Feedback-Controlled Release of Alendronate from Composite Microparticles



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. CaP Particle Formation
Physicochemical Characterization
2.2.2. ALD-Loaded Microspheres
Microspheres’ Physicochemical Characterization and Drug Encapsulation
Size Distribution and Morphology
In Vitro Release
Biocompatibility Evaluation
2.2.3. Statistical Analysis
3. Results
3.1. CaP Particles
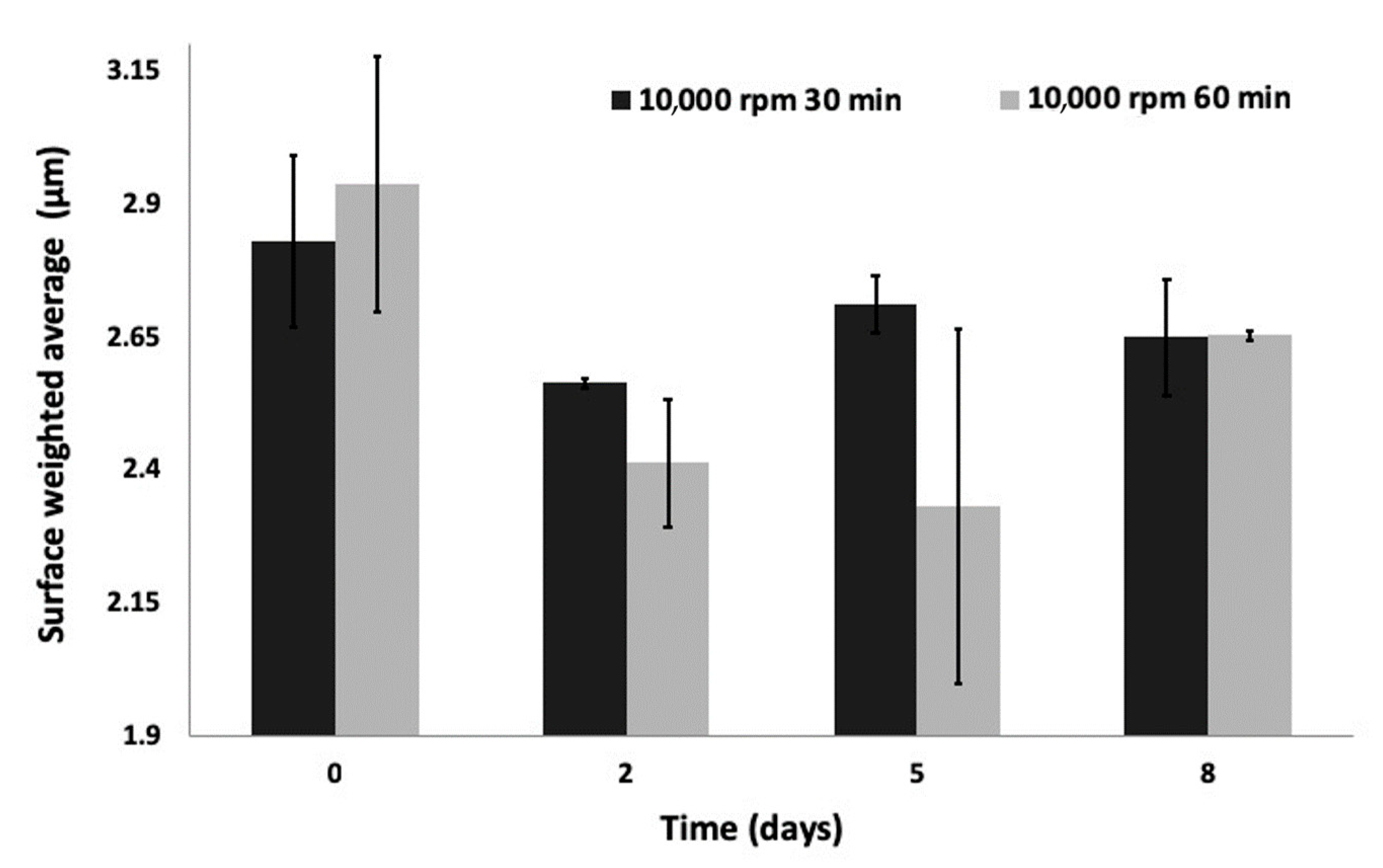
3.2. Physical Properties
3.2.1. Physical Characteristics
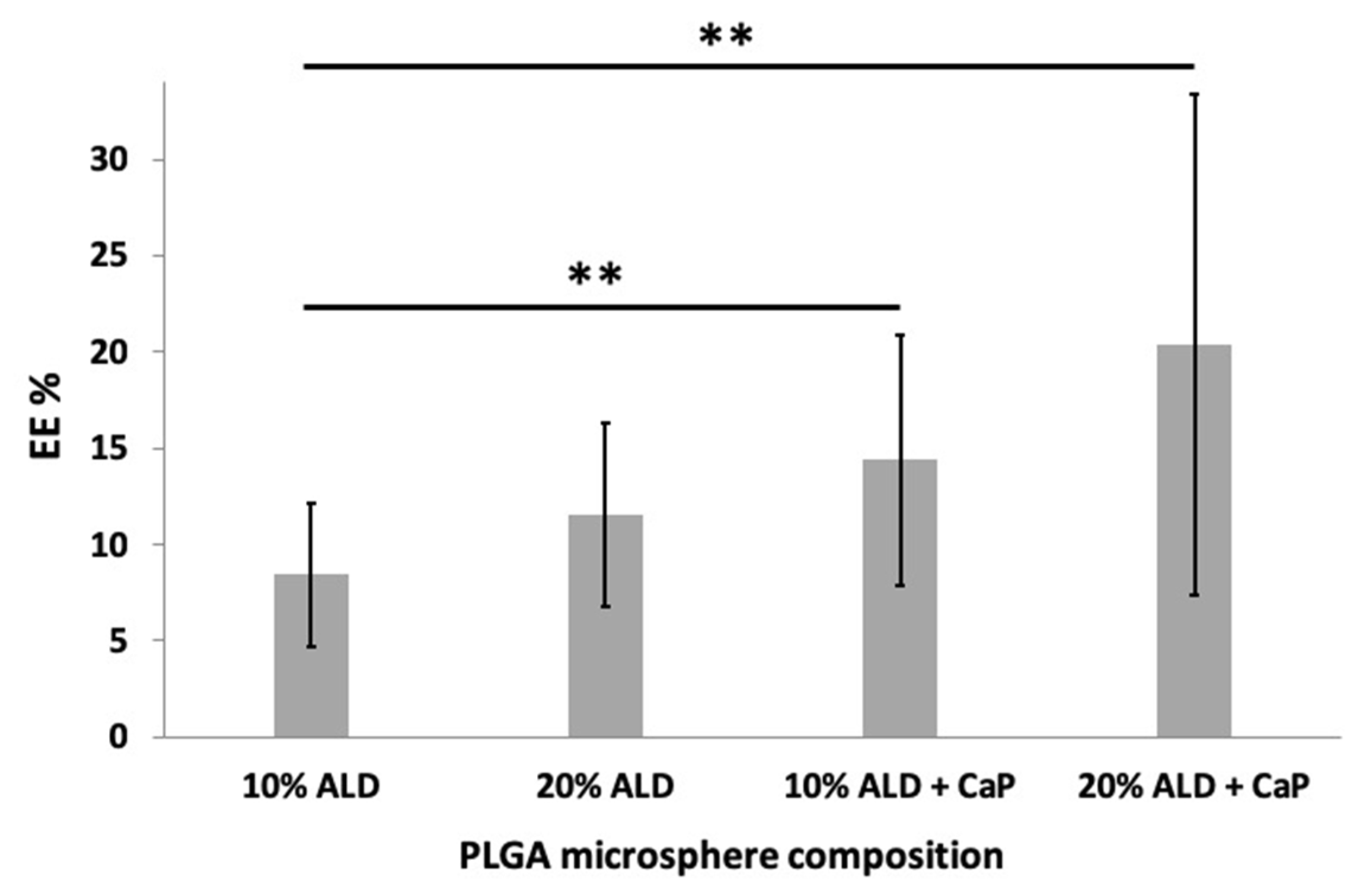
3.2.2. Encapsulation Efficiency (EE%)
3.2.3. In Vitro Release
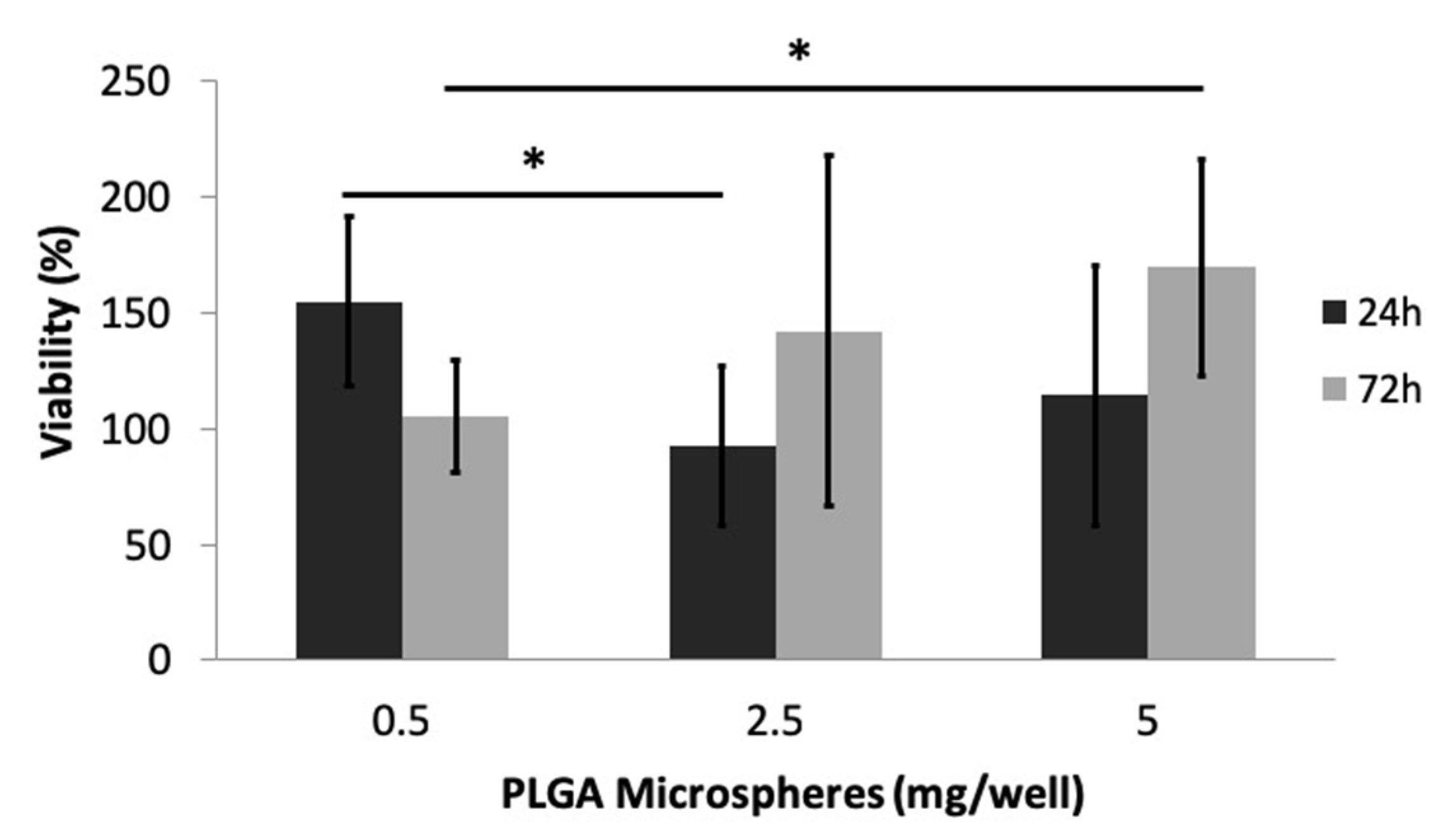
3.2.4. Cytotoxicity Evaluation
4. Discussion
4.1. CaP Particles
4.2. ALD-Loaded PLGA Microspheres
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Porter, J.R.; Ruckh, T.T.; Popat, K.C. Bone Tissue Engineering: A Review in Bone Biomimetics and Drug Delivery Strategies. Biotechnol. Prog. 2009, 25, 1539–1560. [Google Scholar] [CrossRef] [PubMed]
- Bassi, A.; Gough, J.; Zakikhani, M.; Downes, S. Bone tissue regeneration. In Electrospinning for Tissue Regeneration, 1st ed.; Bosworth, L.A., Downes, S., Eds.; Woodhead Publishing Limited: Cambridge, UK, 2011; pp. 93–110. [Google Scholar]
- Klein-Nulend, J.; Bacabac, R.G.; Mullender, M.G. Mechanobiology of bone tissue. Pathol. Biol. 2005, 53, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Awad, H.A.; O’Keefe, R.J.; Lee, C.H; Mao, J.J. Bone Tissue Engineering: Clinical Challenges and Emergent Advances in Orthopedic and Craniofacial Surgery. In Principles of Tissue Engineering; Lanza, R., Langer, R., Vacanti, J., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 1733–1743. [Google Scholar]
- Shrivats, A.R.; Alvarez, P.; Schutte, L.; Hollinger, J.O. Bone Regeneration. In Principles of Tissue Engineering, 4th ed.; Lanza, R., Langer, R., Vacanti, J., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 1201–1221. [Google Scholar]
- Bose, S.; Tarafder, S. Calcium phosphate ceramic systems in growth factor and drug delivery for bone tissue engineering: A review. Acta Biomater. 2012, 8, 1401–1421. [Google Scholar] [CrossRef] [PubMed]
- Khan, Y.; Yaszemski, M.J.; Mikos, A.G.; Laurencin, C.T. Tissue Engineering of Bone: Material and Matrix Considerations. J. Bone Jt. Surg. Am. 2008, 90, 36–42. [Google Scholar] [CrossRef]
- Bassi, A.K.; Gough, J.E.; Zakikhani, M.; Downes, S. The Chemical and Physical Properties of Poly(ε-caprolactone) Scaffolds Functionalised with Poly(vinyl phosphonic acid-co-acrylic acid). J. Tissue Eng. 2011, 2011, 615328. [Google Scholar] [CrossRef]
- Ghag, A.K.; Gough, J.E.; Downes, S. The osteoblast and osteoclast responses to phosphonic acid containing poly(ε-caprolactone) electrospun scaffolds. Biomater. Sci. 2014, 2, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, A.; Sagadevan, S.; Dakshnamoorthy, A. Synthesis and characterization of nano-hydroxyapatite (n-HAP) using the wet chemical technique. Int. J. Phys. Sci. 2013, 8, 1639–1645. [Google Scholar]
- Elbackly, R.M.; Mastrogiacomo, M.; Cancedda, R. Bone Regeneration and Bioengineering. In Regenerative Medicine Applications in Organ Transplantation, 1st ed.; Orlando, G., Lerut, J.P., Soker, S., Stratta, R.J., Eds.; Elsevier: Chicago, IL, USA, 2014; pp. 783–797. [Google Scholar]
- Szpalski, C.; Sagebin, F.; Barbaro, M.; Warren, S.M. The influence of environmental factors on bone tissue engineering. J. Biomed. Mater. Res. B Appl. Biomater. 2013, 101, 663–675. [Google Scholar] [CrossRef]
- Silva, G.A.; Coutinho, O.P.; Ducheyne, P.; Reis, R.L. Materials in particulate form for tissue engineering. 2. Applications in bone. J. Tissue Eng. Regen. Med. 2007, 1, 97–109. [Google Scholar] [CrossRef]
- Garbuz, D.S.; Hu, Y.; Kim, W.Y.; Duan, K.; Masri, B.A.; Oxland, T.R.; Burt, H.; Wang, R.; Duncan, C.P. Enhanced Gap Filling and Osteoconduction Associated with Alendronate-Calcium Phosphate-Coated Porous Tantalum. J. Bone Jt. Surg. Am. 2008, 90, 1090–1100. [Google Scholar] [CrossRef]
- Samdancioglu, S.; Calis, S.; Sumnu, M.; Hincal, A. Formulation and In Vitro Evaluation of Bisphosphonate Loaded Microspheres for Implantation in Osteolysis. Drug Dev. Ind. Pharm. 2006, 32, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wang, Y.; Ren, L.; Gong, Y.; Wang, D. Enhancing alendronate release from a novel PLGA/hydroxyapatite microspheric system for bone repairing applications. Pharm. Res. 2009, 26, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Kyllönen, L.; D’Este, M.; Alini, M.; Eglin, D. Local drug delivery for enhancing fracture healing in osteoporotic bone. Acta. Biomater. 2015, 11, 412–434. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; De Simone, G.; Ascenzi, P. Cysteine-based regulation of redox-sensitive Ras small GTPases. Redox. Biol. 2019, 26, 101282. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Cong, W.; Zhou, S.; Shi, Y.; Dai, W.; Zhang, H.; Wang, X.; He, B.; Zhang, Q. Small GTPases: Stucture, biological function and its interaction with nanoparticles. Asian J. Pharm. Sci. 2019, 14, 30–39. [Google Scholar] [CrossRef]
- Coombes, A.G.A.; Rizzi, S.C.; Williamson, M.; Barralet, J.E.; Downes, S.; Wallace, W.A. Precipitation casting of polycaprolactone for applications in tissue engineering and drug delivery. Biomaterials 2004, 25, 315–325. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Mobasherpour, I.; Soulati Heshajin, M.; Kazemzadeh, A.; Zakeri, M. Synthesis of nanocrystalline hydroxyapatite by using precipitation method. J. Alloy. Compd. 2007, 430, 330–333. [Google Scholar] [CrossRef]
- Nasr, M.; Awad, G.A.; Mansour, S.; Shamy, A.A.; Mortada, N.D. A Reliable Predictive Factorial Model for Entrapment Optimization of a Sodium Bisphosphonate into Biodegradable Microspheres. J. Pharm. Sci. 2011, 100, 612–621. [Google Scholar] [CrossRef]
- Mondal, T.; Sunny, M.C.; Khastgir, D.; Varma, H.K.; Ramesh, P. Poly (l-lactide-co-Є caprolactone) microspheres laden with bioactive glass-ceramic and alendronate sodium as bone regenerative scaffolds. Mater. Sci. Eng. C 2012, 32, 697–706. [Google Scholar] [CrossRef]
- Ostovic, D.; Stelmach, C.; Hulshizer, B. Formation of a Chromophoric Complex Between Alendronate and Copper(II) Ions. Pharm. Res. 1993, 10, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Perugini, P.; Genta, I.; Conti, B.; Modena, T.; Pavanetto, F. Long-term Release of Clodronate from Biodegradable Microspheres. AAPS PharmSciTech 2001, 2, 6–14. [Google Scholar] [CrossRef]
- Cheng, Z.H.; Yasukawa, A.; Kandori, K.; Ishikawa, T. FTIR Study on incorporation of CO2 into calcium hydroxyapatite. J. Chem. Soc. Faraday Trans. 1998, 94, 1501–1505. [Google Scholar] [CrossRef]
- Fu, B.; Sun, X.; Qian, W.; Shen, Y.; Chen, R.; Hannig, M. Evidence of chemical bonding to hydroxyapatite by phosphoric acid esters. Biomaterials 2005, 26, 5104–5110. [Google Scholar] [CrossRef] [PubMed]
- Ślósarczyk, A.; Paszkiewicz, Z.; Paluszkiewicz, C. FTIR and XRD evaluation of carbonated hydroxyapatite powders synthesized by wet methods. J. Mol. Struct. 2005, 744, 657–661. [Google Scholar] [CrossRef]
- Berzina-Cimdina, L.; Borodajenko, N. Research of Calcium Phosphates Using Fourier Transform Infrared Spectroscopy. In Infrared Spectroscopy–Materials Science, Engineering and Technology; Theophanides, T., Ed.; IntechOpen: London, UK, 2012; pp. 123–148. Available online: http://www.intechopen.com/books/infrared-spectroscopy-materials-science-engineering-and-technology/research-of-calcium-phosphates-using-fourier-transformation-infrared-spectroscopy (accessed on 1 May 2020).
- Boudia, S.; Zuddas, P.; Fermane, F.; Fiallo, M.; Sharrock, P. Minerological transformation during hydroxyapatite dissolution in simple aqueous solutions. Chem. Biol. 2018, 477, 85–91. [Google Scholar]
- Abou Neel, E.A.; Ahmed, I.; Pratten, J.; Nazhat, S.N.; Knowles, J.C. Characterisation of antibacterial copper releasing degradable phosphate glass fibres. Biomaterials 2005, 26, 2247–2254. [Google Scholar] [CrossRef]
- Markich, S.J.; Brown, P.L.; Jeffree, R.A. Divalent metal accumulation in freshwater bivalves: An inverse relationship with metal phosphate solubility. Sci. Total Environ. 2001, 275, 27–41. [Google Scholar] [CrossRef]
- Jiang, J.; Tang, A.; Ateshian, G.A.; Guo, E.; Hung, C.T; Lu, H.H. Bioactive Stratified Polymer Ceramic-Hydrogel Scaffold for Integrative Osteochondral Repair. Ann. Biomed. Eng. 2010, 38, 2183–2196. [Google Scholar] [CrossRef]
- Matsumoto, T.; Okazaki, M.; Inoue, M.; Yamaguchi, S.; Kusunose, T.; Toyonaga, T.; Hamada, Y.; Takahashi, J. Hydroxyapatite particles as a controlled release carrier of protein. Biomaterials 2004, 25, 3807–3812. [Google Scholar] [CrossRef]
- Sabokbar, A.; Pandey, R.; Diaz, J.; Quinn, J.M.; Murray, D.W.; Athanasou, N.A. Hydroxyapatite particles are capable of inducing osteoclast formation. J. Mater. Sci. Mater. Med. 2001, 12, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Lebre, F.; Sridharan, R.; Sawkins, M.J.; Kelly, D.J.; O’Brien, F.J.; Lavelle, E.C. The shape and size of hydroxyapatite particles dictate inflammatory responses following implantation. Sci. Rep. 2017, 7, 2922–2935. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Grasser, W.; Endo, N.; Akins, R.; Simmons, H.; Thompson, D.D.; Golub, E.; Rodan, G.A. Bisphosphonate action. Alendronate localization in rat bone and effects on osteoclast ultrastructure. J. Clin. Investig. 1991, 88, 2095–2105. [Google Scholar] [CrossRef]
- Nancollas, G.H.; Tang, R.; Phipps, R.J.; Henneman, Z.; Gulde, S.; Wu, W.; Mangood, A.; Russell, R.G.G.; Ebetino, F.H. Novel insights into actions of bisphosphonates on bone: Differences in interactions with hydroxyapatite. Bone 2006, 38, 617–627. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Homogenization Parameters | Time (h) | Size Distribution (μm) | ||
|---|---|---|---|---|---|
| Speed (rpm) | Duration (min) | SWA | Median Diameter (d50) | ||
| 1 | 7000 | 10 | - | 12.4 +/− 2.274 | 13.8 +/− 2.354 |
| 48 | 5.1 +/− 0.349 | 8.9 +/− 5.322 | |||
| 2 | 7000 | 30 | - | 7 +/− 1.948 | 9.1 +/− 3.398 |
| 48 | 6.0 +/− 0.093 | 7.9 +/− 0.066 | |||
| 3 | 10,000 | 30 | - | 2.8 +/− 0.163 | 3.1 +/− 0.210 |
| 48 | 2.6 +/− 0.009 | 2.8 +/− 0.006 | |||
| 4 | 10,000 | 60 | - | 2.9 +/− 0.239 | 3.2 +/− 0.407 |
| 48 | 2.4 +/− 0.332 | 2.6 +/− 0.057 | |||
| Sample | Density (g/cm3) | |
|---|---|---|
| Mean | SD | |
| CaP | 3.14 | 0.02 |
| PLGA microspheres | 1.51 | 0.13 |
| CaP-loaded PLGA microspheres | 1.53 | 0.01 |
| 10% ALD-loaded PLGA microspheres | 1.48 | 0.02 |
| 10% ALD-CaP-loaded PLGA microspheres | 1.99 | 0.06 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matrali, S.S.H.; Ghag, A.K. Feedback-Controlled Release of Alendronate from Composite Microparticles. J. Funct. Biomater. 2020, 11, 46. https://doi.org/10.3390/jfb11030046
Matrali SSH, Ghag AK. Feedback-Controlled Release of Alendronate from Composite Microparticles. Journal of Functional Biomaterials. 2020; 11(3):46. https://doi.org/10.3390/jfb11030046
Chicago/Turabian StyleMatrali, Sofia S. H., and Anita K. Ghag. 2020. "Feedback-Controlled Release of Alendronate from Composite Microparticles" Journal of Functional Biomaterials 11, no. 3: 46. https://doi.org/10.3390/jfb11030046
APA StyleMatrali, S. S. H., & Ghag, A. K. (2020). Feedback-Controlled Release of Alendronate from Composite Microparticles. Journal of Functional Biomaterials, 11(3), 46. https://doi.org/10.3390/jfb11030046