
Extraction of a One-Particle Reduced Density Matrix from a Quantum Monte Carlo Electronic Density: A New Tool for Studying Nondynamic Correlation


Abstract
:1. Introduction
2. Methodology
2.1. 1-RDM from a Correlated Electron Density
The Fit Procedure
2.2. 1-RDM from Unrestricted KS Computations
2.3. Fit of the Fermi Distribution
3. Computational Details
3.1. Construction of the Electronic Density
3.2. Description of the Fitting Procedure
3.3. Methods and Softwares
4. Results
4.1. Fit of the Electronic Density
4.2. Unrestricted Computations
4.3. Fit of the Fermi Distribution
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Löwdin, P.O. Quantum Theory of Many-Particle Systems. III. Extension of the Hartree-Fock Scheme to Include Degenerate Systems and Correlation Effects. Phys. Rev. 1955, 97, 1509–1520. [Google Scholar] [CrossRef]
- Coulson, P.C.; Fischer, M.I. XXXIV. Notes on the molecular orbital treatment of the hydrogen molecule. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1949, 40, 386–393. [Google Scholar] [CrossRef]
- Heitler, W.; London, F. Wechselwirkung neutraler Atome und homöopolare Bindung nach der Quantenmechanik. Z. Phys. 1927, 44, 455–472. [Google Scholar] [CrossRef]
- Cremer, D. Density functional theory: Coverage of dynamic and non-dynamic electron correlation effects. Mol. Phys. 2001, 99, 1899–1940. [Google Scholar] [CrossRef]
- Garza, A.J.; Jimenez-Hoyos, C.A.; Scuseria, G.E. Capturing static and dynamic correlations by a combination of projected Hartree-Fock and density functional theories. J. Chem. Phys. 2013, 138, 134102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobola, R. Employing broken symmetry effects from unrestricted coupled cluster wave function to determine dynamic and non-dynamic electron correlation during triple bond breaking in the N2 molecule. Int. J. Quantum Chem. 2019, 119, e25865. [Google Scholar] [CrossRef]
- Zhang, D.; Truhlar, D.G. Unmasking Static Correlation Error in Hybrid Kohn-Sham Density Functional Theory. J. Chem. Theory Comput. 2020, 16, 5432–5440. [Google Scholar] [CrossRef]
- Timothy, J. Lee, Martin Head-Gordon, A.P.R. Investigation of a diagnostic for perturbation theory. Comparison to the T1 diagnostic of coupled-cluster theory. Chem. Phys. Lett. 1995, 243, 402–408. [Google Scholar]
- Lee, T.J.; Taylor, P.R. A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 1989, 36, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Fogueri, U.R.; Kozuch, S.; Karton, A.; Martin, J.M.L. A simple DFT-based diagnostic for nondynamical correlation. Theor. Chem. Accounts 2013, 132, 1–9. [Google Scholar] [CrossRef]
- Janssen, C.L.; Nielsen, I.M. New diagnostics for coupled-cluster and Møller–Plesset perturbation theory. Chem. Phys. Lett. 1998, 290, 423–430. [Google Scholar] [CrossRef]
- Nielsen, I.M.; Janssen, C.L. Double-substitution-based diagnostics for coupled-cluster and Møller–Plesset perturbation theory. Chem. Phys. Lett. 1999, 310, 568–576. [Google Scholar] [CrossRef]
- Ziesche, P. Correlation strength and information entropy. Int. J. Quantum Chem. 1995, 56, 363–369. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, H.; Kais, S. Entanglement and electron correlation in quantum chemistry calculations. J. Mod. Opt. 2006, 53, 2543–2558. [Google Scholar] [CrossRef]
- Amovilli, C.; Floris, F.M. Shannon Entropy and Correlation Energy for Electrons in Atoms. In Many-Body Approaches at Different Scales: A Tribute to Norman H. March on the Occasion of their 90th Birthday; Angilella, G., Amovilli, C., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 187–198. [Google Scholar]
- Via-Nadal, M.; Rodríguez-Mayorga, M.; Ramos-Cordoba, E.; Matito, E. Singling Out Dynamic and Nondynamic Correlation. J. Phys. Chem. Lett. 2019, 10, 4032–4037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Cordoba, E.; Salvador, P.; Matito, E. Separation of dynamic and nondynamic correlation. Phys. Chem. Chem. Phys. 2016, 18, 24015–24023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Cordoba, E.; Matito, E. Local Descriptors of Dynamic and Nondynamic Correlation. J. Chem. Theory Comput. 2017, 13, 2705–2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tishchenko, O.; Zheng, J.; Truhlar, D.G. Multireference Model Chemistries for Thermochemical Kinetics. J. Chem. Theory Comput. 2008, 4, 1208–1219. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A. A Practicable Real-Space Measure and Visualization of Static Electron-Correlation Effects. Angew. Chem. Int. Ed. 2015, 54, 12308–12313. [Google Scholar] [CrossRef] [PubMed]
- Pribram-Jones, A.; Pittalis, S.; Gross, E.K.U.; Burke, K. Thermal Density Functional Theory in Context. In Frontiers and Challenges in Warm Dense Matter; Graziani, F., Desjarlais, M.P., Redmer, R., Trickey, S.B., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Pulay, P. Localizability of dynamic electron correlation. Chem. Phys. Lett. 1983, 100, 151–154. [Google Scholar] [CrossRef]
- Lester, W.A., Jr.; Mitas, L.; Hammond, B. Quantum Monte Carlo for atoms, molecules and solids. Chem. Phys. Lett. 2009, 478, 1–10. [Google Scholar] [CrossRef]
- Kent, P.R.C.; Hood, R.Q.; Towler, M.D.; Needs, R.J.; Rajagopal, G. Quantum Monte Carlo calculations of the one-body density matrix and excitation energies of silicon. Phys. Rev. B 1998, 57, 15293–15302. [Google Scholar] [CrossRef] [Green Version]
- Delle Site, L. Levy-Lieb principle: The bridge between the electron density of Density Functional Theory and the wavefunction of Quantum Monte Carlo. Chem. Phys. Lett. 2015, 619, 148–151. [Google Scholar] [CrossRef] [Green Version]
- Assaraf, R.; Caffarel, M.; Scemama, A. Improved Monte Carlo estimators for the one-body density. Phys. Rev. E 2007, 75, 035701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coccia, E.; Chernomor, O.; Barborini, M.; Sorella, S.; Guidoni, L. Molecular Electrical Properties from Quantum Monte Carlo Calculations: Application to Ethyne. J. Chem. Theory Comput. 2012, 8, 1952–1962. [Google Scholar] [CrossRef]
- Coleman, A.J. Structure of Fermion Density Matrices. Rev. Mod. Phys. 1963, 35, 668–686. [Google Scholar] [CrossRef]
- Herbert, J.M.; Harriman, J.E. N-representability and variational stability in natural orbital functional theory. J. Chem. Phys. 2003, 118, 10835–10846. [Google Scholar] [CrossRef] [Green Version]
- Lathiotakis, N.N.; Gidopoulos, N.I.; Helbig, N. Size consistency of explicit functionals of the natural orbitals in reduced density matrix functional theory. J. Chem. Phys. 2010, 132, 084105. [Google Scholar] [CrossRef] [Green Version]
- Mazziotti, D.A. Structure of Fermionic Density Matrices: Complete N-Representability Conditions. Phys. Rev. Lett. 2012, 108, 263002. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.; Schmidt, K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. In Proceedings of the International Conference on Density Functional Theory and its Applications to Materials, Antwerp, Belgium, 8–10 June 2000. [Google Scholar]
- Filippi, C.; Umrigar, C.J. Multiconfiguration wave functions for quantum Monte Carlo calculations of first-row diatomic molecules. J. Chem. Phys. 1996, 105, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, W.M.C.; Mitas, L.; Needs, R.J.; Rajagopal, G. Quantum Monte Carlo simulations of solids. Rev. Mod. Phys. 2001, 73, 33–83. [Google Scholar] [CrossRef] [Green Version]
- Löwdin, P.O. On the Nonorthogonality Problem. In Advances in Quantum Chemistry; Academic Press: Cambridge, MA, USA, 1970; Volume 5, pp. 185–199. [Google Scholar]
- Goddard, W.A.; Dunning, T.H.; Hunt, W.J.; Hay, P.J. Generalized valence bond description of bonding in low-lying states of molecules. Accounts Chem. Res. 1973, 6, 368–376. [Google Scholar] [CrossRef]
- Fracchia, F.; Filippi, C.; Amovilli, C. Size-Extensive Wave Functions for Quantum Monte Carlo: A Linear Scaling Generalized Valence Bond Approach. J. Chem. Theory Comput. 2012, 8, 1943–1951. [Google Scholar] [CrossRef]
- Burkatzki, M.; Filippi, C.; Dolg, M. Energy-consistent pseudopotentials for quantum Monte Carlo calculations. J. Chem. Phys. 2007, 126, 234105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CHAMP. A Quantum Monte Carlo Program Written by C. J. Umrigar and C. Filippi; The Code Is Not Freely Distributed, Contact the Authors for More Information. Available online: https://www.utwente.nl/en/tnw/ccp/research/CHAMP.html (accessed on 7 December 2021).
- Schmidt, M.; Baldridge, K.; Boatz, J.; Elbert, S.; Gordon, M.; Jensen, J.; Koseki, S.; Matsunaga, N.; Nguyen, K.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Accounts 2008, 120, 215–241. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Zulfikri, H.; Amovilli, C.; Filippi, C. Multiple-Resonance Local Wave Functions for Accurate Excited States in Quantum Monte Carlo. J. Chem. Theory Comput. 2016, 12, 1157–1168. [Google Scholar] [CrossRef]
- Chan, G.K.L.; Sharma, S. The Density Matrix Renormalization Group in Quantum Chemistry. Annu. Rev. Phys. Chem. 2011, 62, 465–481. [Google Scholar] [CrossRef] [Green Version]
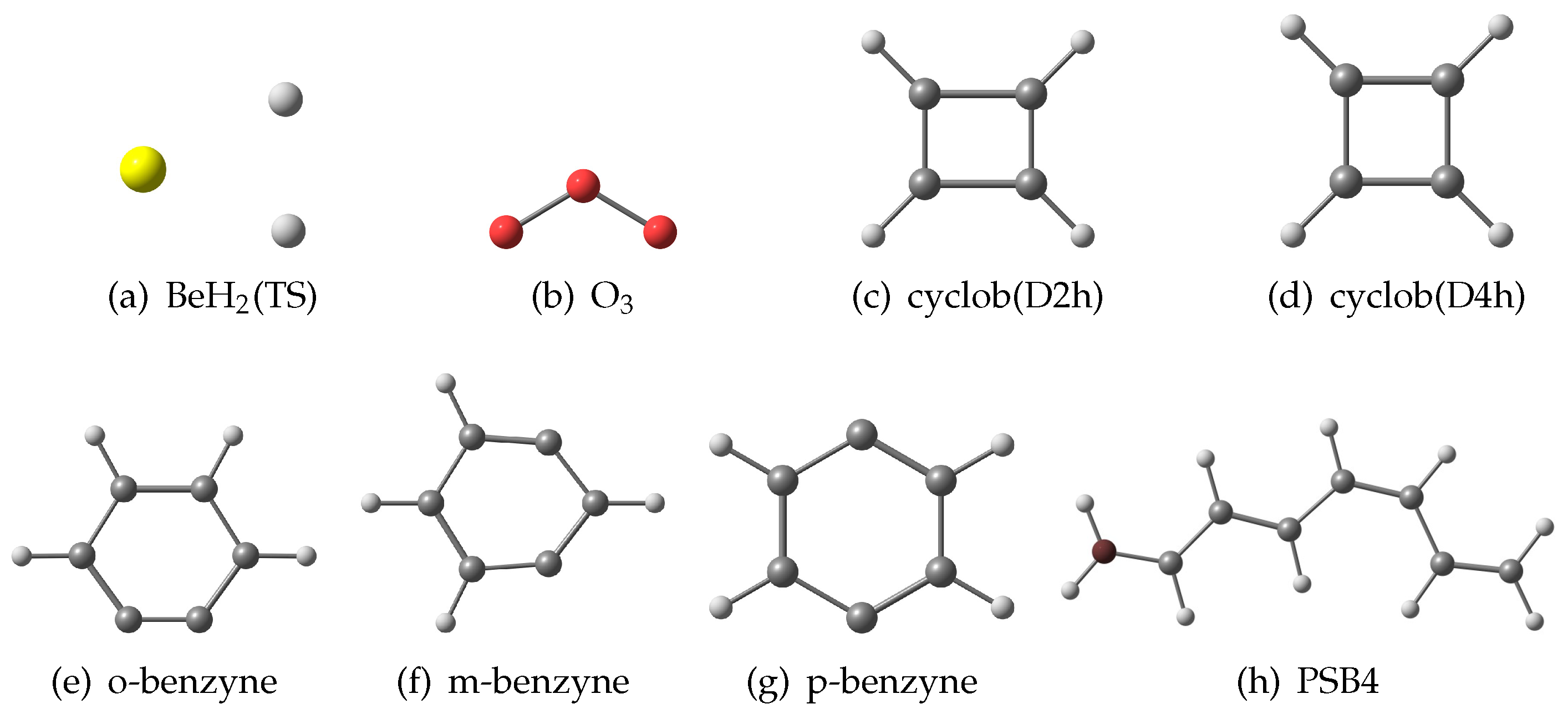



{kind=link}
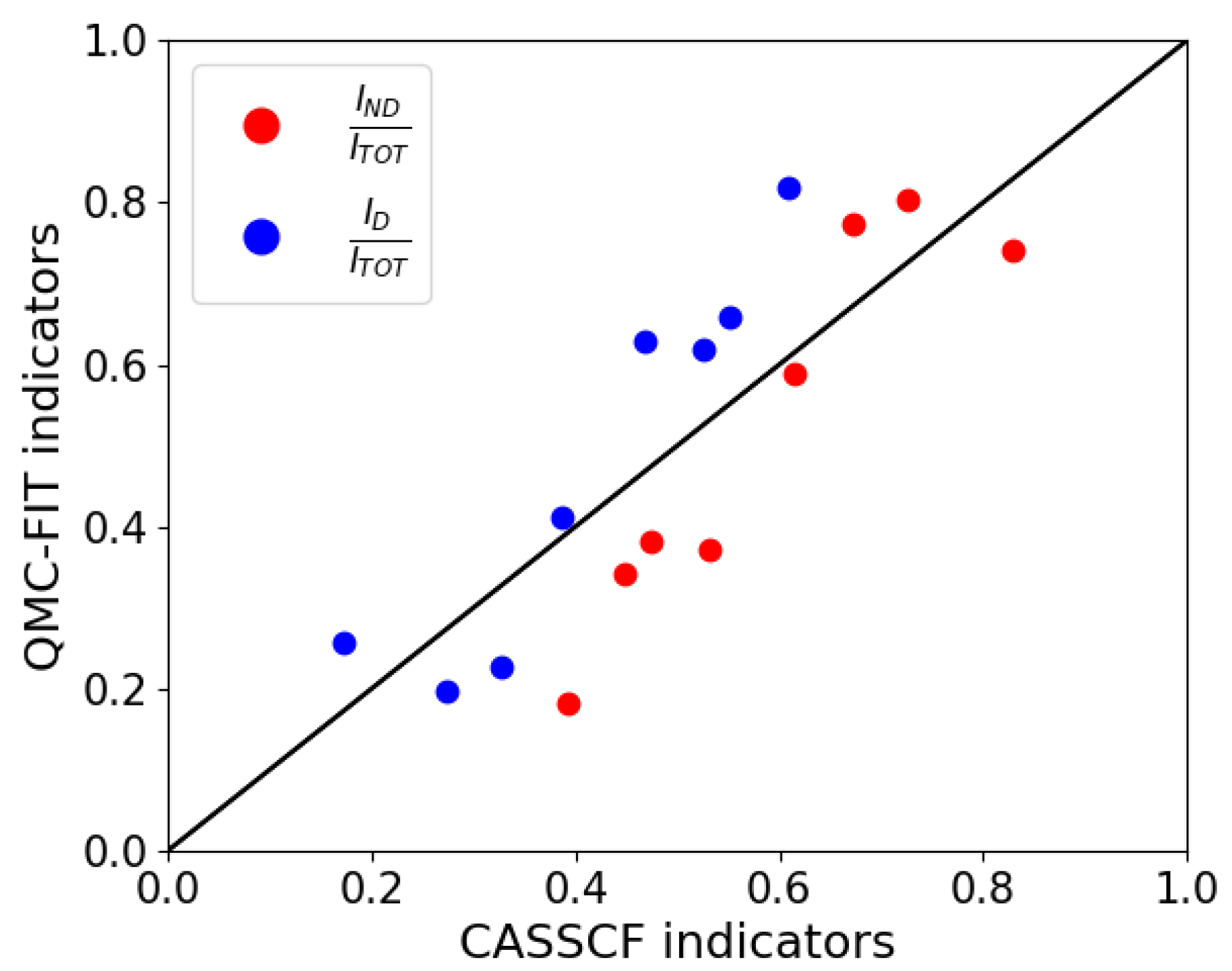{kind=link}
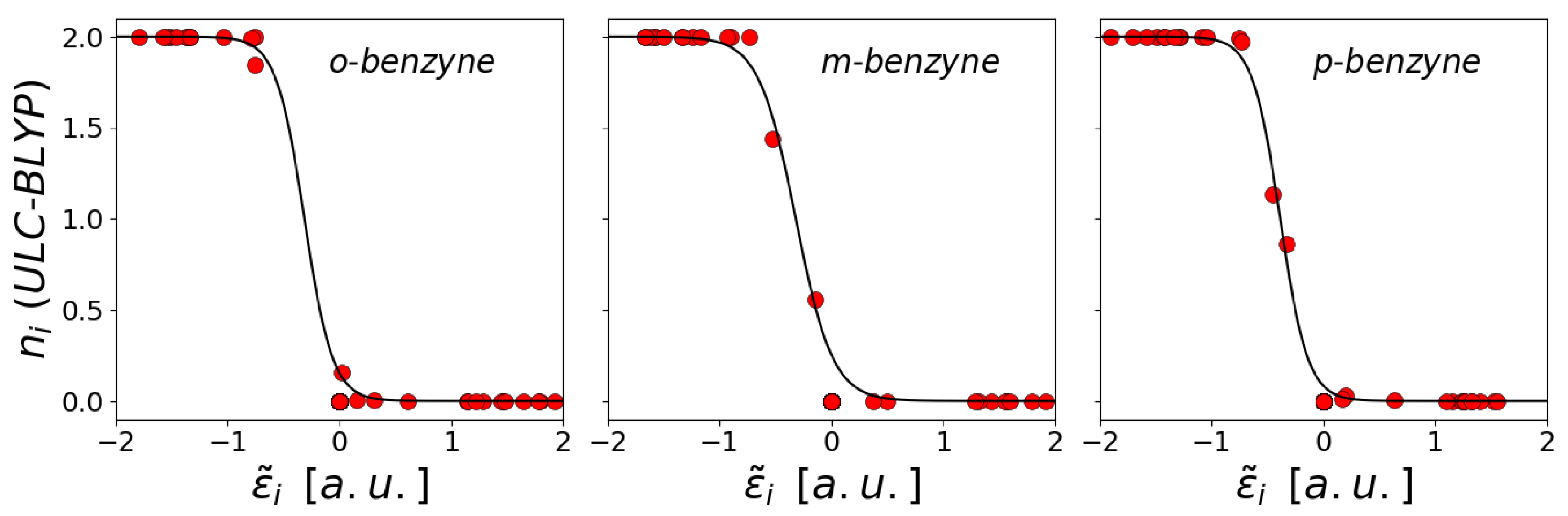{kind=link}
| Molecule | (m,n) | |||
|---|---|---|---|---|
| Be + H2 (TS) | −1.53204 | −2.00598 | (4,6) | −2.01200(5) |
| O | −47.04377 | −47.14969 | (4,4) | −47.8447(6) |
| cyclobutadiene (D2h) | −24.26855 | −24.33294 | (4,4) | −24.9648(2) |
| cyclobutadiene (D4h) | −24.21910 | −24.32268 | (4,4) | −24.9501(4) |
| o−benzyne | −35.39269 | −35.45937 | (8,8) | −36.3440(2) |
| m−benzyne | −35.36251 | −35.43193 | (8,8) | −36.3198(2) |
| p−benzyne | −35.20607 | −35.40144 | (8,8) | −36.2877(2) |
| PSB4 | −53.97069 | −54.06574 | (8,8) | −55.3412(3) |
| Molecule | Kinetic Energies [a.u.] | |||
|---|---|---|---|---|
| Be + H2 (TS) | 1.25440 | 1.3555 | 1.3928 (7) | 0.06 |
| O | 35.28944 | 35.690 | 35.764 (4) | 2.12 |
| cyclobutadiene (D2h) | 18.74213 | 18.815 | 19.203 (4) | 0.84 |
| cyclobutadiene (D4h) | 18.83450 | 19.016 | 19.397 (5) | 1.15 |
| o-benzyne | 27.17912 | 27.308 | 27.849 (3) | 2.24 |
| m-benzyne | 26.85206 | 27.050 | 27.585(3) | 2.00 |
| p-benzyne | 26.66995 | 26.963 | 27.482 (3) | 1.85 |
| PSB4 | 42.25434 | 42.462 | 42.468 (3) | 7.56 |
| (a) | ||||||
| Molecule | UHF | UB3LYP | UBH&HLYP | UCAM | ULC-BLYP | QMC-fit |
| Be + H2 (TS) | 0.54 | 0.50 | 0.50 | 0.50 | 0.51 | 0.50 |
| cyclo (D2h) | 0.48 | 0.08 | 0.13 | 0.12 | 0.17 | 0.17 |
| cyclo (D4h) | 0.62 | 0.53 | 0.54 | 0.54 | 0.55 | 0.52 |
| o-benzyne | 0.69 | 0.01 | 0.12 | 0.08 | 0.15 | 0.27 |
| m-benzyne | 0.47 | 0.30 | 0.37 | 0.36 | 0.41 | 0.38 |
| p-benzyne | 0.91 | 0.49 | 0.52 | 0.51 | 0.54 | 0.43 |
| MAE | 0.24 | 0.08 | 0.05 | 0.06 | 0.05 | |
| MAPE [%] | 38.19 | 31.08 | 17.83 | 21.01 | 14.22 | |
| (b) | ||||||
| Molecule | UHF | UB3LYP | UBH&HLYP | UCAM | ULC-BLYP | QMC-fit |
| Be + H2 (TS) | 0.83 | 0.89 | 0.87 | 0.87 | 0.86 | 0.81 |
| cyclob (D2h) | 0.53 | 0.27 | 0.33 | 0.33 | 0.38 | 0.39 |
| cyclob (D4h) | 0.74 | 0.71 | 0.69 | 0.70 | 0.69 | 0.74 |
| o-benzyne | 0.52 | 0.08 | 0.32 | 0.28 | 0.36 | 0.34 |
| m-benzyne | 0.44 | 0.62 | 0.67 | 0.68 | 0.71 | 0.37 |
| p-benzyne | 0.60 | 0.71 | 0.66 | 0.69 | 0.64 | 0.77 |
| MAE | 0.10 | 0.13 | 0.10 | 0.10 | 0.10 | |
| MAPE [%] | 22.44 | 32.46 | 21.14 | 23.54 | 21.09 | |
| ] | ||||||
|---|---|---|---|---|---|---|
| Molecule | UHF | UB3LYP | UBH&HLYP | UCAM | ULC-BLYP | QMC-Fit |
| Be + H (TS) | 3.3 (4) | 2.38 (3) | 2.51 (7) | 2.42 (6) | 2.48 (1) | 2.2 (7) |
| cyclob (D2h) | 3.59 (6) | 1.841 (6) | 2.088 (8) | 2.057 (8) | 2.30 (1) | 2.46 (2) |
| cyclob (D4h) | 3.8 (1) | 2.83 (8) | 2.99 (7) | 2.98 (8) | 3.17 (9) | 2.87 (2) |
| o-benzyne | 3.5 (2) | 1.13 (4) | 2.0 (1) | 1.86 (9) | 2.2 (1) | 2.91 (4) |
| m-benzyne | 3.1 (2) | 2.5 (1) | 2.7 (1) | 2.7 (1) | 2.9 (2) | 2.75 (7) |
| p-benzyne | 3.0 (2) | 2.1 (1) | 2.0 (1) | 2.1 (1) | 2.1 (2) | 1.96 (7) |
| MAPE [%] | 35.6 | 18.3 | 11.8 | 12.2 | 11.1 | |
| MAE * [Kelvin · 10] | 8.5 | 4.9 | 3.0 | 3.2 | 2.9 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naim, C.; Amovilli, C. Extraction of a One-Particle Reduced Density Matrix from a Quantum Monte Carlo Electronic Density: A New Tool for Studying Nondynamic Correlation. Computation 2021, 9, 135. https://doi.org/10.3390/computation9120135
Naim C, Amovilli C. Extraction of a One-Particle Reduced Density Matrix from a Quantum Monte Carlo Electronic Density: A New Tool for Studying Nondynamic Correlation. Computation. 2021; 9(12):135. https://doi.org/10.3390/computation9120135
Chicago/Turabian StyleNaim, Carmelo, and Claudio Amovilli. 2021. "Extraction of a One-Particle Reduced Density Matrix from a Quantum Monte Carlo Electronic Density: A New Tool for Studying Nondynamic Correlation" Computation 9, no. 12: 135. https://doi.org/10.3390/computation9120135
APA StyleNaim, C., & Amovilli, C. (2021). Extraction of a One-Particle Reduced Density Matrix from a Quantum Monte Carlo Electronic Density: A New Tool for Studying Nondynamic Correlation. Computation, 9(12), 135. https://doi.org/10.3390/computation9120135