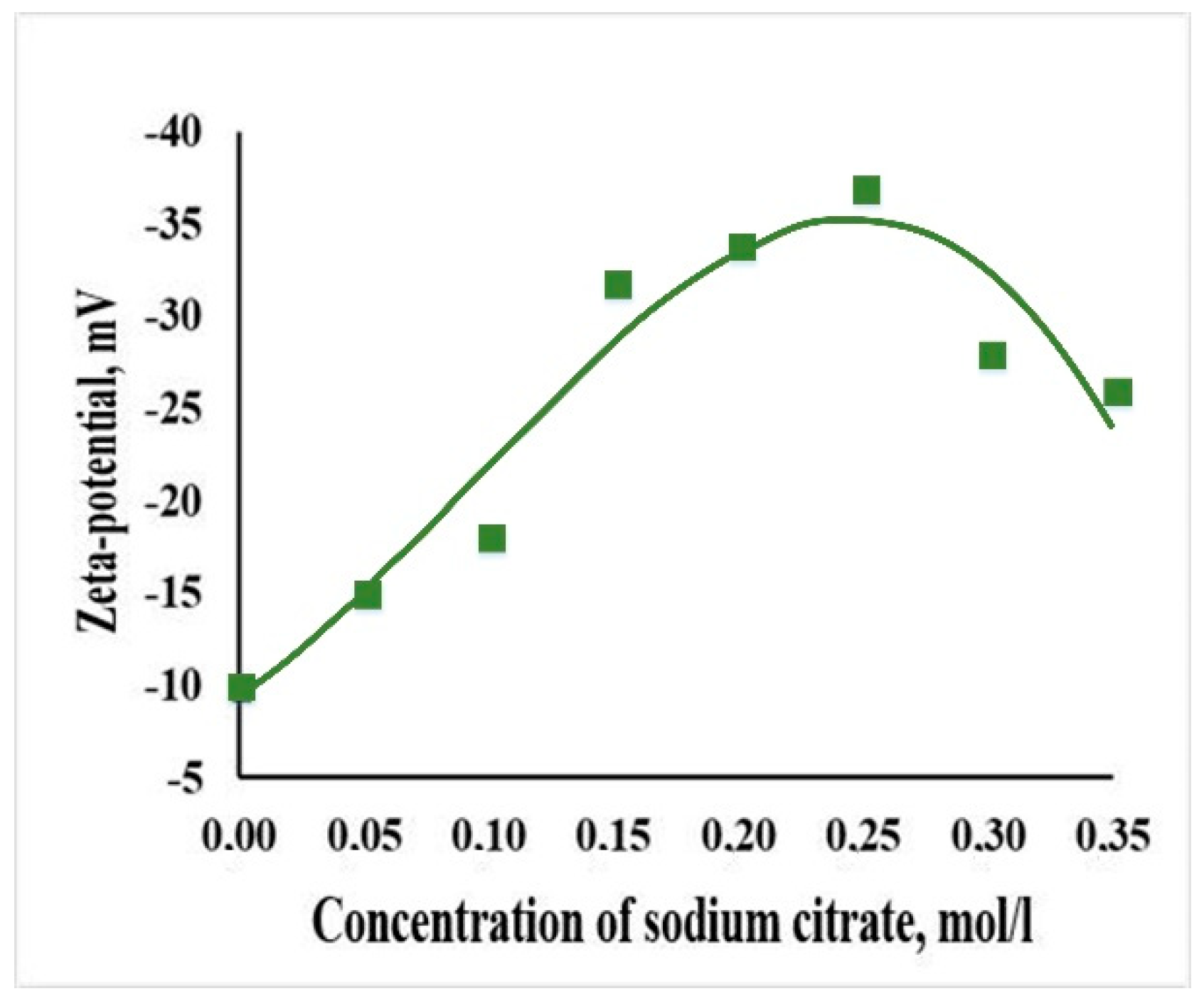
2.4. Measurement of the ζ-Potential of Hydroxyapatite Nanoparticles
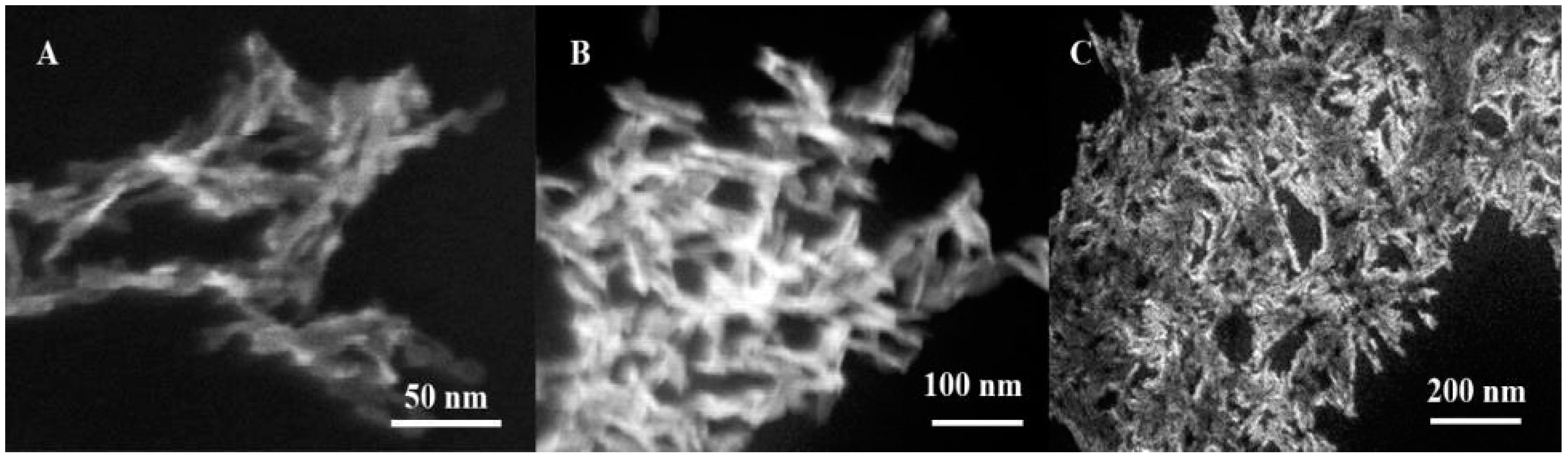
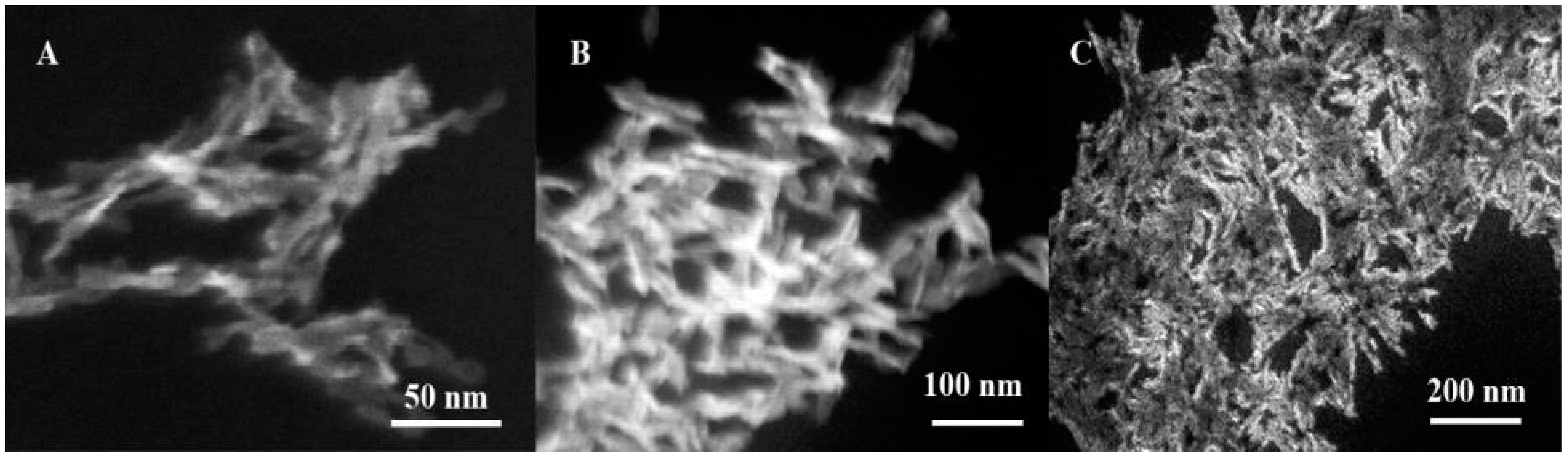
2.5. Scanning Electron Microscopy
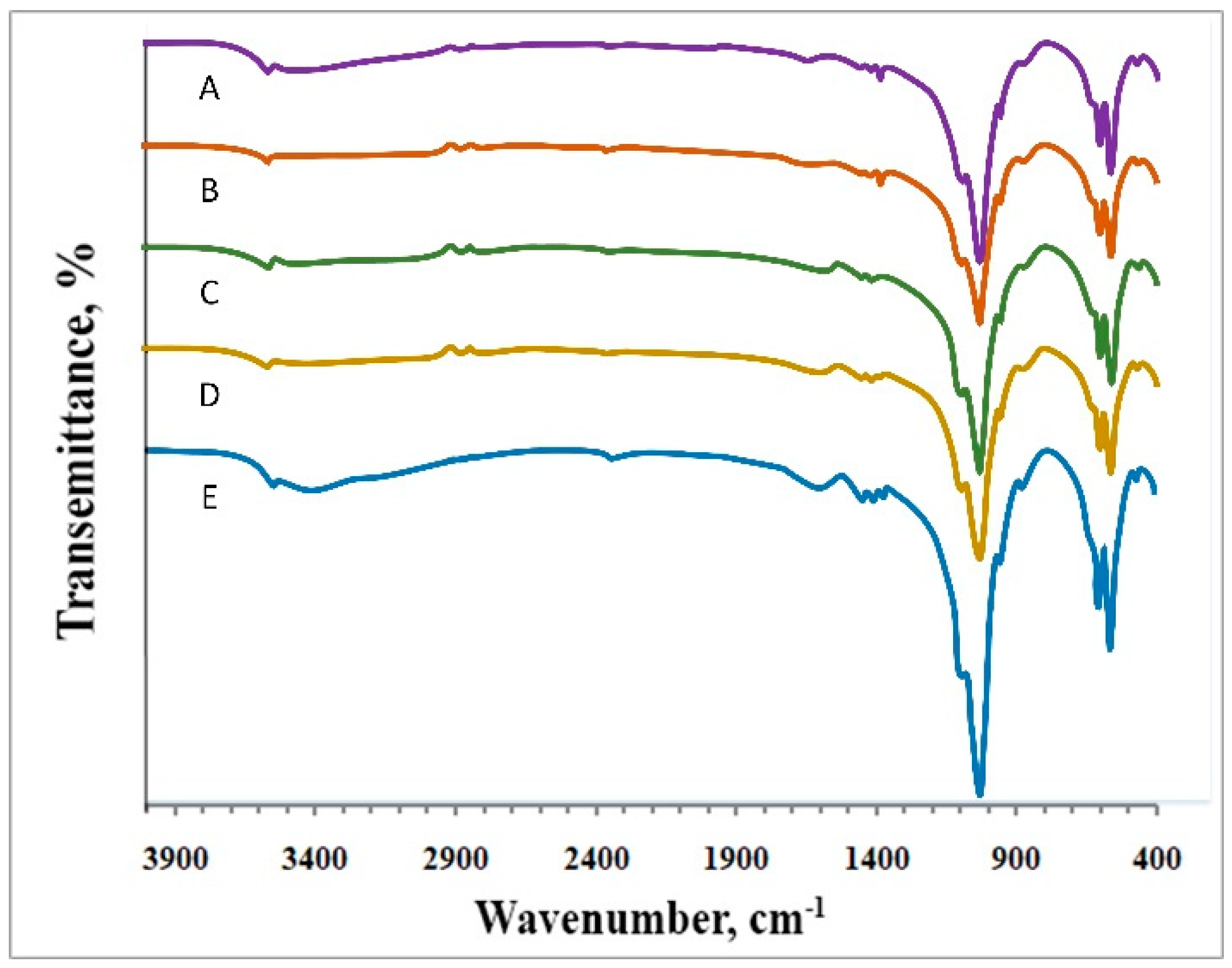
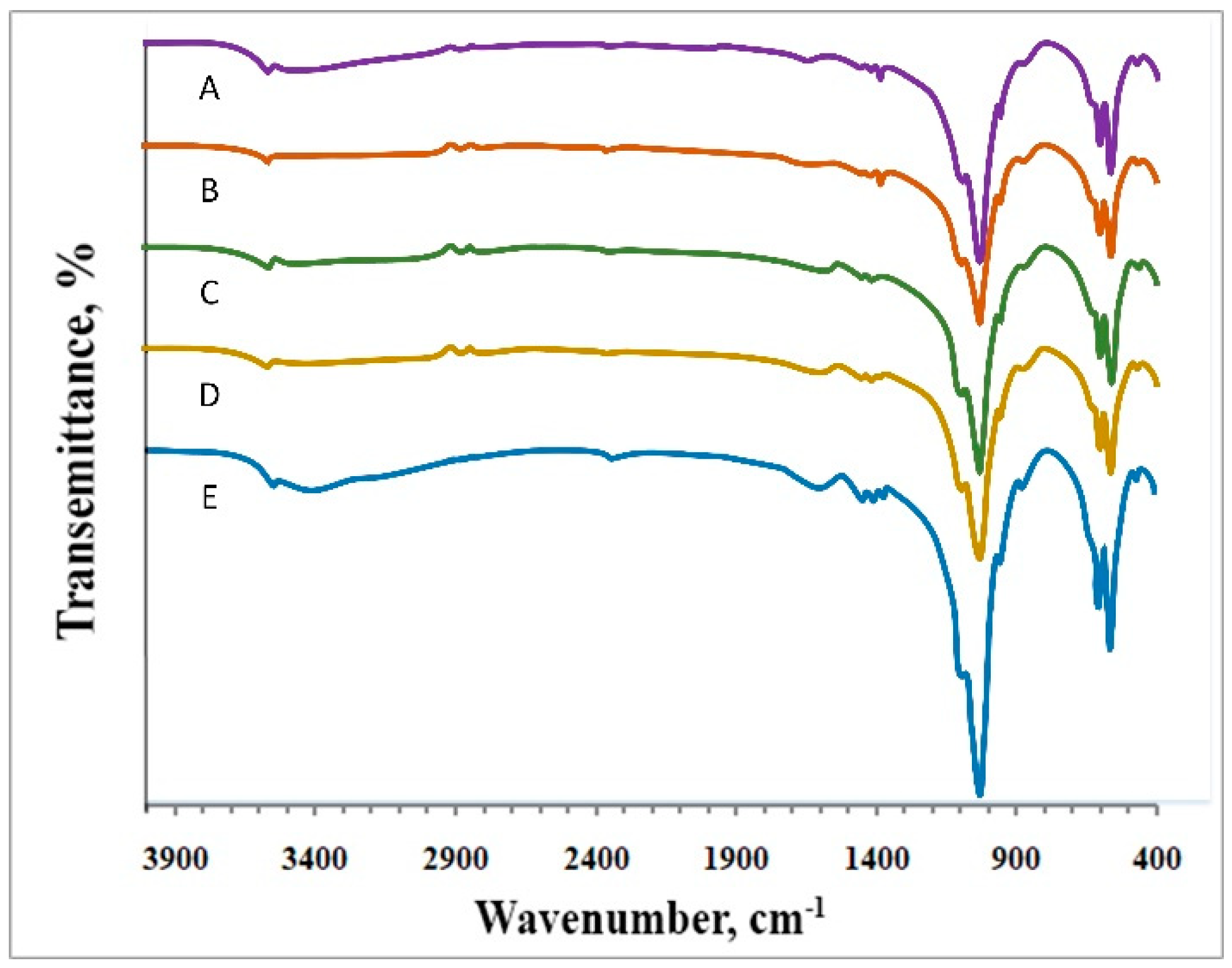
2.6. Fourier-Transform Infrared (FT-IR) Spectra
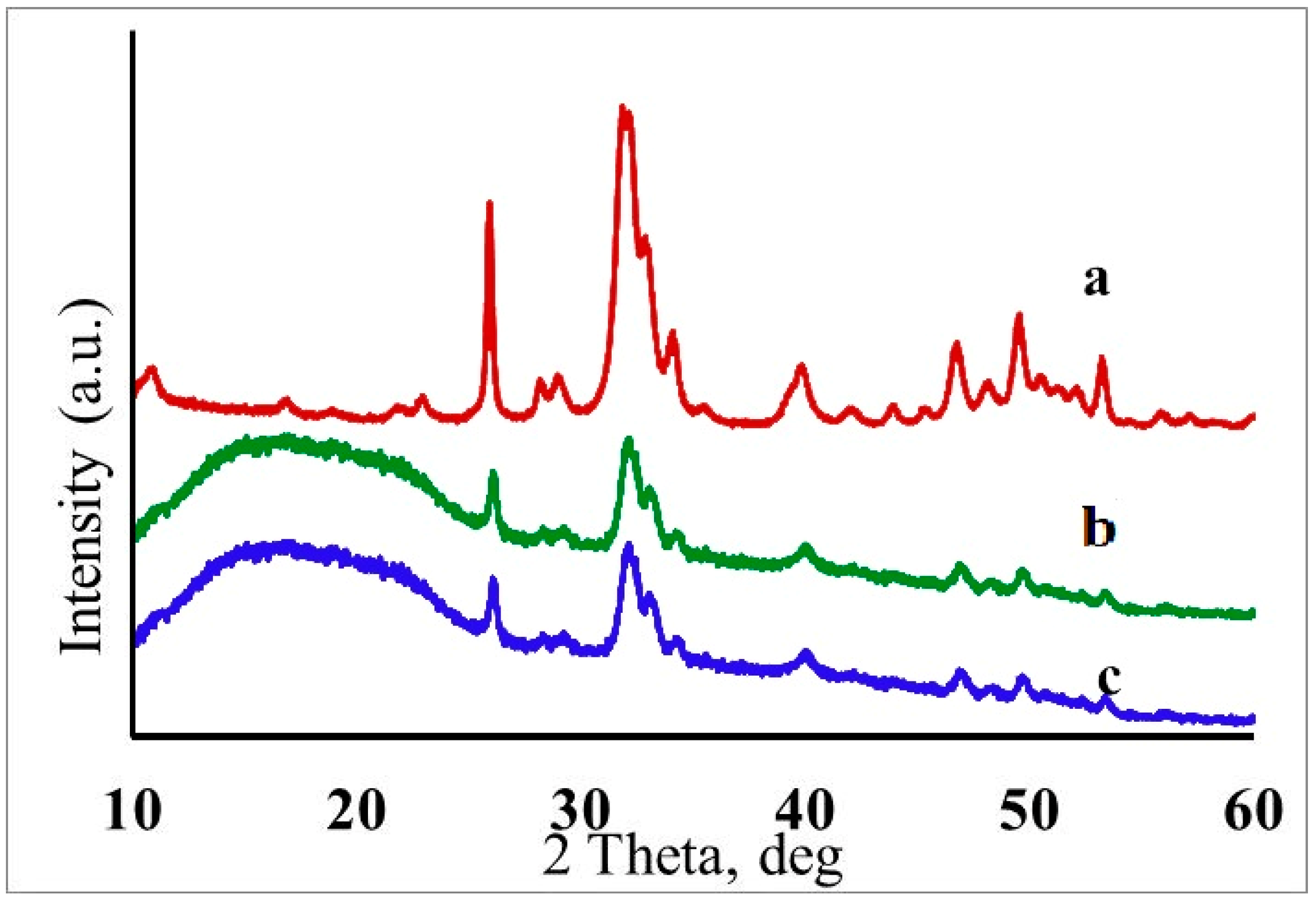
2.7. X-ray Phase Analysis
2.8. Energy-Dispersive Microanalysis
2.9. Assessment of the Mechanical Stability of Hydrophobic Polymer(s)/Alginate/HAP NPs Matrices
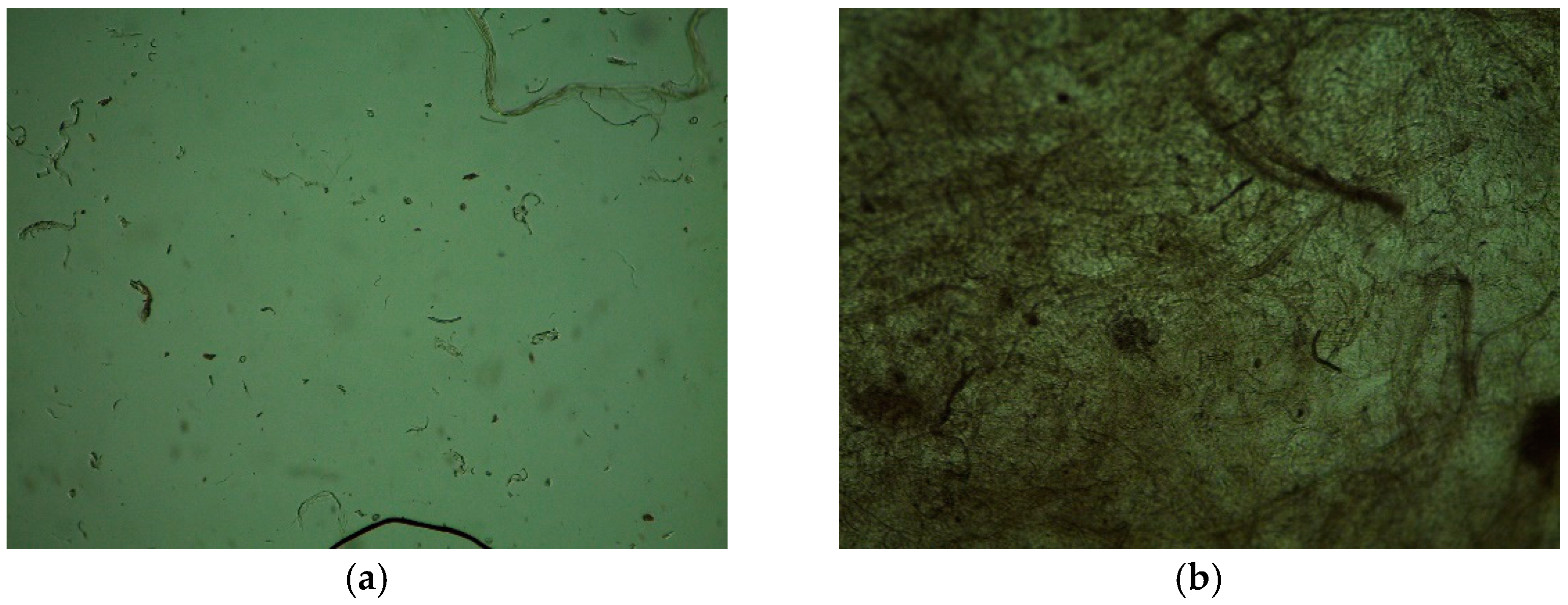
2.10. Assessment of Cytotoxicity and Cell Adhesion of Hydrophobic Polymer(s)/Alginate/HAP NPs Matrices
2.11. Assessment of Acute Toxicity of Hydrophobic Polymer(s)/Alginate/HAP NPs Matrices
3. Results
3.1. Characterization of Hydroxyapatite Nanoparticles
3.2. Characterization of Hydrophobic Polymer(s)/Alginate/HAP NPs Matrices
3.3. Mechanical Stability and Cytotoxicity of Hydrophobic Polymer(s)/Alginate/HAP NPs Matrices
3.4. Assessment of Acute Toxicity of Hydrophobic Polymer(s)/Alginate/HAP NPs Matrices
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jeon, O.; Song, S.J.; Kang, S.-W.; Putnam, A.J.; Kim, B.-S. Enhancement of ectopic bone formation by bone morphogenetic protein-2 released from a heparin-conjugated poly (L-lactic-co-glycolic acid) scaffold. Biomaterials 2007, 28, 2763–2771. [Google Scholar] [CrossRef]
- Ashton, R.S.; Banerjee, A.; Punyani, S.; Schaffer, D.V.; Kane, R.S. Scaffolds based on degradable alginate hydrogels and poly (lactide-co-glycolide) microspheres for stem cell culture. Biomaterials 2007, 28, 5518–5525. [Google Scholar] [CrossRef]
- Ghorbani, F.; Nojehdehian, H.; Zamanian, A. Physicochemical and mechanical properties of freeze cast hydroxyapatite-gelatin scaffolds with dexamethasone loaded PLGA microspheres for hard tissue engineering applications. Mater. Sci. Eng. C 2016, 69, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Dzierzkowska, E.; Scisłowska-Czarnecka, A.; Kudzin, M.; Boguń, M.; Szatkowski, P.; Gajek, M.; Kornaus, K.; Chadzinska, M.; Stodolak-Zych, E. Effects of process parameters on structure and properties of melt-blown poly(lactic acid) nonwovens for skin regeneration. J. Funct. Biomater. 2021, 12, 16. [Google Scholar] [CrossRef]
- Sadeghi, A.; Nokhasteh, S.; Molavi, A.; Khorsand-Ghayeni, M.; Naderi-Meshkin, H.; Mahdizadeh, A. Surface modification of electrospun PLGA scaffold with collagen for bioengineered skin substitutes. Mater. Sci. Eng. C 2016, 66, 130–137. [Google Scholar] [CrossRef]
- Zhao, W.; Li, J.; Jin, K.; Liu, W.; Qiu, X.; Li, C. Fabrication of functional PLGA-based electrospun scaffolds and their applications in biomedical engineering. Mater. Sci. Eng. C 2016, 59, 1181–1194. [Google Scholar] [CrossRef]
- Nguyen, T.-H.; Lee, B.-T. The effect of cross-linking on the microstructure, mechanical properties and biocompatibility of electrospun polycaprolactone–gelatin/PLGA–gelatin/PLGA–chitosan hybrid composite. Sci. Technol. Adv. Mater. 2012, 13, 035002. [Google Scholar] [CrossRef]
- Vasconcellos, L.; Santana-Melo, G.; Silva, E.; Pereira, V.; Araújo, J.; Silva, A.; Furtado, A.; Elias, C.; Viana, B.; Marciano, F.; et al. Electrospun poly(butylene-adipate-co-terephthalate)/nano-hydroxyapatite/graphene nanoribbon scaffolds improved the in vivo osteogenesis of the neoformed bone. J. Funct. Biomater. 2021, 12, 11. [Google Scholar] [CrossRef]
- Jubeli, E.; Khzam, A.; Yagoubi, N. Cells integration onto scaffolds prepared from polyester based polymers–importance of polymer thermal properties in addition to hydrophilicity. Int. J. Polym. Mater. 2018, 68, 1068–1077. [Google Scholar] [CrossRef]
- Koroleva, M.Y.; Shcherbakov, V.A.; Khasanova, L.K.; Rakitin, A.I.; Shirokikh, S.A.; Yurtov, E.V. The stability of highly concentrated water-in-oil emulsions and structure of highly porous polystyrene produced from them. Colloid J. 2018, 80, 272–281. [Google Scholar] [CrossRef]
- Ma, H.; Feng, C.; Chang, J.; Wu, C. 3D-printed bioceramic scaffolds: From bone tissue engineering to tumor therapy. Acta Biomater. 2018, 79, 37–59. [Google Scholar] [CrossRef]
- De La Lastra, A.A.; Hixon, K.R.; Aryan, L.; Banks, A.N.; Lin, A.Y.; Hall, A.F.; Sell, S.A. Tissue engineering scaffolds fabricated in dissolvable 3D-printed molds for patient-specific craniofacial bone regeneration. J. Funct. Biomater. 2018, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Liu, Y.; Li, X.; Wen, P.; Zhang, Y.; Long, Y.; Wang, X.; Guo, Y.; Xing, F.; Gao, J. Preparation of aligned porous gelatin scaffolds by unidirectional freeze-drying method. Acta Biomater. 2010, 6, 1167–1177. [Google Scholar] [CrossRef]
- Ho, M.-H.; Kuo, P.-Y.; Hsieh, H.-J.; Hsien, T.-Y.; Hou, L.-T.; Lai, J.-Y.; Wang, D.-M. Preparation of porous scaffolds by using freeze-extraction and freeze-gelation methods. Biomaterials 2004, 25, 129–138. [Google Scholar] [CrossRef]
- Deville, S.; Saiz, E.; Tomsia, A.P. Freeze casting of hydroxyapatite scaffolds for bone tissue engineering. Biomaterials 2006, 27, 5480–5489. [Google Scholar] [CrossRef] [Green Version]
- Lytkina, D.; Fedorishin, D.; Kalachikova, P.; Plyaskina, A.; Babeshin, A.; Kurzina, I. Cryo-structured materials based on polyvinyl alcohol and hydroxyapatite for osteogenesis. J. Funct. Biomater. 2021, 12, 18. [Google Scholar] [CrossRef]
- Loh, Q.L.; Choong, C. Three-dimensional scaffolds for tissue engineering applications: Role of porosity and pore size. Tissue Eng. Part B Rev. 2013, 19, 485–502. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Fan, Y.; Dunne, N.; Li, X. Effect of microporosity on scaffolds for bone tissue engineering. Regen. Biomater. 2018, 5, 115–124. [Google Scholar] [CrossRef]
- Prakasam, M.; Locs, J.; Salma-Ancane, K.; Loca, D.; Largeteau, A.; Berzina-Cimdina, L. Biodegradable materials and metallic implants—A review. J. Funct. Biomater. 2017, 8, 44. [Google Scholar] [CrossRef] [Green Version]
- Park, G.E.; Pattison, M.A.; Park, K.; Webster, T.J. Accelerated chondrocyte functions on NaOH-treated PLGA scaffolds. Biomaterials 2005, 26, 3075–3082. [Google Scholar] [CrossRef]
- Habraken, W.; Wolke, J.; Jansen, J. Ceramic composites as matrices and scaffolds for drug delivery in tissue engineering. Adv. Drug Deliv. Rev. 2007, 59, 234–248. [Google Scholar] [CrossRef]
- Mondal, S.; Hoang, G.; Manivasagan, P.; Moorthy, M.S.; Nguyen, T.P.; Phan, T.T.V.; Kim, H.H.; Kim, M.H.; Nam, S.Y.; Oh, J. Nano-hydroxyapatite bioactive glass composite scaffold with enhanced mechanical and biological performance for tissue engineering application. Ceram. Int. 2018, 44, 15735–15746. [Google Scholar] [CrossRef]
- Liu, X.; Ma, P.X. Polymeric scaffolds for bone tissue engineering. Ann. Biomed. Eng. 2004, 32, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, S.M.; Thomas, M.; Reddy, K.K.; Sooraparaju, S.G.; Asthana, A.; Bhatnagar, I. Chitosan as biomaterial in drug delivery and tissue engineering. Int. J. Biol. Macromol. 2018, 110, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Zarintaj, P.; Manouchehri, S.; Ahmadi, Z.; Saeb, M.R.; Urbanska, A.M.; Kaplan, D.L.; Mozafari, M. Agarose-based biomaterials for tissue engineering. Carbohydr. Polym. 2018, 187, 66–84. [Google Scholar] [CrossRef]
- Nazir, R.; Bruyneel, A.; Carr, C.; Czernuszka, J. Mechanical and degradation properties of hybrid scaffolds for tissue engineered heart valve (TEHV). J. Funct. Biomater. 2021, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, L.; Zhang, P.; Wang, X.; Chen, X.; Jing, X.; Su, Z. Surface modification of poly (L-lactic acid) to improve its cytocompatibility via assembly of polyelectrolytes and gelatin. Acta Biomater. 2006, 2, 155–164. [Google Scholar] [CrossRef]
- Espinoza, S.M.; Patil, H.I.; Martinez, E.S.M.; Pimentel, R.C.; Ige, P.P. Poly-ε-caprolactone (PCL), a promising polymer for pharmaceutical and biomedical applications: Focus on nanomedicine in cancer. Int. J. Polym. Mater. 2019, 69, 85–126. [Google Scholar] [CrossRef]
- Yang, W.-F.; Long, L.; Wang, R.; Chen, D.; Duan, S.; Xu, F.-J. Surface-modified hydroxyapatite nanoparticle-reinforced polylactides for three-dimensional printed bone tissue engineering scaffolds. J. Biomed. Nanotechnol. 2018, 14, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Nazeer, M.A.; Yilgor, E.; Yilgor, I. Electrospun polycaprolactone/silk fibroin nanofibrous bioactive scaffolds for tissue engineering applications. Polymer 2019, 168, 86–94. [Google Scholar] [CrossRef]
- Overby, R.J.; Feldman, D.S. Influence of poly(ethylene glycol) end groups on poly(Ethylene Glycol)-albumin system properties as a potential degradable tissue scaffold. J. Funct. Biomater. 2018, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotturi, H.; Abuabed, A.; Zafar, H.; Sawyer, E.; Pallipparambil, B.; Jamadagni, H.; Khandaker, M. Evaluation of polyethylene glycol diacrylate-polycaprolactone scaffolds for tissue engineering applications. J. Funct. Biomater. 2017, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ates, B.; Koytepe, S.; Ulu, A.; Gürses, C.; Thakur, V.K. Chemistry, structures, and advanced applications of nanocomposites from biorenewable resources. Chem. Rev. 2020, 120, 9304–9362. [Google Scholar] [CrossRef] [PubMed]
- Iovene, A.; Zhao, Y.; Wang, S.; Amoako, K. Bioactive polymeric materials for the advancement of regenerative medicine. J. Funct. Biomater. 2021, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Roointan, A.; Kianpour, S.; Memari, F.; Gandomani, M.; Hayat, S.M.G.; Mohammadi-Samani, S. Poly (lactic-co-glycolic acid): The most ardent and flexible candidate in biomedicine! Int. J. Polym. Mater. 2018, 67, 1028–1049. [Google Scholar] [CrossRef]
- Vargha-Butler, E.I.; Kiss, E.; Lam, C.N.C.; Keresztes, Z.; Kálmán, E.; Zhang, L.; Neumann, A.W. Wettability of biodegradable surfaces. Colloid Polym. Sci. 2001, 279, 1160–1168. [Google Scholar] [CrossRef]
- Wang, L.; Abedalwafa, M.; Wang, F.; Li, C. Biodegradable poly-epsilon-caprolactone (PCL) for tissue engineering applications: A review. Rev. Adv. Mater. Sci. 2013, 34, 123–140. [Google Scholar]
- Mondal, D.; Griffith, M.; Venkatraman, S.S. Polycaprolactone-based biomaterials for tissue engineering and drug delivery: Current scenario and challenges. Int. J. Polym. Biomater. 2016, 65, 255–265. [Google Scholar] [CrossRef]
- Bahcecioglu, G.; Hasirci, N.; Hasirci, V. Cell behavior on the alginate-coated PLLA/PLGA scaffolds. Int. J. Biol. Macromol. 2019, 124, 444–450. [Google Scholar] [CrossRef]
- Dinoro, J.; Maher, M.; Talebian, S.; Jafarkhani, M.; Mehrali, M.; Orive, G.; Foroughi, J.; Lord, M.S.; Dolatshahi-Pirouz, A. Sulfated polysaccharide-based scaffolds for orthopaedic tissue engineering. Biomaterials 2019, 214, 119214. [Google Scholar] [CrossRef]
- Valente, J.F.A.; Valente, T.A.M.; Alves, P.; Ferreira, P.; Silva, A.; Correia, I.J. Alginate based scaffolds for bone tissue engineering. Mater. Sci. Eng. C 2012, 32, 2596–2603. [Google Scholar] [CrossRef]
- Yan, J.; Miao, Y.; Tan, H.; Zhou, T.; Ling, Z.; Chen, Y.; Xing, X.; Hu, X. Injectable alginate/hydroxyapatite gel scaffold combined with gelatin microspheres for drug delivery and bone tissue engineering. Mater. Sci. Eng. C 2016, 63, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-P.; Chang, Y.-S. Preparation and characterization of composite nanofibers of polycaprolactone and nanohydroxyapatite for osteogenic differentiation of mesenchymal stem cells. Colloids Surf. B Biointerfaces 2011, 86, 169–175. [Google Scholar] [CrossRef]
- Miculescu, F.; Maidaniuc, A.; Voicu, S.I.; Thakur, V.K.; Stan, G.E.; Ciocan, L.T. Progress in hydroxyapatite—Starch based sustainable biomaterials for biomedical bone substitution applications. ACS Sustain. Chem. Eng. 2017, 5, 8491–8512. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, C.M.; Athanasiou, K.A. Technique to control pH in vicinity of biodegrading PLA-PGA implants. J. Biomed. Mater. Res. 1997, 38, 105–114. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, Y.; Yan, W.; Hu, Q.; Tao, J.; Zhang, M.; Shi, Z.; Tang, R. Role of hydroxyapatite nanoparticle size in bone cell proliferation. J. Mater. Chem. 2007, 17, 3780–3787. [Google Scholar] [CrossRef]
- Gómez-Morales, J.; Torrent-Burgues, J.; Boix, T.; Fraile, J.; Rodríguez-Clemente, R. Precipitation of stoichiometric hydroxyapatite by a continuous method. Cryst. Res. Technol. J. Exp. Ind. Crystallogr. 2021, 36, 15–26. [Google Scholar] [CrossRef]
- Drouet, C.; Bosc, F.; Banu, M.; Largeot, C.; Combes, C.; Dechambre, G.; Estournes, C.; Raimbeaux, G.; Rey, C. Nanocrystalline apatites: From powders to biomaterials. Powder Technol. 2009, 190, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Olivier, F.; Picard, Q.; Delpeux-Ouldriane, S.; Chancolon, J.; Warmont, F.; Sarou-Kanian, V.; Fayon, F.; Bonnamy, S. Influence of electrochemical parameters on the characteristics of sono-electrodeposited calcium phosphate-coated carbon fiber cloth. Surf. Coatings Technol. 2020, 389, 125507. [Google Scholar] [CrossRef]
- Grunenwald, A.; Keyser, C.; Sautereau, A.; Crubézy, E.; Ludes, B.; Drouet, C. Revisiting carbonate quantification in apatite (bio)minerals: A validated FTIR methodology. J. Archaeol. Sci. 2014, 49, 134–141. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Hydrophobic Polymer | Mass of Hydrophobic Polymer (onto 3 mL of Dichloromethane), g | Sodium Alginate Mass (onto 9 mL Water, g | Type of HAP NPs (Shape and Sizes) | Mass of HAP NPs, g |
|---|---|---|---|---|---|
| I-PLGA-0.05 | PLGA | 0.09 | 0.18 | plate-shaped nanoparticles with an average thickness of 4 nm and a length of 30 to 50 nm | 0.05 |
| I-PLGA-0.10 | 0.09 | 0.10 | |||
| II-PLGA-0.05 | 0.09 | rod-shaped nanoparticles with an average diameter of 10 nm and a length of 150–180 nm | 0.05 | ||
| II-PLGA-0.10 | 0.09 | 0.10 | |||
| III-PLGA-0.05 | 0.09 | rod-shaped nanoparticles with an average diameter of 10 nm and a length of 110–130 nm | 0.05 | ||
| III-PLGA-0.10 | 0.09 | 0.10 | |||
| III-PLGA-0.05-Ca | 0.09 | 0.05 | |||
| III-PLGA-0.10-Ca | 0.09 | 0.10 | |||
| III-PLGA-0.15-Ca | 0.09 | 0.15 | |||
| III-PCL-0.05-Ca | PCL | 0.09 | 0.05 | ||
| III-PCL-0.10-Ca | 0.09 | 0.10 | |||
| III-PCL-0.15-Ca | 0.09 | 0.15 | |||
| III-PLGA/PCL-0.05-Ca | PLGA | 0.045 | 0.05 | ||
| PCL | 0.045 | ||||
| III-PLGA/PCL-0.10-Ca | PLGA | 0.045 | 0.10 | ||
| PCL | 0.045 | ||||
| III-PLGA/PCL-0.15-Ca | PLGA | 0.045 | 0.15 | ||
| PCL | 0.045 |
| Sample | Mechanical Stability | pH | Cytotoxicity | Cell Adhesion (12 Day) |
|---|---|---|---|---|
| I-PLGA-0.05 | non-stable | acidic medium | cytotoxic | absent |
| I-PLGA-0.10 | non-stable | acidic medium | cytotoxic | absent |
| II-PLGA-0.05 | non-stable | acidic medium | cytotoxic | absent |
| II-PLGA-0.10 | non-stable | acidic medium | cytotoxic | absent |
| III-PLGA-0.05 | partial non-stable | neutral | low-cytotoxic | medium |
| III-PLGA-0.10 | partial non-stable | neutral | low-cytotoxic | medium |
| III-PLGA-0.05-Ca | stable | neutral | non-cytotoxic | high |
| III-PLGA-0.10-Ca | stable | neutral | non-cytotoxic | high |
| III-PLGA-0.15-Ca | stable | neutral | non-cytotoxic | high |
| III-PCL-0.05-Ca | stable | neutral | non-cytotoxic | high |
| III-PCL-0.10-Ca | stable | neutral | non-cytotoxic | high |
| III-PCL-0.15-Ca | stable | neutral | non-cytotoxic | high |
| III-PLGA/PCL-0.05-Ca | stable | neutral | non-cytotoxic | high |
| III-PLGA/PCL-0.10-Ca | stable | neutral | non-cytotoxic | high |
| III-PLGA/PCL-0.15-Ca | stable | neutral | non-cytotoxic | high |
| Sample | Number of Animals | Body Weight, g | ||
|---|---|---|---|---|
| Initial | 8 Day | 14 Day | ||
| Male mouse | ||||
| Control | 5 | 24.7 ± 1.7 | 29.7 ± 2.5 | 36.7 ± 2.2 |
| III-PLGA-0.10-Ca | 5 | 24.4 ± 1.9 | 29.3 ± 2.4 | 35.6 ± 2.9 |
| III-PCL-0.10-Ca | 5 | 24.2 ± 1.8 | 30.7 ± 2.4 | 36.9 ± 1.7 |
| III-PLGA/PCL-0.10-Ca | 5 | 25.0 ± 1.4 | 30.9 ± 1.8 | 35.5 ± 2.3 |
| Female mouse | ||||
| Control | 5 | 21.9 ± 1.2 | 24.8 ± 1.2 | 29.0 ± 1.0 |
| III-PLGA-0.10-Ca | 5 | 21.7 ± 1.6 | 24.5 ± 1.3 | 28.0 ± 2.4 |
| III-PCL-0.10-Ca | 5 | 21.5 ± 1.8 | 24.2 ± 1.9 | 28.4 ± 2.4 |
| III-PLGA/PCL-0.10-Ca | 5 | 21.4 ± 1.5 | 24.0 ± 1.8 | 28.1 ± 1.2 |
| Male rats | ||||
| Control | 5 | 204.0 ± 13.7 | 223.8 ± 12.9 | 251.4 ± 16.5 |
| III-PLGA-0.10-Ca | 5 | 206.2 ± 14.0 | 225.8 ± 10.7 | 255.4 ± 13.6 |
| III-PCL-0.10-Ca | 5 | 205.2 ± 14.1 | 232.6 ± 5.0 | 260.4 ± 15.0 |
| III-PLGA/PCL-0.10-Ca | 5 | 207.4 ± 14.0 | 229.2 ± 14.2 | 259.4 ± 17.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordienko, M.; Karakatenko, E.; Menshutina, N.; Koroleva, M.; Gilmutdinova, I.; Eremin, P. Composites Composed of Hydrophilic and Hydrophobic Polymers, and Hydroxyapatite Nanoparticles: Synthesis, Characterization, and Study of Their Biocompatible Properties. J. Funct. Biomater. 2021, 12, 55. https://doi.org/10.3390/jfb12040055
Gordienko M, Karakatenko E, Menshutina N, Koroleva M, Gilmutdinova I, Eremin P. Composites Composed of Hydrophilic and Hydrophobic Polymers, and Hydroxyapatite Nanoparticles: Synthesis, Characterization, and Study of Their Biocompatible Properties. Journal of Functional Biomaterials. 2021; 12(4):55. https://doi.org/10.3390/jfb12040055
Chicago/Turabian StyleGordienko, Mariia, Elena Karakatenko, Natalia Menshutina, Marina Koroleva, Ilmira Gilmutdinova, and Petr Eremin. 2021. "Composites Composed of Hydrophilic and Hydrophobic Polymers, and Hydroxyapatite Nanoparticles: Synthesis, Characterization, and Study of Their Biocompatible Properties" Journal of Functional Biomaterials 12, no. 4: 55. https://doi.org/10.3390/jfb12040055
APA StyleGordienko, M., Karakatenko, E., Menshutina, N., Koroleva, M., Gilmutdinova, I., & Eremin, P. (2021). Composites Composed of Hydrophilic and Hydrophobic Polymers, and Hydroxyapatite Nanoparticles: Synthesis, Characterization, and Study of Their Biocompatible Properties. Journal of Functional Biomaterials, 12(4), 55. https://doi.org/10.3390/jfb12040055