1. Introduction
The gene expression analysis has been widely used to determine the mechanism of certain diseases in their early stages, and in determining gene expression profiles [
1,
2,
3]. Data Mining (DM) techniques focus on extracting knowledge from large databases. Clustering algorithms within gene expression databases have been widely exploited in unsupervised learning techniques, whose objective is to segment data to obtain gene
clusters with correlated behavior [
4]. These types of algorithms describe gene functionality, regulation, and involvement in cellular processes [
5]. The main objective of this analysis is to create clusters such that the intragroup variance is minimal and the extragroup variance is maximum. The algorithm defines the variance through a similarity metric and verifies whether a pair of objects are closely related. The clustering analysis can be achieved based on genes and experimental samples. The grouping in genes describes sets of genes with a level of expression correlated in all experimental conditions; on the other hand, grouping in experimental conditions presents a correlated expression profile in all genes [
6]. The group can be partial or complete; the partial clustering tends to be more flexible since all the elements should not necessarily be grouped, while in the complete group, all the elements must be assigned to a specific group. In the gene expression analysis, the partial grouping allows excluding genes that could represent a source of noise derived from the biological experiment. The cluster can be classified in a hard or soft clustering [
7]. The hard clustering assigns a gene to the group with the dominant degree of belonging, while the smooth pool allows the assignment of a gene to more than one group. There is no clustering algorithm that presents the best performance for any type of problem [
8,
9,
10].
There is a wide range of approaches. The standard hard-partitioned clustering algorithms, such as K-means (KM) [
11], are simple and easy to implement, but they have drawbacks when clustering biological due to high dimensional data parsing [
12]. In [
13], a joint K-means-based Co-Clustering algorithm (KCC) is proposed for sparse and high-dimensionality data. KCC uses higher-order statistics in order to the assignment of objects to each group, as well as for the estimation of centroids in the iterative process. The fuzzy C-Means Algorithm (FCM) [
14] is one of the most used diffuse grouping methods for the gene expression analysis due to it carrying out a smooth grouping in which each object has a value of belonging to each group. The Self-Organized Map (SOM) [
15] was introduced as an algorithm based on single-layer neural network methods, which generate an intuitively attractive map of a high-dimensional dataset in a two-dimensional space. Although SOM performs better than KM and FCM when analyzing noisy data sets, merging different objects in a cluster can make the algorithm ineffective at finding clustering limits and balanced solutions [
16]. However, it is difficult to come up with appropriate input nodes and may result in a non-convergence problem of the algorithm. Furthermore, SOM is sensitive to choice of number of nodes such as KM [
17].
Algorithms based on Hierarchical Clustering (HC) [
18] group genes are naturally co-expressed through a graphical representation. However, a small disturbance in the data or a poor decision performed in the initial steps may greatly modify the final results. The authors of [
15,
16,
19] stated that HC lacks robustness because in each iteration the groups are mixed locally by the distance between peers as an alternative of a global criterion. Another important limitation of HC is its high computational complexity [
20]. The Self-Organized Tree Algorithm (SOTA) [
21,
22] was proposed as an alternative to obtaining robustness in the analysis of data prone to various sources of noise. SOTA combines the advantages of both SOM and HC [
16]. It allows a visual representation of the clusters and their structure and is also more flexible than the other clustering methods. Agglomerative Nesting (AGNES) has been proposed in [
23]. AGNES is a bottom-up approach. Compute all pair-wise pattern likeness coefficients and merge two interconnected clusters into one. The AGNES rigidity is vital for its success since it allows to obtain a solution in a relatively short period of time [
24]. On the other hand, the Divisive Analysis algorithm (DIANA) [
25] performs a hierarchical division due to starting with the entire population and ending once all the groups are separated following a top-down approach. This method has the same deficiency as AGNES; that is, it will never be able to repair what was performed in a previous step [
26]. However, top-down clustering is more complex and efficient; also, it is more accurate, because a flat clustering is needed as a subroutine and for a fixed number of top levels. Grouping algorithms based on density approaches have been proposed in [
27,
28,
29], whose objective is to identify high-density regions that are surrounded by low-density areas. Each of the density-identified regions is called a Cluster.
Robust algorithms based on factoring methods have been proposed [
30] to reduce the data dimensionality and to apply conventional clustering algorithms. The most common algorithms to reduce dimensionality are the Roust Principal Components Analysis (PCA) [
31], Singular Values Decomposition (SVD) [
32], Independent Components Analysis (ICA) [
33], and Non-negative Matrix Factorization (NMF) [
34]. Even though PCA captures the variance of the dataset from a small group of genes, its application is limited to a linearity assumption [
35,
36]. ICA identifies additional substructures by projection vectors that represent independent features; however, its application implies complex cancellations between positive and negative values, entailing that gene inhibition is hard to interpret [
37]. Moreover, the factors and coefficients must be restricted to a non-negative environment because a gene can be either present with a variable intensity or absent. NMF is useful since it allows to describe biological data as the product of two positive matrices with a semantic consistent with the biological context [
38,
39,
40]. Despite the advantages offered by NMF, most of the implementations [
41,
42,
43,
44] have become obsolete due to the constant data increase defined by the curse of dimensionality [
45].
The authors of [
46,
47,
48,
49,
50] demonstrated that Clustering algorithms based on NMF, as well as software tools developed for this purpose, are efficient. However, these implementations have a high computational complexity and they are computationally demanding on processing units (CPUs), so their applicability is limited in many circumstances [
51,
52]. In this way, current research should focus on improving computational performance by creating efficient algorithms from the already effective ones. To overcome this problem, High-Performance Computing (HPC) mechanisms based on general-purpose Graphics Processing Units (GPUs) are starting to be used within this area [
53,
54,
55], as they can considerably reduce the execution time required in a CPU and allow more intensive investigations of biological systems.
The objective of this work is to propose a parallel clustering algorithm in a GPU for a gene expression analysis, such that it reduces the necessary computation time to obtain a result, without losing biological importance. The algorithm approach should focus on matrix factoring methods. In this way, the algorithms used in the proposal are NMF and KM. NMF is expected to reduce the dimensionality of biological data and then be able to apply KM, taking advantage of its high performance in low-dimensional data. The hypothesis of this work is based on the idea that the parallelization of NMF through High Performance Computing (HPC) mechanisms will help to reduce the required calculation time and expand its use in the high dimensional data to obtain a computational result according to the biological context of the study. To confirm or refute our hypothesis, we define NMF implementation as the most intensive computational task. The parallelization of this task on a GPU is proposed. We add the Consensus Clustering algorithm (CC) [
56] to evaluate the stability of NMF and refine the selection of centroids in the implementation of KM. Our proposal is compared with the proposal described in [
57]. The results show that our parallel implementation achieves a speed-up of 6.22× versus our sequential version. Moreover, our proposal is invariant to the classes number and the size of the gene expression matrix.
The document is organized as follows:
Section 2 describes the problem statement.
Section 3 contains the proposed clustering algorithm. The sequential NMF analysis is given in
Section 4 and the obtained maximum theoretical acceleration is exposed. In
Section 5, the parallelization is explained.
Section 7 contains the experimental evaluation of the implementation compared to other proposals in the state of art.
Section 8 includes a discussion of the work, some implications and some limitations. Finally, in
Section 9, we describe the main implications of the results from a biological point of view.
2. Problem Statement
Let be a pattern or vector of features defined by a single observation in m-dimensional space (in the gene expression analysis, each gene can be seen as a pattern). Thus, a feature or attribute is the individual component of a pattern. A cluster is defined as a set of patterns whose features are described as similar based on a similarity metric.
The problem of clustering is formally defined below.
Let be a set of patterns, where , , is a pattern in the m-th feature dimension space and n is the number of patterns in E; therefore, the clustering problem is defined as the partitioning of E into k clusters , such that the following conditions are satisfied:
Each pattern must be assigned to a cluster; that is, .
Each cluster has at least one pattern assigned; that is, .
Each pattern is assigned to one and only one cluster, such that . This property is called hard clustering.
The clustering problem in gene expression data, in most cases, involves the analysis of high dimensional data. Therefore, the local feature relevance problem [
58] and the curse of dimensionality must be considered during its resolution [
59].
3. Materials and Methods
In gene expression analysis, matrix factorization (MF) focuses on reducing the dimensionality of the biological data in order to figure out pathways of genetic regulation in a specific genome [
60,
61] through linear combinations.
Factorization Problem. Given matrix , express it as with and .
This problem may be modified in the following form:
Approximated Factorization Problem. Given matrix , construct with and , such that .
Naturally, the approximation “≈” should be estimated with respect to a given metric in the space .
If , then a gain in storage can be obtained, but most importantly, the dimension of the columns in V; namely, n, is reduced to the dimension k of the factor matrices columns. In this sense, a dimensionality reduction is obtained.
With such factorization, each entry of V can be expressed as a linear combination .
3.1. NMF for the Analysis of Gene Expression Data
Let be a genetic expression matrix, namely, its rows correspond to genes and its columns to conditions with respective cardinalities n and m. Each entry indicates the level of expression of the i-th gene under the j-th experimental condition. The i-th row is the expression profile of the i-th gene, and the j-th column is the j-th experiment profile.
The
Non-negative Matrix Factorization (NMF) Problem is the above stated
Approximated Factorization Problem with the requirement that
W and
H have non-negative entries, i. e.,
and
, and, moreover:
Condition (
1) aims at data reduction [
62].
If the matrix Euclidean norm
is considered as the metric to estimate approximations, then the NMF Problem is posed equivalently as:
Euclidean approximation. Given matrix , minimize with and , such that and .
The solution
can be iteratively updated through the rule of Lee and Seung [
63]:
There is no unique solution for the NMF problem, but some computationally efficient algorithms for minimizing the difference between
V and
for different error functions have been proposed [
64].
It might as well be considered [
65], that the
squared Frobenius norm:Frobenius approximation. Given matrix , minimize with and , such that and .
The solution
can be iteratively updated through the rules [
66,
67]:
The interpretation of NMF within biological context is the clustering of genes or conditions into
k clusters whose expression level is similar [
68]. The
i-th column of the factor marix
W describes the degree of membership of the
i-th gene within the
k-th group. The
j-th row of
H indicates the degree of influence that the
k-th group of genes has on the
m-th condition. The subset of rows of
H with high values in the same column of
W represents a
centroid. The set of genes and conditions with relatively high values in the same column of
W and the corresponding rows of
H is called
bicluster [
69]. Finally, whether some entries in a row of
H have coefficients greater than zero, the conditions are influenced by local characters in the data. A detailed exposition of NMF appears in [
70].
3.2. Hybrid Clustering Algorithm
CC is an algorithm that emerged as an important contribution of classical clustering and refers to the situation in which
T different partitions were obtained
for a dataset
E, where
consists of a clusters set
that complies with the restrictions imposed in
Section 2; we wanted to find a consensus clustering
, such that:
We defined the distance between two partitions
,
as
, such that:
where
means that
i and
j belong to the same cluster in partition
and
represents that
i and
j belong to different clusters in partition
.
A consensus matrix
M can be constructed, such that each input is defined as:
Then, we could easily say that
, since
. Consequently, the Equation (
9) can be rewritten as:
Let
denote the solution to the optimization problem of CC (see Equation (
9)).
U is the consensus matrix. Let the consensus (average) association between
i and
j be
. Define the average squared difference from the consensus association
. Clearly, the smaller
, the closer to each other the partitions are. This quantity was a constant. We had:
Therefore, consensus clustering took the form of the following optimization problem:
where the matrix norm is the Frobenius norm. Therefore, consensus clustering was equivalent to clustering the consensus association. Then, it was easy to show that:
To solve Equation (
13), NMF was used (see
Section 3.1). The result would be the high dimensional biological dataset to a low dimensional dataset (
W) and the candidate set of centroids (
H). CC was applied directly to the
H matrix in order to refine the centroids in a given
t partition (
).
In order to solve the clustering problem in the low dimensional dataset, we implemented KM, taking into account the set of elements in
E mapped to a
k-dimensional space (rows in
W) and the set of
k centroids described by the columns in
H. KM groups elements through a set of numerical properties in such a way that the elements within a cluster are more similar than the elements in different clusters. This is known as the default Sum of Squared Error (SSE) or total cohesion if a cosine-based metric of similarity is used. Therefore, it was necessary to provide a criterion to measure the similarity of the elements. Generally, KM uses Euclidean distance [
71] as a similarity metric. However, it is not effective for analyzing high-dimensional datasets because, as the dimensionality of the features increases, the elements tend to move evenly away from each other; that is, the relationship between the closest and furthest elements tends to be 1 in all cases.
In [
72], it is shown that the Manhattan distance has better performance for high-dimensional datasets. The use of fractional norms has been proposed as similarity metrics in high-dimensional data, which exhibit the property of increasing the contrast between the most distant and closest elements. This can be useful in some contexts, but it should be considered that they are not distance metrics because they violate the triangle inequality. In the end, the type of metric to use should depend on the features of the data and the type of analysis that is being carried out [
73,
74,
75].
Finally,
Silhouette coefficient [
76] was used in order to quantitatively evaluate the clustering result. A maximum value indicated the stability of the results through the evaluation of intragroup and extragroup variance. The optimal value of
k corresponded to the maximum silhouette value that reflected a better partition. It is worth mentioning that the estimation of the parameter k was considered an NP-complete problem [
77].
Given a value of
k and based on CC, we proceeded with Algorithm 1. The core of this procedure was the execution of NMF until a convergence criterion was fulfilled. We obtained two matrices
W and
H. The
-th entry of the
i-th row in the
W array represented the degree of membership of the
i-th gen to the
-th cluster. On the other hand, the
-th column of
H represented the choice of the centroid. A set of
k clusters was obtained to be analyzed (line 5 of Algorithm 1) to generate a consensus matrix
. The probability
corresponded to the event in which two conditions fell in the same cluster in two partitions
such that
(Algorithm 2). Then, to obtain the class label of each condition (
), KM was run on the matrix
W, which contained the biological data mapped to a low dimensional space (
features in the worst case) and taking account the matrix
C that represented the centroids obtained by NMF. Finally, the Silhouette coefficient was calculated for each cluster to determine the resulting quality. The result was given as a vector
. The implementation of the algorithms
KM and
silhouette followed [
76].
| Algorithm 1 Proposal of hybrid clustering algorithm based on NMF, KM, and CC. |
- Require:
: gene expression matrix; k: number of clusters; T: maximum number of partitions for NMF execution as part of CC; : convergence threshold. - Ensure:
: class label vector of each gene; : Silhoutte coefficients of clusters. - 1:
procedurehybClust() - 2:
▹ Initialization of consensus matrix. - 3:
for do - 4:
NMF() ▹ See Algorithm 4. - 5:
consensusMatrix() ▹ See Algorithm 2. - 6:
end for - 7:
KM() ▹ Class label vector. - 8:
silhouette() ▹ Silhouette coefficients vector. - 9:
return ▹ Return the class label and silhouette coefficients vectors. - 10:
end procedure
|
| Algorithm 2 Consensus matrix update for the i-th iteration. |
- Require:
: right factor in NMF; k: number of clusters; T: maximum number of partitions for NMF executions as part of CC. - Ensure:
: updated consensus matrix. - 1:
procedureconsensusMatrix() - 2:
- 3:
▹ Initialization of consensus matrix. - 4:
for do - 5:
for do - 6:
▹ Compute the probability. - 7:
for do - 8:
if or then ▹ Verifies whether all entries are greater or equal to 0. - 9:
▹ No belongs to the same group. - 10:
break - 11:
end if - 12:
end for - 13:
end for - 14:
end for - 15:
return C ▹ Return the consensus matrix. - 16:
end procedure
|
Algorithm 1 was executed for different values of k as described in Algorithm 3 in order to obtain their optimal value. For each of the many executions, NMF carried out a large number of costly matrix operations. Therefore, line 4 in Algorithm 1 was the critical step.
| Algorithm 3 Gene expression analysis. |
- Require:
: gene expression matrix; : lower bound to test k; : upper limit to test k; T: maximum number of iterations for NMF executions as part of CC; : convergence threshold. - Ensure:
Return 0 if an exception occurs, return 1 otherwise. - 1:
proceduregeneExpressionAnalysis() - 2:
if or then - 3:
return 0 ▹ Fails if K is too large (Equation ( 1)). - 4:
end if - 5:
hybClust() - 6:
- 7:
for do - 8:
hybClust() ▹ See Algorithm 1. - 9:
if S is bether than then ▹ Update values according to the best value of the S. - 10:
- 11:
- 12:
- 13:
end if - 14:
end for - 15:
return 1 - 16:
end procedure
|
Algorithm 4 is, properly, an NMF algorithm. Given three parameters,
, representing a gene expression dataset,
k such that
represented the current number of clusters, and the convergence threshold
produced two matrices,
and
, containing non-negative values such that
. The random initialization of
W and
H (line 2 of Algorithm 4) indicates the start of the algorithm; next, the algorithm calculates the inner products
in line 3 of Algorithm 4. In NMF, the factor dimension (
k) determines the quality of results as long as it determines the centroids of each cluster. The optimum value depends on the biological data type. If
k tends to be too small, groups will be pretty generic to provide information, whilst, if
k tends to be too large, although bounded by (
1), the result will be pretty detailed and hard to interpret. CC is an algorithm to estimate an optimum value for
k by consensus of the results obtained from the multiple executions of a clustering algorithm [
56].
Finally, it enters an iterative process (lines 6–23 of Algorithm 4).
W and
H were updated based on (
4) and (
5). The number of iterations performed by Algorithm 4 would vary depending on the input data, on the initial values, as well as on the used convergence method. The iterative process stopped when the convergence criterion was met.
| Algorithm 4 Non-negative matrix factorization (NMF) |
- Require:
such that all rows and columns of V contain at least a value greater than zero, and : a convergence threshold. - Ensure:
and such that . - 1:
procedureNMF() - 2:
initialize() ▹ Random values non-negatives. - 3:
- 4:
- 5:
isConvergence() ▹ See Algorithm 5. - 6:
while do - 7:
- 8:
for do ▹ Update W. - 9:
for do - 10:
- 11:
- 12:
end for - 13:
end for - 14:
▹ Update X. - 15:
for do ▹ Update H. - 16:
for do - 17:
- 18:
- 19:
end for - 20:
end for - 21:
▹ Update X. - 22:
isConvergence() - 23:
end while - 24:
return - 25:
end procedure
|
| Algorithm 5 Compute NMF convergence |
- Require:
. All rows and columns of V contain at least a value greater than zero and . - Ensure:
. - 1:
procedureisConvergence() - 2:
- 3:
for do - 4:
for do - 5:
- 6:
end for - 7:
end for - 8:
return - 9:
end procedure
|
4. Sequential Analysis of NMF Algorithm
The performance analysis of the sequential algorithm was helpful to confirm information about the segment of code that could potentially be parallelized. We implemented the sequential NMF in C language according to Algorithm 4. The experiments were run on an Intel Xeon E5-1603 v3 processor with four cores and 32 Gb of RAM. An ext4 filesystem and a SATA HDD were used. These features could change the speed-up of the read and write operations. The operating system was Ubuntu 18.04 and the compiler was GCC 9.2. Two dataset groups in silico (generated through a computational simulation.) were generated. Group A was used to analyze the computational time when the matrix size increased, whilst Group B was used to determine the most expensive operation in terms of computational time. The experiments allowed to detect the code section potentially parallelizable.
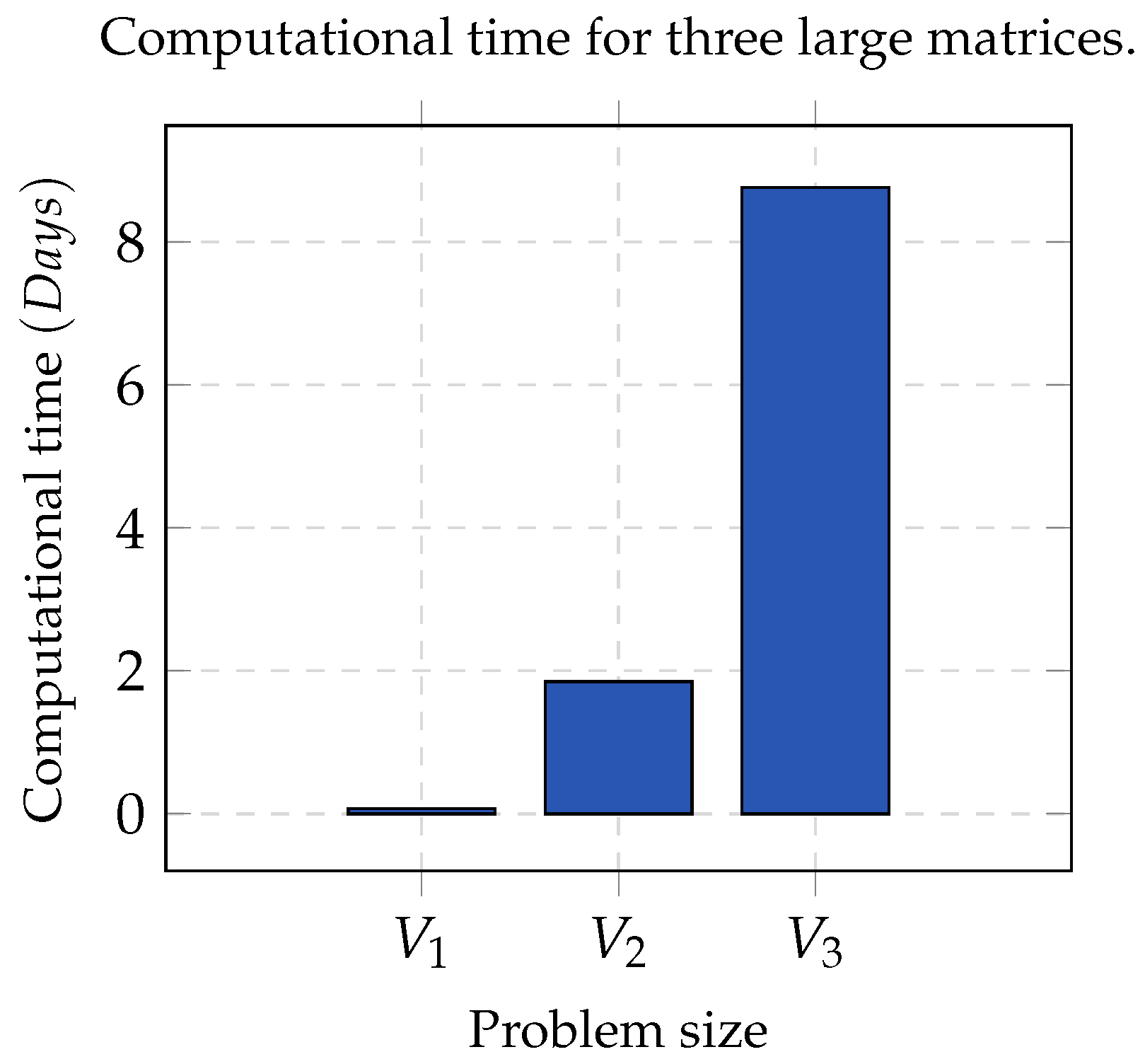
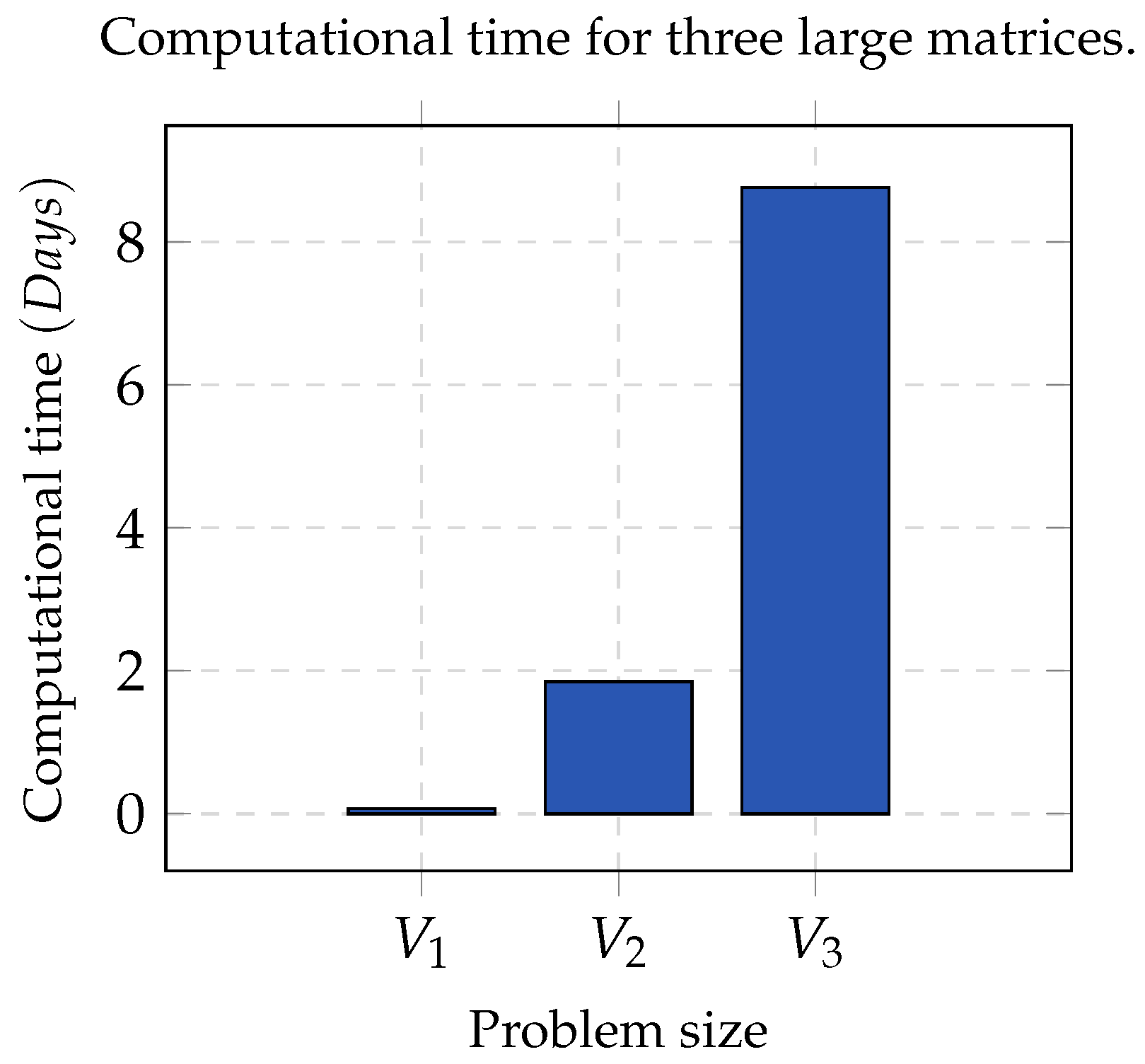
Group A. It consists of three matrices
,
, and
, whose contents were partitioned into five, seven and nine clusters, respectively. In all cases, the error threshold was
and
. In order to make a statistically significant analysis, the whole process was run 32 times per each chosen value of
K, as well as for each matrix. This dataset was optimal since the clusters contained contiguous elements and were pairwise disjointed.
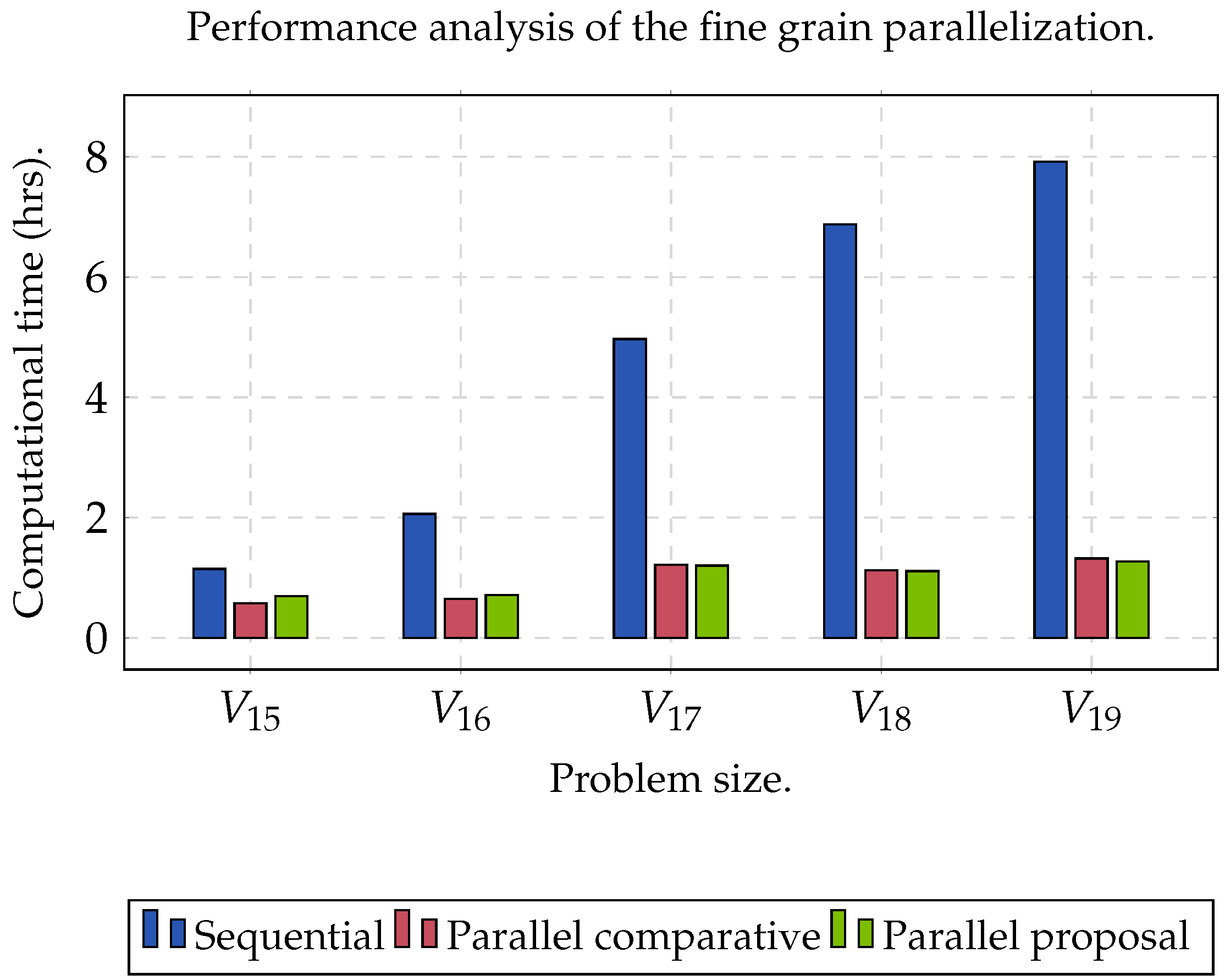
Figure 1 shows the computational time used by NMF. As the size of the matrix increased, the computation time tended to be infeasible; hence, sequential NMF was unfeasible in biological data processing due to a high dimensionality.
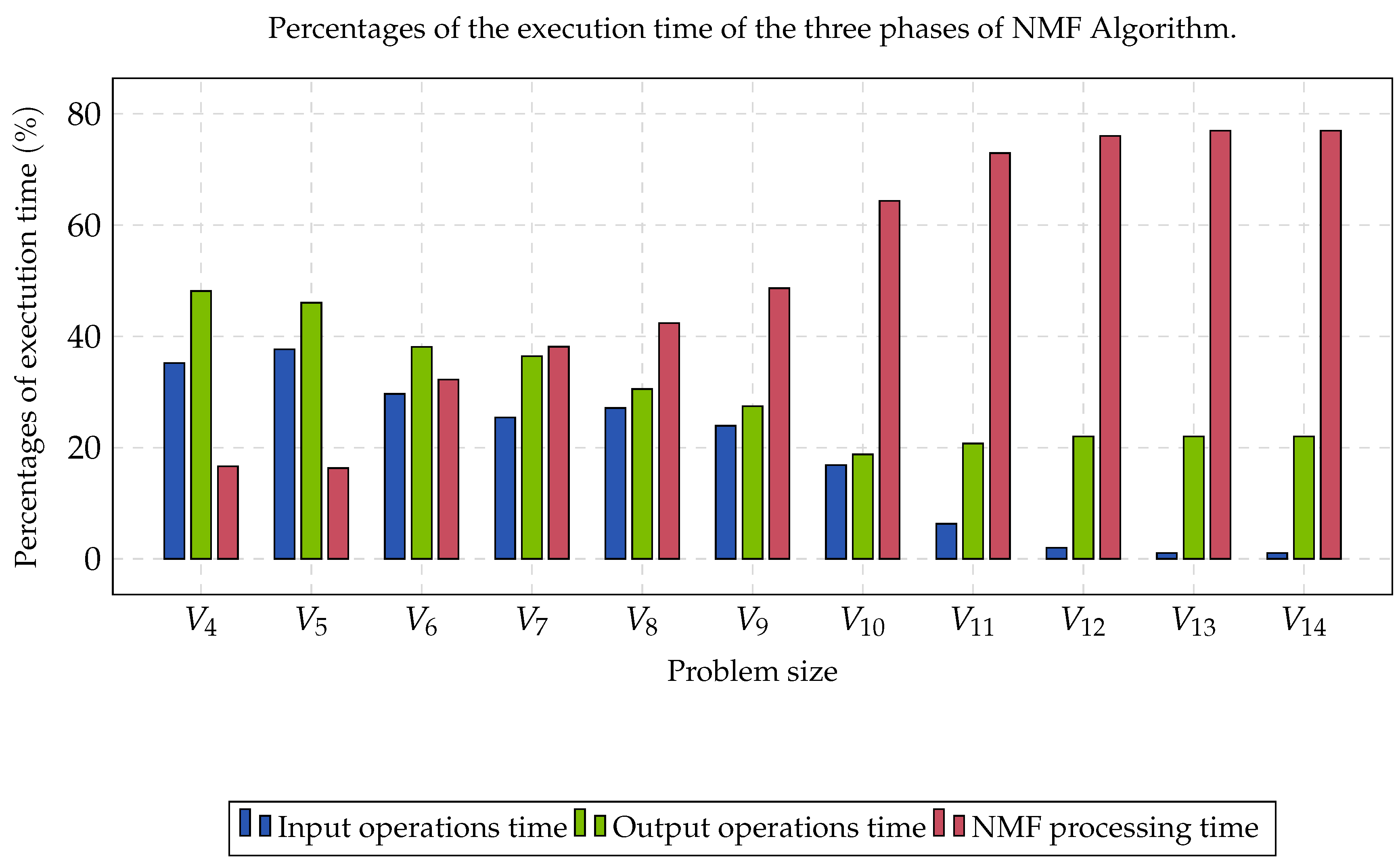
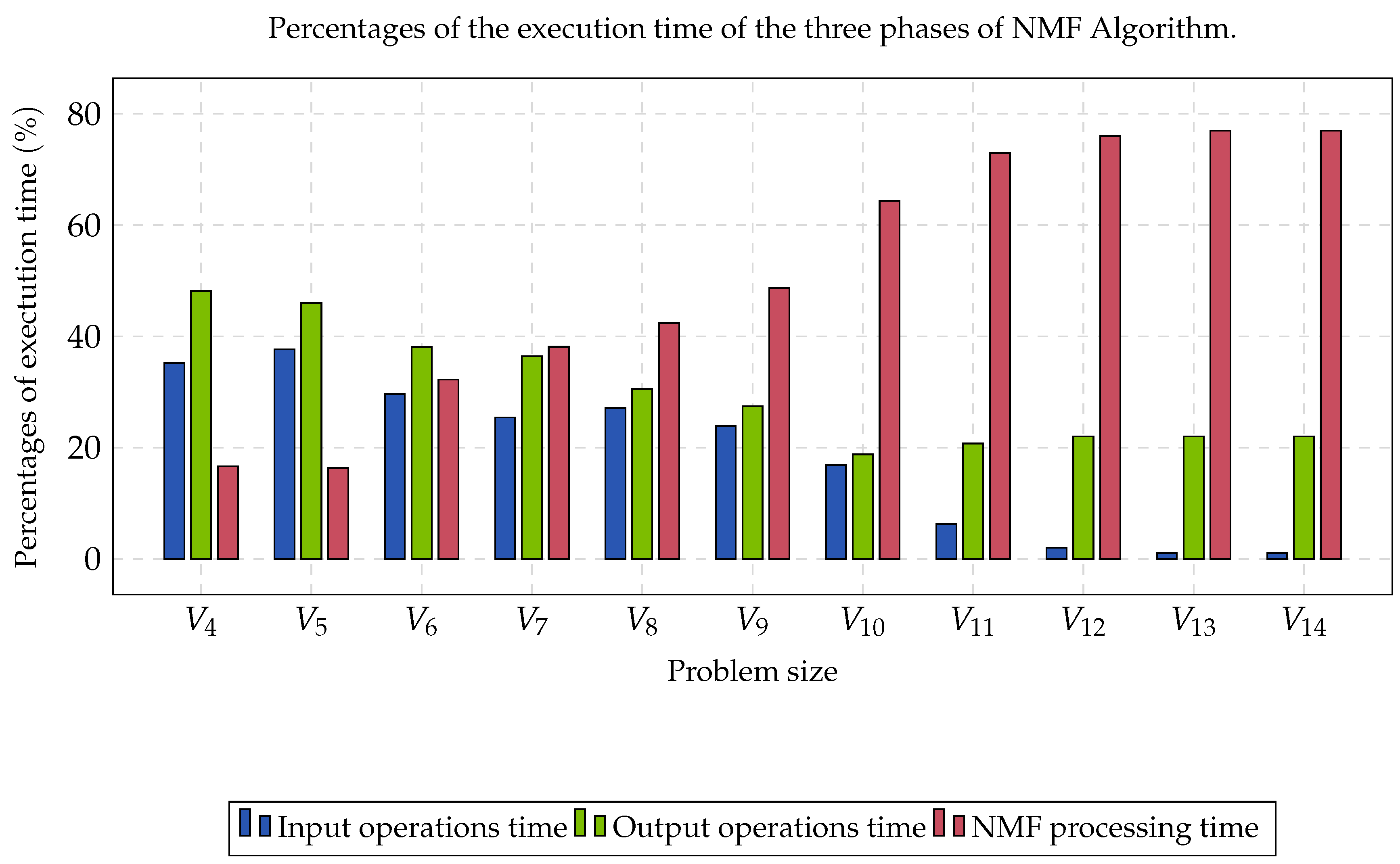
Group B was compound by eleven matrices whose contents were partitioned into three clusters. Each matrix was perturbated by a noise with a Gaussian distribution, and rows and columns were randomly switched, aiming to obtain the worst case instances. The computational time required to read data, process NMF, and write solutions, is shown in
Figure 2. In the first three cases, the percentage of reading and writing was more significant than the processing time. As the size of the matrix increased, the portion of the processing time began to converge at approximately 77%. This percentage represented the limit of the algorithm’s processing time when parallelized.
5. Strategies for Parallelizing NMF Algorithm
We discussed in detail the parallelization of NMF (see Algorithm 6) in a GPU through the CUDA library in line with NMF+HC [
57,
78,
79]. However, as we mentioned in
Section 1, a small disturbance in the data or a poor decision determined in the initial steps of the algorithm can profoundly modify the final results. The solution strategy is to implement parallelism at the data level [
80], namely, to divide the data set into
b blocks of size
r, such that each processing unit on the GPU solves the problem by applying the same sequence of operations to a specific subset of data. Ideally, this simultaneous execution of operations resulted in a global acceleration of the computation.
The proposed algorithm started with
initializeGPU, the initialization of CUDA and CUBLAS [
81]. The amount of global memory was returned or the execution was finished if a failure occurred (line two of Algorithm 6). Next,
setupGPU checked the matrix dimensions and estimated the parameter
r and the convenient size of
W and
H. Memory allocation for
W and
H was performed, and the matrices were initialized with non-negative random values (line five of Algorithm 6). Then,
V,
H, and
W were sent from the host to the device. Finally, iteration was performed by updating
W and
H (lines 9–13 of the Algorithm 6). Given the nature of these functions, they were executed directly in the device. When the algorithm met the convergence criterion, the whole procedure stopped.
Algorithm 7 describes the algorithm to update
H according to relation (
5). Products of submatrices were involved (line three of the Algorithm 7) and the function
CUBLAS_R_GEMM predefined in the CUBLAS library was employed. For line four of Algorithm 7, we implemented a
kernel:
elementWiseDiv. The
kernel reduce (line seven of the Algorithm 7) computed the reduction in column vectors. The last step of the algorithm was conducted by the
kernel elementWiseMultDiv.
The algorithm operated in the same way to update W. To optimize the memory consumption, we directly modified H and W to avoid allocating too many memory regions.
| Algorithm 6 Parallel NMF |
- Require:
; . . k: factorization rank; T is the maximum number of iterations that NMF will be executed as part of CC; as a convergence threshold. - Ensure:
and contains non-negative values such that and . - 1:
procedurenmfGPU() - 2:
=initializeGPU() - 3:
setupGPU() - 4:
allocateMemoryHost() - 5:
initialize() ▹ Random values non-negative. - 6:
copyToDevice() - 7:
- 8:
- 9:
while do ▹ Verify the convergence criterion. - 10:
- 11:
updateW() ▹ See Algorithm 6. - 12:
updateH() - 13:
isConvergenceGPU() - 14:
end while - 15:
return - 16:
end procedure
|
| Algorithm 7 Parallel NMF |
- Require:
as submatrix of V; as submatrix of H; contains non-negative values. - Ensure:
contains non-negative update values. - 1:
procedureupdateH() - 2:
for do - 3:
CUBLAS_R_GEMM() ▹ Matrix product to obtain . - 4:
elementWiseDiv() - 5:
CUBLAS_R_GEMM() ▹ Matrix product. - 6:
reduce() ▹ Reduce columns of . - 7:
elementWiseMultDiv() - 8:
end for - 9:
end procedure
|
6. Experiment
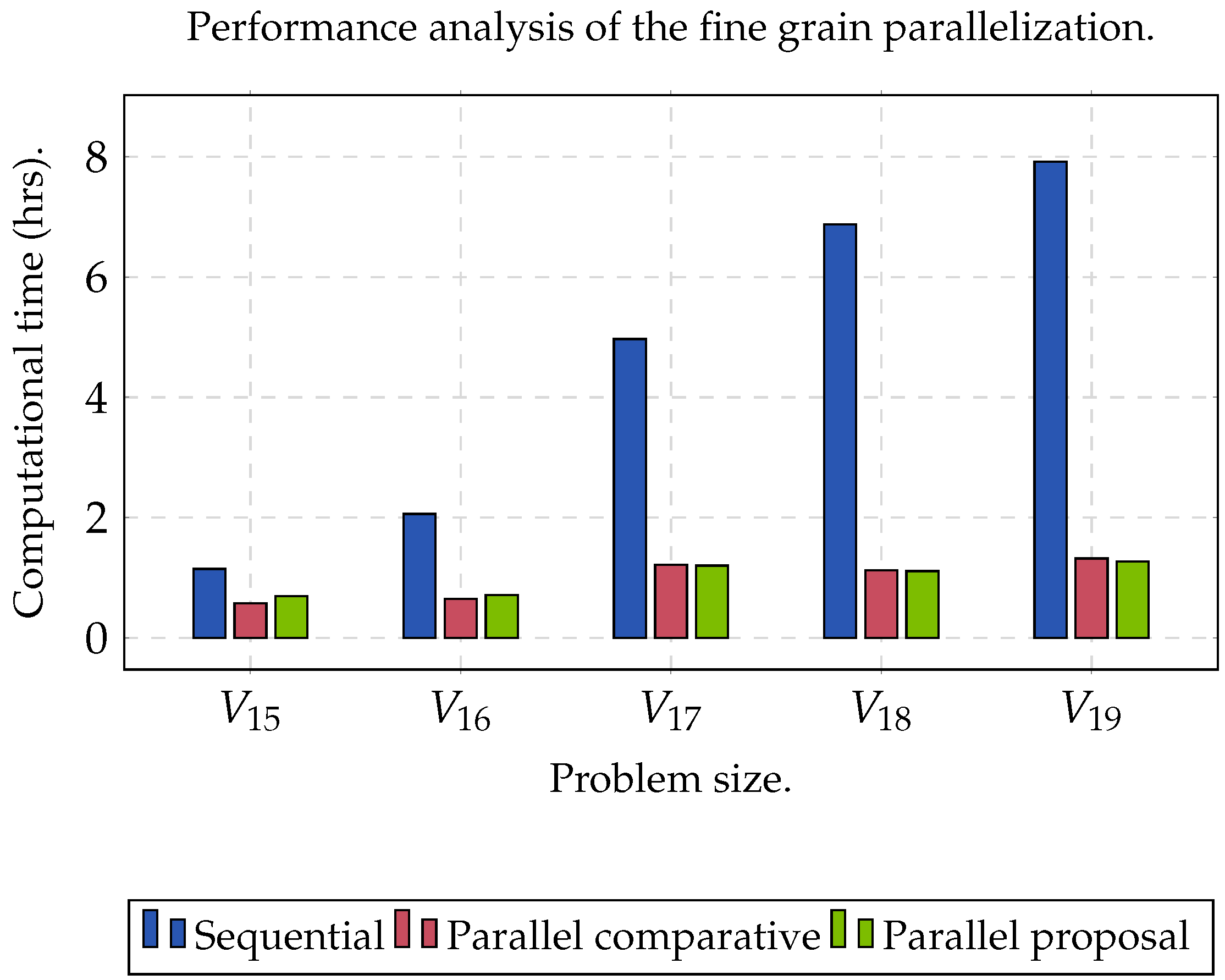
Two datasets were used to evaluate the performance of the proposed algorithm.
Group C was used to evaluate the performance of the algorithm when the size of the matrix increased (
,
,
,
,
), as well as obtaining the maximum acceleration when the algorithm was executed sequentially (CPU) and in parallel (GPU).
Group D was used to evaluate the efficiency of the algorithm with three datasets described in [
82,
83].
Table 1 summarizes the information for each data set in
Group D. The dataset was sufficiently representative since both the number of classes as well as the number of entries varied significantly [
84].
The experiment was planned in two stages. The first one focused on finding the maximum speed-up that we could obtain; the second one was used to measure the precision of the clustering analysis. In order to obtain the maximum speed-up () that we could achieve when executing the proposed algorithm on a GPU, a comparison was determined between the sequential time () against parallel time (), such that . On the other hand, we evaluated the clusters set (C) obtained through the Rand Index (RI) and Accuracy (Acc). For both experiments, and was set. In addition, we established the range of k between . Specifically, for KM, we used the Manhattan distance metric. In order to obtain statistically significant results, we ran the algorithm 32 times for each dataset. In this sense, the reported result represented the arithmetic mean of the 32 results obtained. Finally, we used a processor Xeon E5-1603 v3 with four cores, with Ubuntu v.18.04 as the operating system and CUDA v.10.2. Consider that an ext4 filesystem and a SATA HDD were used. Moreover, we used a NVIDIA Quadro RTX 4000 GPU with 2304 cores and 8 GB of DDR6 RAM (Turing microarchitecture).
8. Discussion
A wide range of clustering algorithms was proposed which showed to have a good performance within the biological area. However, these implementations had a high computational complexity and were computationally demanding on the CPU, so their applicability is limited in many circumstances. A parallel clustering algorithm was proposed based on matrix factorization. The results obtained showed that it was possible to obtain a speed-up of at least 6.22× during the execution of the proposed algorithm in a GPU; thus, achieving to reduce the execution time. This analysis supported the theory that it is possible to increase the efficiency of current algorithms, which are already effective, through high-performance computing techniques. Likewise, it can be affirmed that any improvement conducted in NMF would have a very important impact on the overall performance of any proposal that uses this type of matrice factorization.
The reported speed-up was obtained through the comparison of the performance rate between the sequential version and the parallel version. In
Figure 2, it was observed that the processing time of NMF increased considerably as the size of the gene expression matrix increased. The above caused the processing time to begin to be a preponderant factor in the execution of this type of algorithms (≈77%). The NMF selection, as the most intensive computational task, was suitable due to us being able to explore the fine-grain parallelism. This type of parallelism corresponds to matricial products and other algebraic operations. Finally, the parallelization in a GPU was justified with the existence of highly parallel codes and the almost absence of synchronization operations that may derive from blockages and barriers, which are very expensive in the GPU (thousands of threads competing for a padlock or waiting for a barrier)
On the other hand, as we could see in
Figure 2, the computational time for the input and output operations required at least 33% of the total computational time for this experiment. We can conclude that the type of HDD and the type of filesystem that was being used influenced the read and write operations. In this sense, if a file system based on Lustre [
87] was used as a parallel file system and, in turn, SAS HDD was used, instead of SATA HDD, this computation time would have reduced considerably due to us having improved the performance of input and output operations. The most important part, then, is to focus on the data processing performance.
Regarding clustering algorithms based on factoring methods, PCA, SVD, ICA, and NMF were exposed for a gene expression analysis. For the purpose of this study, NMF was selected because it is capable of decomposing the matrix into two sub-matrices, which represent a compressed version of the set of original data without losing the original features. In this way, NMF allows reducing the dimensionality of the data while preserving the biological context, and KM presents good performance when applied to low dimensional datasets. As shown in
Table 2, the clustering NMF+KM performed quite well on the three datasets described in
Table 1. The dimensionality reduction improved the clustering performance NMF+HC in all cases. However, it is appreciated that, as the number of classes increased, the results began to be unsatisfactory. This implied that irrelevant or misleading genes may not be taken into account during the dimensionality reduction phase.
Another important aspect to mention is the similarity metrics used by KM. The selection of the best metric will depend on the features of the data. Furthermore, based on the proposal described in [
56], we managed to establish a methodology that, in addition to providing an estimate of the parameter
k, was capable of inducing the initialization of the centroids through NMF factorization. Finally, it should be considered that there are a variety of ways of measuring multivariable differences or distances between elements, which provide various possibilities for analysis. The use of them, as well as the features of the clustering algorithms, or different mathematical rules to assign the elements to different clusters, depends on the case study and the prior knowledge of biological data. In addition, and because the use of the cluster analysis already implies a lack of knowledge or incomplete knowledge of the data grouping, the researcher must be aware of the need to use several methods, none of them unquestionable, to contrast the results.
8.1. Implications
Results confirmed that we could combine simple algorithms in order to propose a solution to the gene expression analysis. If we wanted to obtain more reliable conclusions about this, we would have to investigate more. We could think that clustering algorithms are limited to solve the gene expression analysis, without taking into account limitations imposed by the computational architectures and computational performance. Then, we could conclude that scientist’s expectations are obtaining results. Scientists have proposed clustering algorithms, but they did so to improve computational performance. Nowadays, computational time and memory consumption optimization could become the decisive factor in choosing the clustering algorithm. The aforementioned showed that when proposing a clustering algorithm, more attention should be given to improving the computational yield of the algorithm, as well as allowing the user with little or null knowledge in computing, to perform a genetic expression analysis in less time, and focus their attention on more practical aspects.
Although previous research has focused on clustering analysis through sequential algorithms [
45,
49,
82,
83], the results described in
Table 2 showed that the cluster’s quality was competitive compared to the results described in the state of art [
83,
88,
89]. Therefore, we can confirm the main idea in [
53], which mentioned that it is possible to improve computational performance to reduce the execution time of the clustering algorithms. It was evident that much has been conducted to propose clustering algorithms capable of obtaining results according to the biological context, but little to improve their computational performance. In our opinion, the current vision of Computer Science should consider the improvement of the computational performance of the algorithms described in the state of the art through high-performance computing. Furthermore, the research carried out may allow us to increase the objectivity for the extraction of genes, which can be used for analyzing the gene set involved in a biological process. We will use the results obtained to make a Genetic Ontology analysis(GO). This is the perspective of our research.
8.2. Limitations
The experience of the scientist in the gene expression analysis limits the interpretation and quality of results. To obtain better results, scientists ought to have experience and knowledge about the dataset features as well as of the biological context in order to choose the clustering algorithm which suits perfectly to solve the problem. Nowadays, a significant challenge is having a clustering algorithm general enough in order to analyze any biological datasets, invariant to the initial parameters, and with an optimal computational performance representing a significant challenge. We created the dataset in silico under controlled conditions, so we came to know better. Similarly, the limitations imposed by the high dimensionality of biological data made it difficult to obtain more specialized knowledge. In this work, we mitigated this situation through the parallelization of the algorithm. The development of methods that would allow the scientist to use them in an almost transparent way is still required, without posing an additional concern.
The acceleration obtained from the parallel algorithm was limited by the features of the available computational architecture. The computational performance of the proposed algorithm will depend directly on the number of available processing units, the type of operating system, the type of filesystem available, the type of hard disk, and the features of the GPU available. The experiment in this work was carried out on a Quadro-type GPU; however, the computational performance can improve if a Tesla, Pascal (P100), or volta (V100)-type GPU is used. In this sense, it must be considered that the algorithm was parallelized considering a specific computational architecture. An improvement in the architecture, for example, a distributed architecture, will mean that the algorithm has to be parallelized to continue making efficient use of the computational resources.
The parallelization of the proposed algorithm basically focused on NMF since it was identified as the most computationally intensive task speaking. In this work, we focused on fine-grained parallelization; however, adding medium-grain or coarse-grained parallelization should improve the overall performance of the algorithm.




{kind=link}
{kind=link}
{kind=link}