Prediction of Drug Potencies of BACE1 Inhibitors: A Molecular Dynamics Simulation and MM_GB(PB)SA Scoring


Abstract
1. Introduction
2. Methods
2.1. Molecular Dynamics Simulations
2.2. The Generalized Born/Surface Area Model
3. Results and Discussion
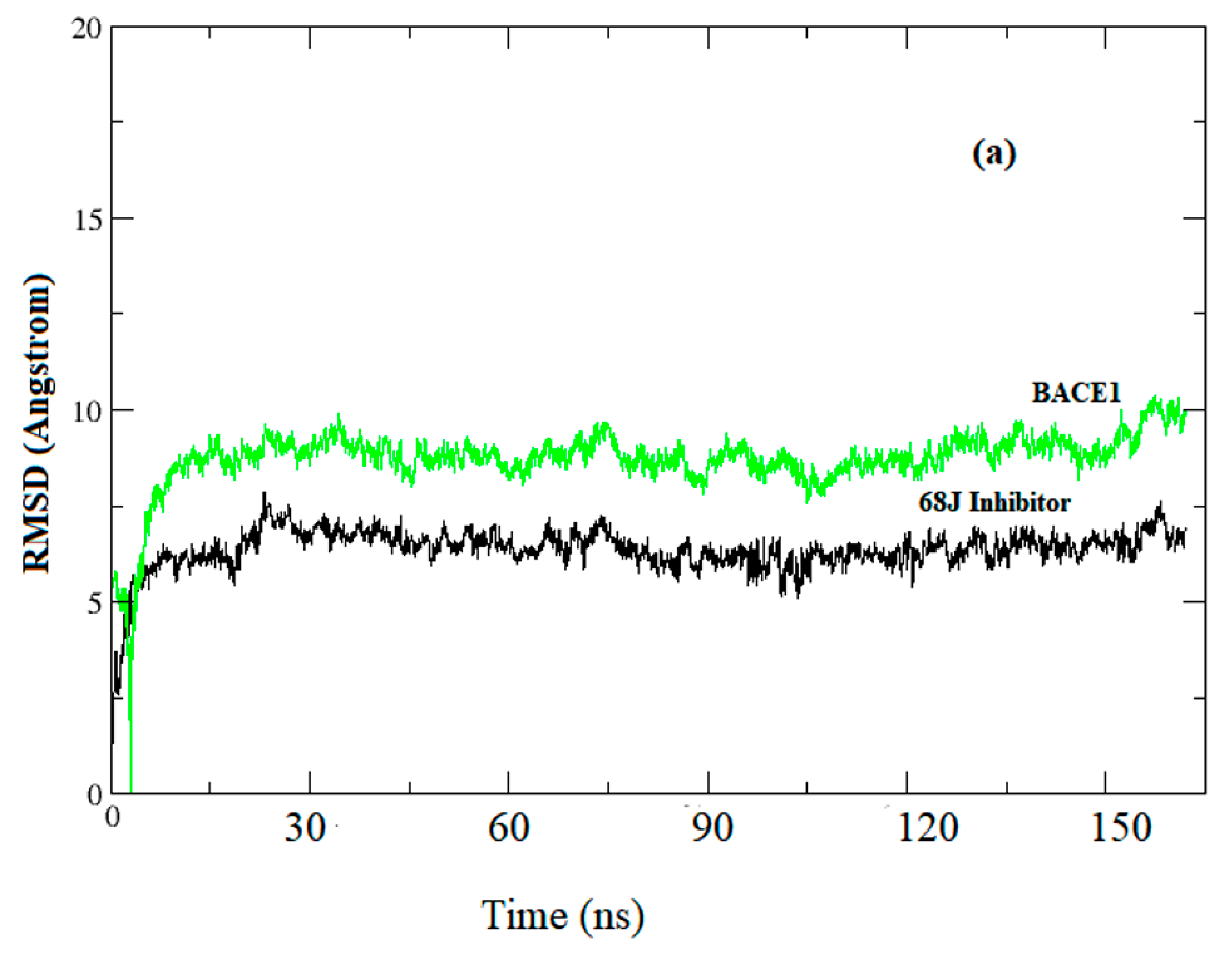
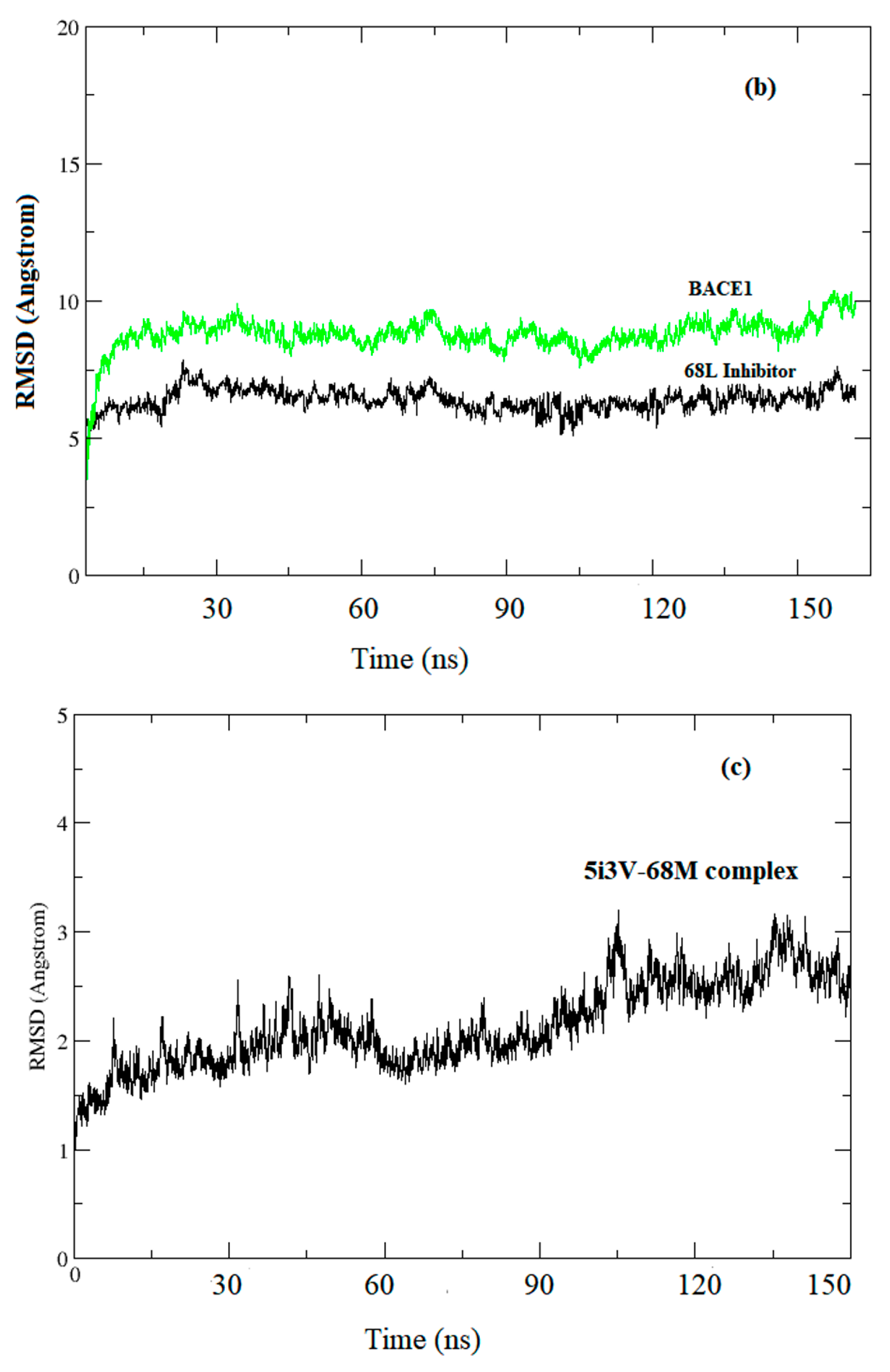
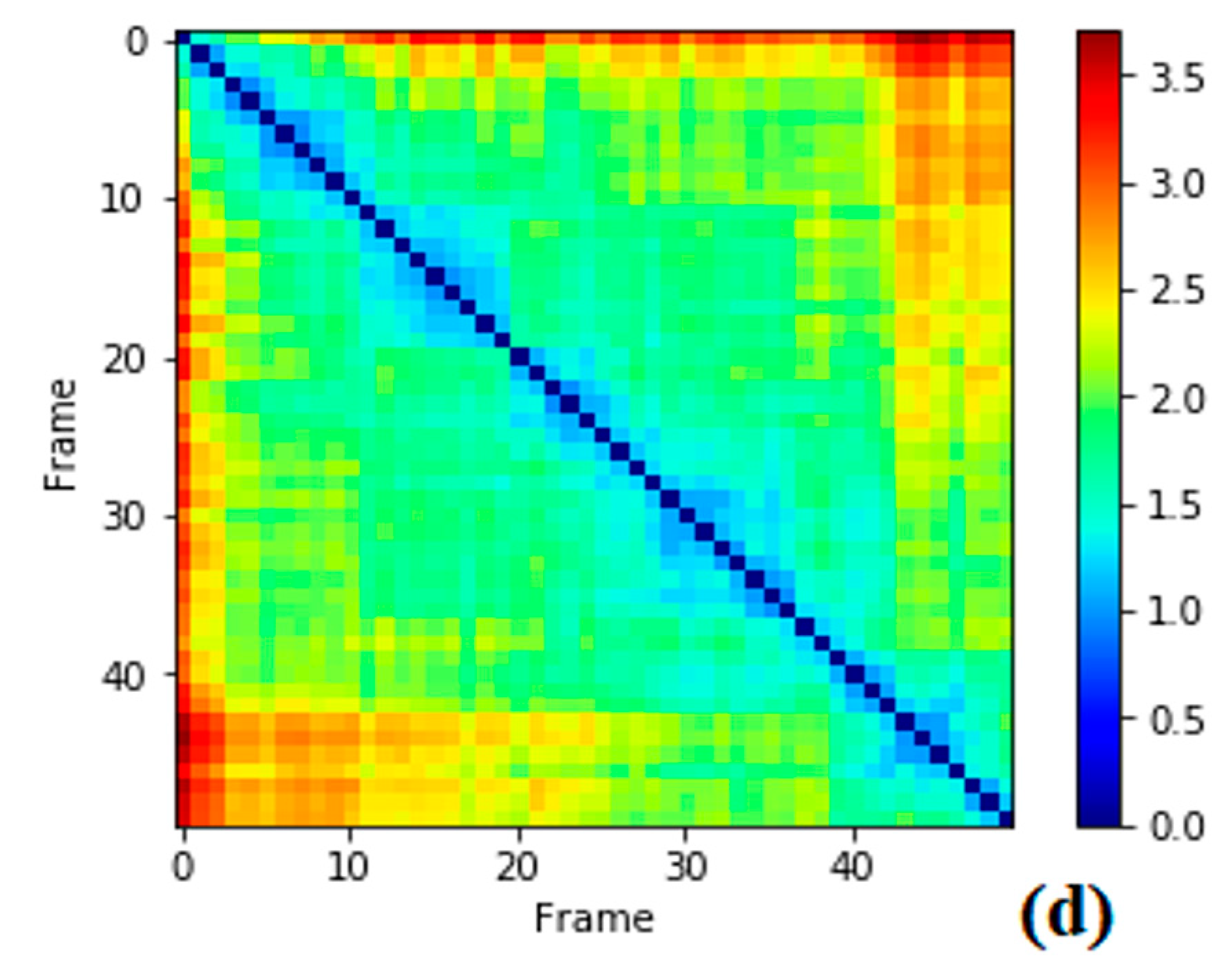
3.1. Data Analysis
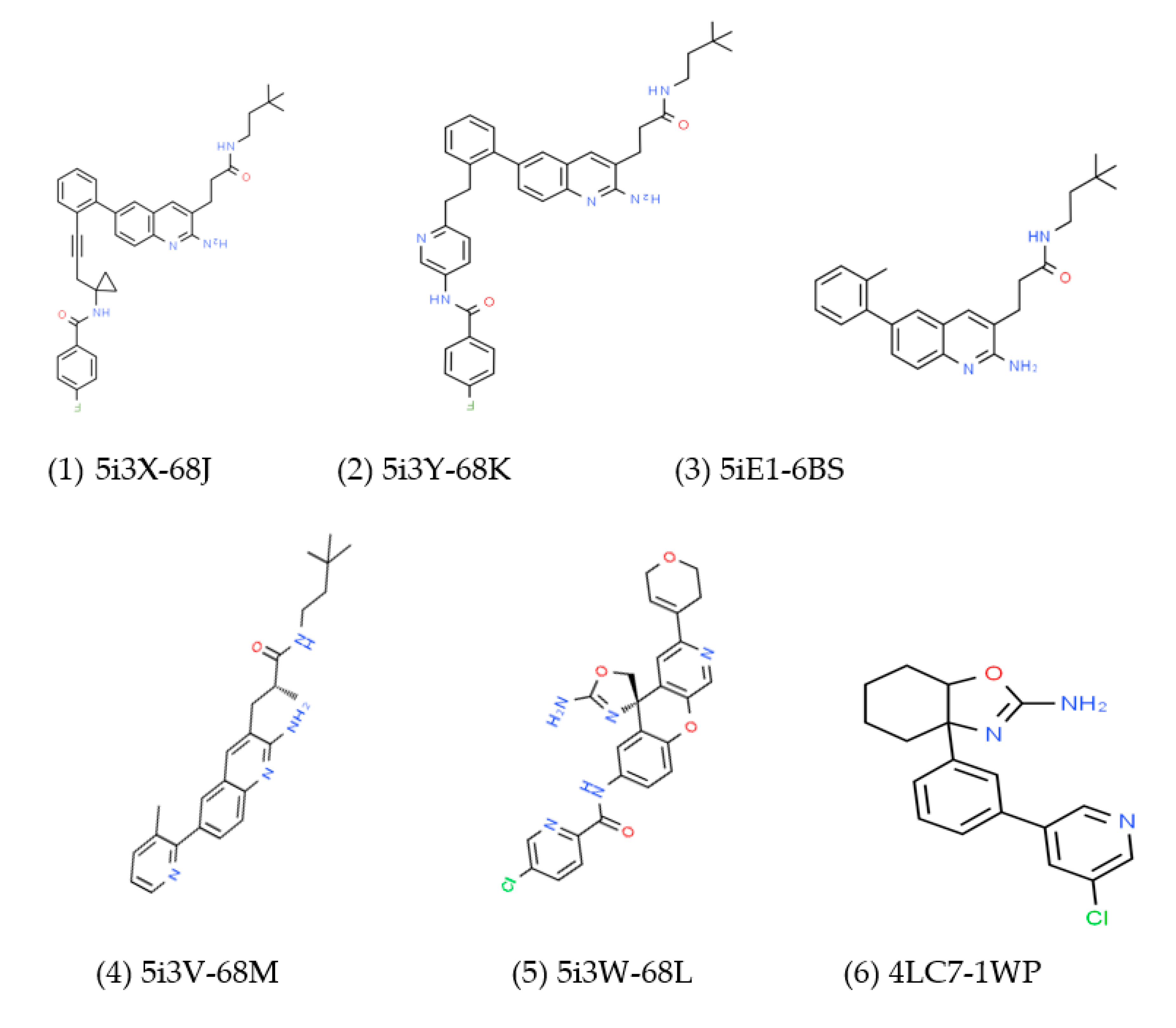
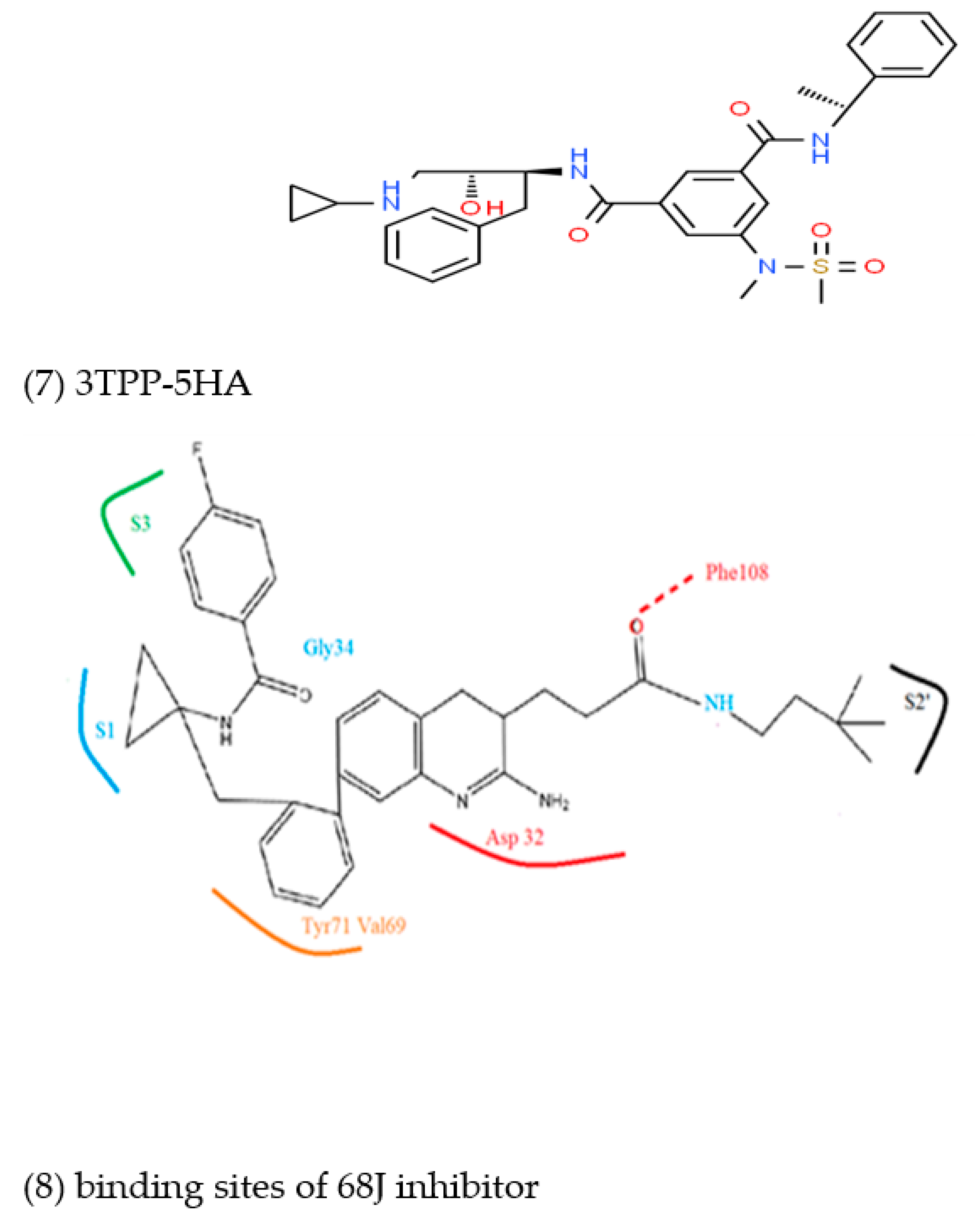
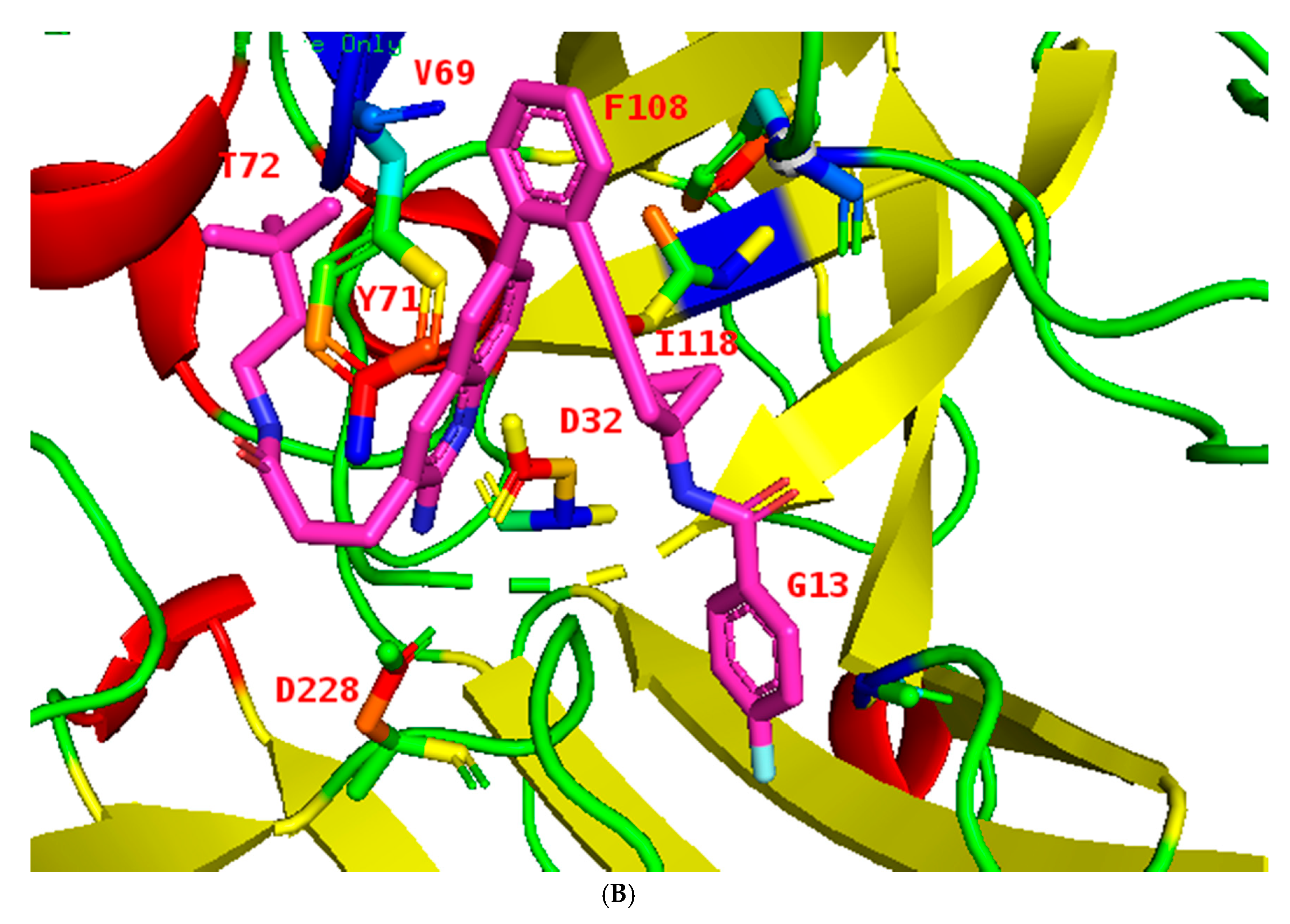
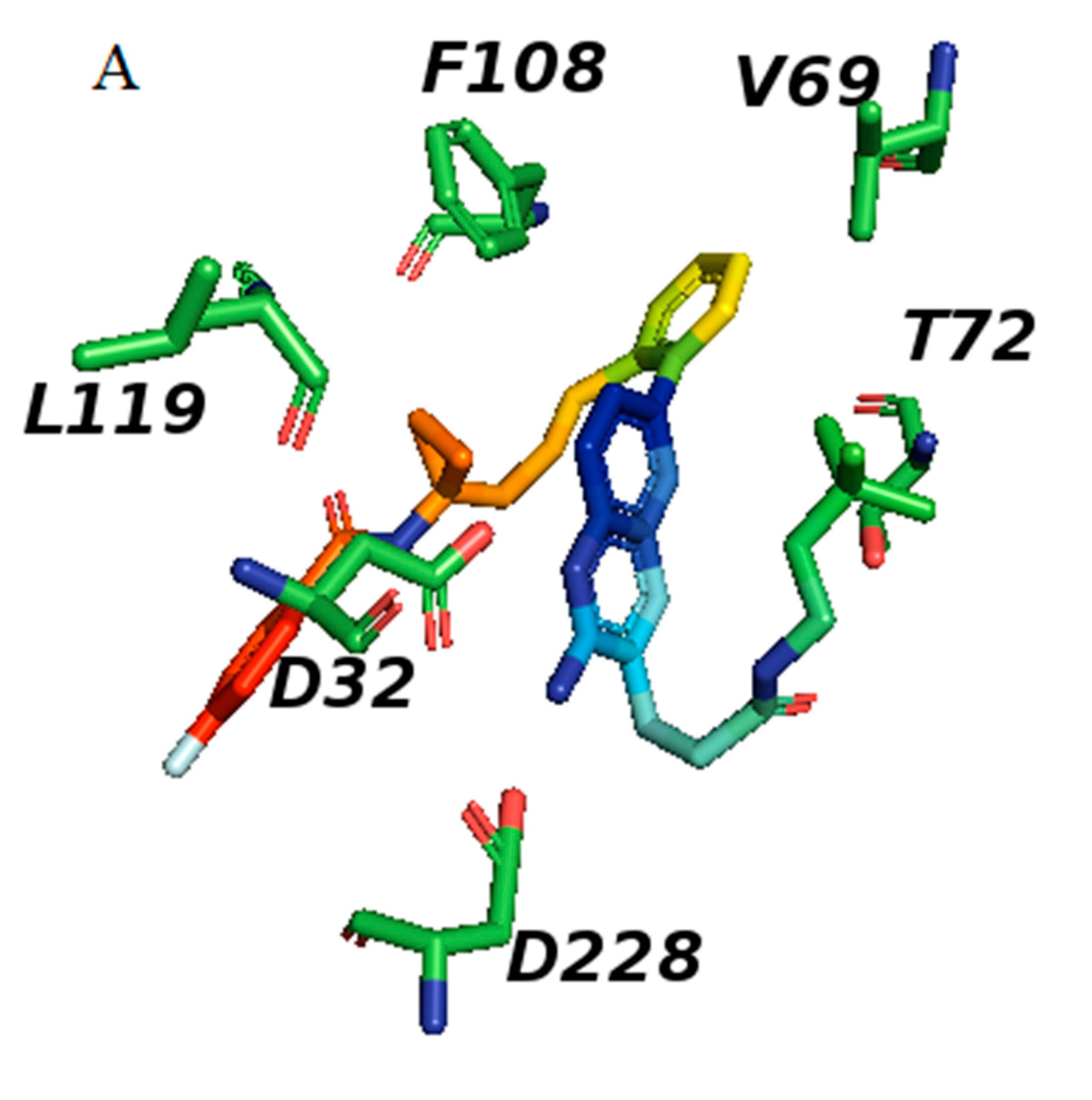
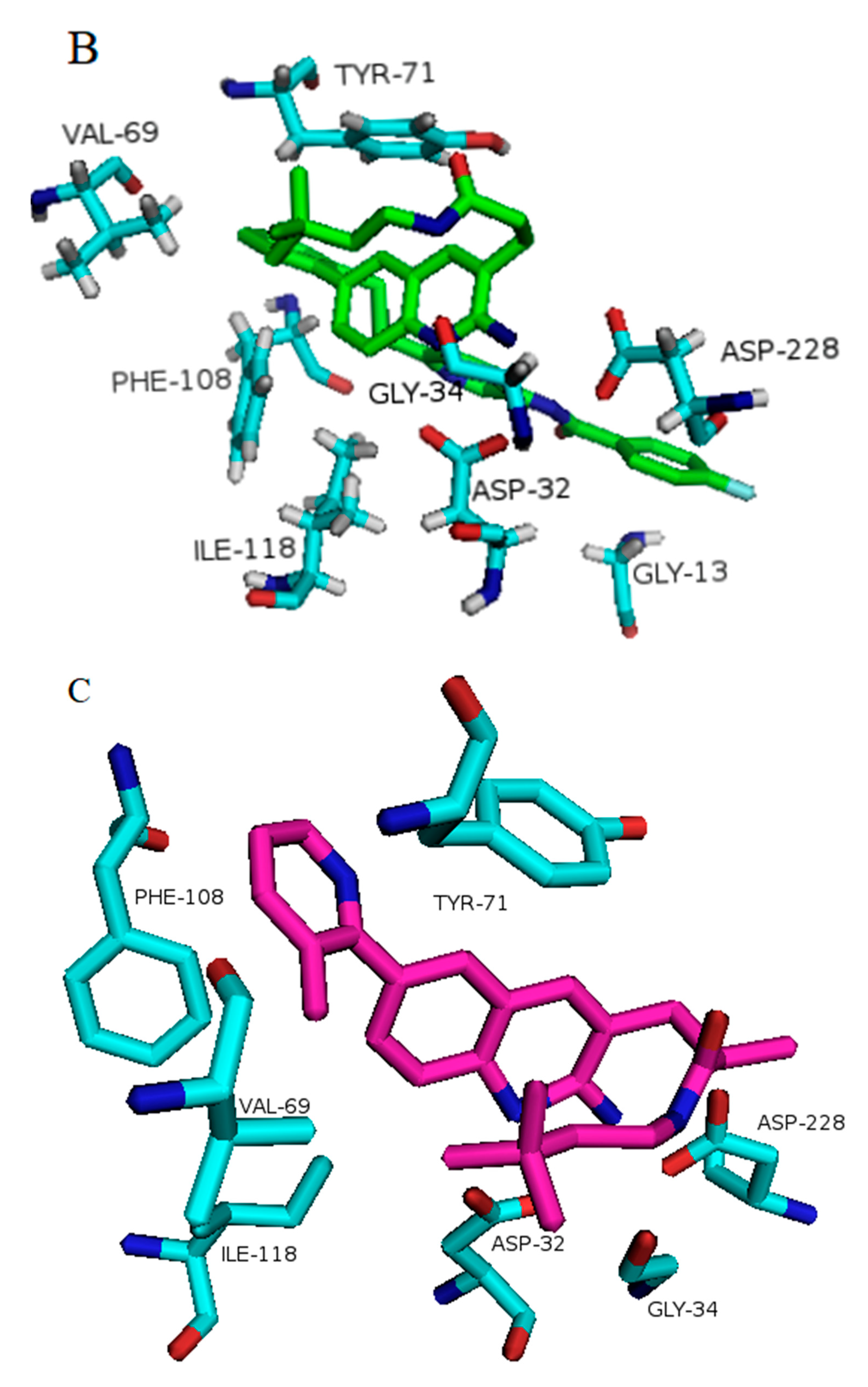
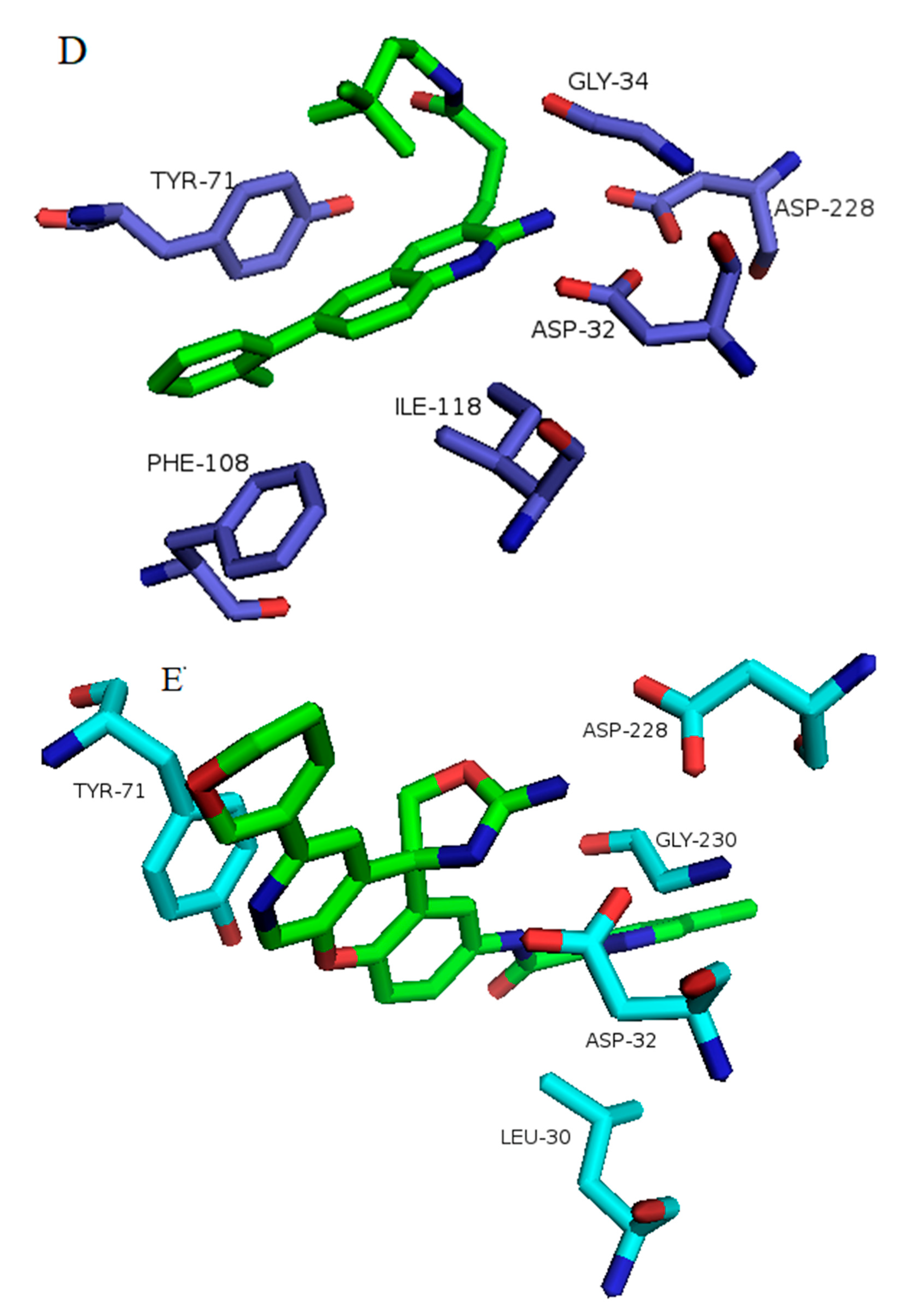
3.2. Prediction of Binding Mode and Key Interactions of Inhibitors to BACE1
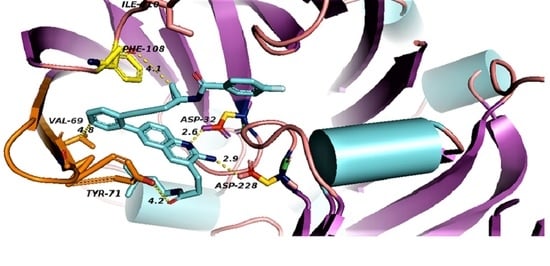
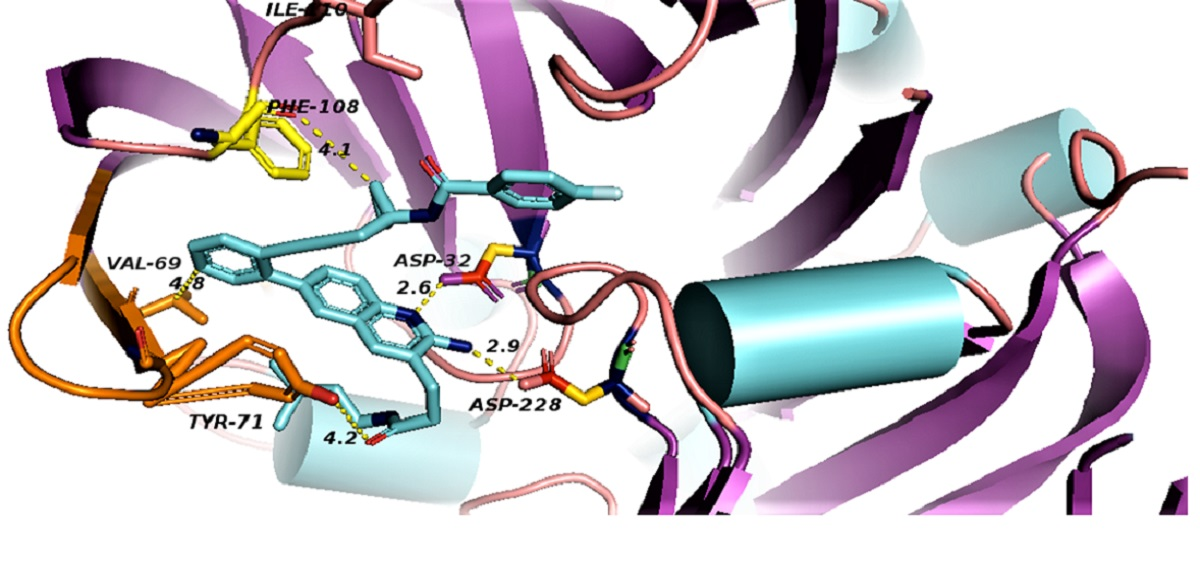
 binds Asp32 and Leu30. The attachment of the phenyl ring could lead to a significant hydrophobic interaction, which would increase the probability of permeability into the brain. Thus, many BACE1 inhibitors were designed using phenyl -based analogs. binds Asp93 and Asp289. This feature is shared with 5i3W (Figure 9E) in which the same ring binds Asp32 and Asp228. Inhibitor 5 (5i3W) has an extra phenyl group that binds the hydrophobic pocket (near Tyr71) which enhanced its binding over 4LC7. Inhibitor 6 (3TPP) has a different structure but shares an aryl ring with other inhibitors and it showed enhanced binding (Figure 6F. The sulfate group binds Asn233 and the attached aryl group interacts with Gln73, the fragment
binds Asp32 and Leu30. The attachment of the phenyl ring could lead to a significant hydrophobic interaction, which would increase the probability of permeability into the brain. Thus, many BACE1 inhibitors were designed using phenyl -based analogs. binds Asp93 and Asp289. This feature is shared with 5i3W (Figure 9E) in which the same ring binds Asp32 and Asp228. Inhibitor 5 (5i3W) has an extra phenyl group that binds the hydrophobic pocket (near Tyr71) which enhanced its binding over 4LC7. Inhibitor 6 (3TPP) has a different structure but shares an aryl ring with other inhibitors and it showed enhanced binding (Figure 6F. The sulfate group binds Asn233 and the attached aryl group interacts with Gln73, the fragment  cyclopropane ring-NH binds the other end of Asn233 and Thr231. The Asp 32, Asp 228, Gly230, Gly34, and the other side of Thr231 all make hydrogen bonds with the oxygen and nitrogen on the polar end (Figure 9F).
cyclopropane ring-NH binds the other end of Asn233 and Thr231. The Asp 32, Asp 228, Gly230, Gly34, and the other side of Thr231 all make hydrogen bonds with the oxygen and nitrogen on the polar end (Figure 9F). 
3.3. Drug Likeness
- -
- No more than five hydrogen bond donors (total H-N, H-O bonds);
- -
- No more than 10 hydrogen bond acceptors (all N+O atoms);
- -
- Molecular mass less than 500;
- -
- LogP value less than 5 (octanol-water partition coefficient);
- -
- Drug likeness improved LogP (−0.4 to 5.6), molecular weight 180 to 480, total atoms 20 to 70, including N and O,
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Gu, T.; Wu, W.Y.; Dong, Z.X.; Yu, S.P.; Sun, Y.; Zhong, Y.; Lu, Y.T.; Li, N.G. Development and Structural Modification of BACE1 Inhibitors. Molecules 2017, 22, 4. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, F.J.R.; Alexander, R.; Cleiren, E.; De Groot, A.; Carpentier, M.; Dijkmans, J.; Fierens, K.; Masure, S.; Moechars, D.; Palomino-Schätzlein, M.; et al. Fragment Binding to β-Secretase 1 without Catalytic Aspartate Interactions Identified via Orthogonal Screening Approaches. ACS Omega 2017, 2, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Brindisi, M.; Tang, J. Developing $β$-secretase inhibitors for treatment of Alzheimer’s disease. J. Neurochem. 2012, 120, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Koelsch, G.; Lin, X.; Wu, S.; Terzyan, S.; Ghosh, A.K.; Zhang, X.C.; Tang, J. Structure of the protease domain of memapsin 2 ($β$-secretase) complexed with inhibitor. Science 2000, 290, 150–153. [Google Scholar] [CrossRef]
- Malamas, M.S.; Erdei, J.; Gunawan, I.; Turner, J.; Hu, Y.; Wagner, E.; Fan, K.; Chopra, R.; Olland, A.; Bard, J.; et al. Design and synthesis of 5,5′-disubstituted aminohydantoins as potent and selective human β-secretase (BACE1) inhibitors. J. Med. Chem. 2010, 53, 1146–1158. [Google Scholar] [CrossRef]
- Steele, T.G.; Hills, I.D.; Nomland, A.A.; de León, P.; Allison, T.; McGaughey, G.; Colussi, D.; Tugusheva, K.; Haugabook, S.J.; Espeseth, A.S.; et al. Identification of a small molecule $β$-secretase inhibitor that binds without catalytic aspartate engagement. Bioorg. Med. Chem. Lett. 2009, 19, 17–20. [Google Scholar] [CrossRef]
- Ren, Z.; Tam, D.; Xu, Y.Z.; Wone, D.; Yuan, S.; Sham, H.L.; Cheung, H.; Regnstrom, K.; Chen, X.; Rudolph, D.; et al. Development of a novel β-secretase binding assay using the alphascreen platform. J. Biomol. Screen. 2013, 18, 695–704. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Lu, N.; Kofke, D.A. Accuracy of free-energy perturbation calculations in molecular simulation. II. Heuristics. J. Chem. Phys. 2001, 115, 6866–6875. [Google Scholar] [CrossRef]
- Veselovsky, A.V.; Ivanov, A.S. Strategy of Computer-Aided Drug Design. Curr. Drug Target -Infectious Disord. 2003, 3, 33–40. [Google Scholar] [CrossRef]
- Temiz, N.A.; Trapp, A.; Prokopyev, O.A.; Camacho, C.J. Optimization of minimum set of protein--DNA interactions: A quasi exact solution with minimum over-fitting. Bioinformatics 2010, 26, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Miller, J.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of RNA hairpin loops and helices. J. Biomol. Struct. Dyn. 1998, 16, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate- DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Vassar, R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Ben-Shalom, I.Y.; Lin, C.; Kurtzman, T.; Walker, R.C.; Gilson, M.K. Simulating water exchange to buried binding sites. J. Chem. Theory Comput. 2019, 15, 2684–2691. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Rother, K. Introduction to PyMOL. Methods Mol. Biol. Clift. Nj 2005, 635, 1–32. [Google Scholar] [CrossRef]
- Binding Constants. Available online: http://www.bindingdb.org/pdb/1o86 (accessed on 23 December 2018).
- Antechamber Tutorial. Available online: http://ambermd.org/tutorials/basic/tutorial4b/ (accessed on 23 December 2018).
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Shanti, P.; Kwon, J.B. How to Cite Amber. Am. Ethnol. 2020, 47, 209. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Miller, B.R.; Mcgee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kollman, P.A. Computational study of protein specificity: The molecular basis of HIV-1 protease drug resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 14937–14942. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Narang, S.S.; Goyal, D.; Goyal, B. Inhibition of Alzheimer’s amyloid-β42 peptide aggregation by a bi-functional bis-tryptoline triazole: Key insights from molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 38, 1598–1611. [Google Scholar] [CrossRef]
- Hermansson, A. Calculating Ligand-Protein Binding Energies from Molecular Dynamics Simulations-Thesis in Physical Chemistry. Master’s Thesis, KTH Royal Institute of Technology, Stockholm, Sweden, July 2015. Available online: https://www.diva-portal.org/smash/record.jsf?pid=diva2%3A839581&dswid=6921 (accessed on 23 December 2018).
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular synamics trajectory data. J. Chem. Theory Com. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A.; Thompson, J.J. Solvent interaction energy calculations on molecular dynamics trajectories: Increasing the efficiency using systematic frame selection. J. Chem. Inf. Model. 2011, 51, 2680–2689. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Roe, D.R.; Cheatham, T.E. Convergence and reproducibility in molecular dynamics simulations of the DNA duplex d(GCACGAACGAACGAACGC). Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 1041–1058. [Google Scholar] [CrossRef] [PubMed]
- RCSB PDB: Search. Available online: https://www.rcsb.org/search (accessed on 23 December 2018).
- Binding MOAD. Available online: http://bindingmoad.org/ (accessed on 23 December 2018).
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Irannejad, H. Lipophilic Ligand Efficiency as a Useful Metric in Hit and Lead Optimization. J. Med. Chem. Drug Des. 2019, 2, 9–10. [Google Scholar] [CrossRef]
- Wang, C.; Nguyen, P.H.; Pham, K.; Huynh, D.; Le, T.B.N.; Wang, H.; Ren, P.; Luo, R. Calculating protein-ligand binding affinities with MMPBSA: Method and error analysis. J. Comput. Chem. 2016, 37. [Google Scholar] [CrossRef]
- Li, S.; Zhao, H.; Li, J.; Hao, J.; Yu, H. A series of molecular modeling techniques to reveal selective mechanisms of inhibitors to β-Site amyloid precursor protein cleaving enzyme 1 (BACE1) and β-site amyloid precursor protein cleaving enzyme 2 (BACE2). J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB id-Inhibitor | Kd Exp [21] | IC50 [21] | ΔHGBSA kcal/mol | T∆S | ∆Gbinding Calculated kcal/mol | ΔG exp | Kd from Calculated ∆Gbind ** |
|---|---|---|---|---|---|---|---|
| 5i3x-(1) | 8 nM, 0.8 nM | 191 nM, 9 nM | −44.5 (4) | −25.2 (5) | −19.3 (5) | −11.34 | 0.000139 nM |
| 5i3y-(2) | 0.4000 nM | 16 nM, 0.8 nM | −37.4 (3) | −24.96 (6) | −12.4 (7) | −13.16 | 1.39 nM |
| 5ie1-(3) | 140 nM | 140 nM | −30.5 (3) | −22.98 (6.2) | −7.5 (6) | −9.60 | 4.39 mM |
| 5i3w-(5) | 0.6 nM | −32.2 (2.6) | −24 (4) | −8.15 (4) | −12.9 | 1.39 mM | |
| 5i3v-(4) | 16 nM | 35 nM | −32.92 (5.2) | −22.26 (4) | −10.66 (4) | −10.92 | 24.3 nM |
| 3tpp-(6) | 233 nM | 15 nM, 15 nM | −35.6 (6) | −26.21 (5) | −9.4 (4) | −9.28 | 193.07 nM |
| 4lc7-(7) | 11800 nM, 14 nM | −24.64 (5) | −22.5 (5) | −2.15 (5) | −6.8 | 29.13 mM |
| Number (Figure 2) | PDB ID | vdW | E EL | E GB | Esurf | Esolv | ΔHGBSA |
|---|---|---|---|---|---|---|---|
| 1 | 5i3x | −67.1 (3.1) | −26.99 (6.1) | 58.3 (4.9) | −8.72 (0.24) | 49.6 (4.83) | −44.52 (4) |
| 2 | 5i3y | −59.13 (3.4) | −16.8 (3.4) | 45.8 (4.2) | −7.2 (0.5) | 38.6 (3.8) | −37.36 (3) |
| 3 | 5ie1 | −39.15 (2.96) | −36.21 (2.9) | 50.77 (1.5) | 3.76 (0.02) | 44.9 (0.7) | −30.48 (2.8) |
| 4 | 5i3v | −43.69 (3.4) | −21.62 (7.7) | 38 (5.6) | −5.6 (0.52) | 32.4 (5.4) | −32.92 (5.2) |
| 5 | 5i3w | −55.34 (2.86) | −14.12 (3.1) | 44.1 (2.6) | −6.8 (0.19) | 37.3 (2.5) | −32.15 (2.6) |
| 6 | 4lc7 | −34.04 (2.9) | −13.2 (13) | 26.8 (11) | −4.3 (0.3) | 22.6 (10.9) | −24.64 (5.02) |
| 7 | 3ttp | −10.73 (0.9) | −66.97 (1.9) | −55.9 (1) | 5.6 (0.03) | −40.26 (1.02) | −35.6 (6) |
| Inhibitor | ASP32 Oxygen Å | ASP228 Oxygen Å | Gly 13 Å | Ser35 Å | Hydrophobic: Tyr71 Å | Hydrph:Val69 Å | |
|---|---|---|---|---|---|---|---|
| 5i3X | N of pyridine ring | 2.6, 3.6 | 4.9, 5.1 | 3.0–3.9 | 4.0–4.9 | ||
| NH2 | 2.9, 3.6 | 2.9, 3.0 | |||||
| 5i3Y | N of pyridine ring | 3.5 | 5.0, 5.2 | 3.8 | 4.1–5 | 4.2–4.3 | 3.9–4.4 |
| NH2 | 2.6 | 3.0, 3.1 | |||||
| 3TPP | 2.7, 3.5 | 2.7, 3.9 | 3.4 Gly230: 3.1 | Gln 73: 3.2 | |||
| 4LC7 | Asp93: 2.7, 2.7 | Asp289: 2.8, 2.8 | Leu91: 4.3 | Tyr132: 3.6 |
| Inhibitors Number (from Figure 2) | vdW | EEL Electrostatic | EGB Polar | Esurf Surface Area | Esolv Desolvation |
|---|---|---|---|---|---|
| 1, 2, 3, 4 | 0.95 | 0.1 | 0.41 | 0.63 | 0.29 |
| 1, 2, 3, 4, 5 | 0.76 | 0.01 | 0.43 | 0.44 | 0.33 |
| 1, 2, 3, 4, 5, 6 | 0.85 | 0.075 | 0.68 | 0.29 | 0.62 |
| 1, 2, 3, 4, 5, 6, 7 | 0.23 | 0.05 | 0.01 | 0.1 | 0.011 |
| Protein-Inhibitor Complex PDB Code [37] | ∆H Kcal/mol | Inhibitor | Binding Sites to the Protein |
|---|---|---|---|
| 5i3x I = 68J | −44.5 | N-(1-{3-[2-(2-amino-3-{3-[(3,3-dimethylbutyl)amino]-3-oxopropyl} quinolin-6-yl)phenyl]prop-2-yn-1-yl}cyclopropyl)-4-fluorobenzamide | N-O:Asp228, Asp32, Gly13 Hydrph:Tyr71, Val69, Ile118, Leu30, Phe108 |
| 5i3y I = 68K | −37.4 | N-(6-{2-[2-(2-amino-3-{3-[(3,3-dimethylbutyl)amino] -3-oxopropyl}quinolin-6-yl)phenyl]ethyl}pyridin-3-yl)-4-fluorobenzamide | N-O:Asp 228, Asp32, Gly34, Gly230Hydrph:Gly13, Ser35, Tyr71, Val69, Ile118, Phe108 |
| 5i3v I = 68M | −32.9 | (2R)-3-[2-amino-6-(3-methylpyridin-2-yl)quinolin-3-yl] -N-(3,3-dimethylbutyl)-2-methylpropanamide | N(L)-O(rec):Asp228, Asp32, Gly34, Hydrph:Tyr71, Phe108 |
| 5i3w I = 68L | −32.15 | N-[(5S)-2’-amino-3-(5,6-dihydro-2H-pyran-3-yl)-5’H -spiro [1-benzopyrano [2 ,3-c]pyridine-5,4’-[1,3]oxazol]-7-yl]-5-chloropyridine-2-carboxamideC25 H20 Cl N5 O4 | Asp 32, Asp 228, Gly 230, Tyr 71 Leu 30, Gly 13 |
| 5ie1 6BS | −30.5 | 3-[2-amino-6-(2-methylphenyl)quinolin-3-yl]-N-(3,3-dimethylbutyl)propanamide | N-O:Asp228, Asp32, Gly34 Hydrph:Tyr71, Val69, Ile118, Leu30, Phe108 |
| 3tpp 5HA | −35.6 | N-[(1S,2R)-1-BENZYL-3-(CYCLOPROPYLAMINO)-2-HYDROXYPROPYL]-5-[METHYL(METHYLSULFONYL)AMINO] -N’-[(1R)-1-PHENYLETHYL]ISOPHTHALAMIDEC31 H38 N4 O5 S | Asp 32, Asp 228 Gln 73 Phe 108, Gly 34 Asn 233 Gly 230, Leu 30 Trp 115, Thr231Gly230, Gln12 Thr232 Gly 13 |
| 4lc7 1WP | −24.64 | (3aR,7aR)-3a-[3-(5-chloropyridin-3-yl) phenyl]-3a,4,5,6,7,7a-hexahydro-1,3-benzoxazol-2-amine | Asp93, Asp289, Tyr 132 Leu 91 |
| Inhibitor | PSA/∆H | PSA/Evdw | PSA/EGB | PSA/EEL | PSA/Esurface | PSA/Esolv |
|---|---|---|---|---|---|---|
| 1, 2, 3, 4, 5, 6, 7 | 0.23 | 0.23 | 0.32 | 0.24 | 0.014 | 0.31 |
| 1, 2, 3, 4, 5, 6 | 0.3 | 0.14 | 0.17 | 0.14 | 0.4 | 0.13 |
| 1, 2, 3, 4, 5 | 0.07 | 0.5 | 0.006 | 0.76 | 0.54 | 0.03 |
| 1, 2, 3, 4 | 0.5 | 0.8 | 0.02 | 0.64 | 0.69 | 0.003 |
| PDB ID-inhibitor | M.Wt <500 | LogP <5 | PSA Å2 [38] | No. H-bond Acceptor Atoms <5 | No. H-bond Donor Atoms <5 | N&O <10 | Number of Rotatable Bonds | No. Rings >3 |
|---|---|---|---|---|---|---|---|---|
| 5i3x-68J | 590.730 | 7.16 ** 8.18 ++ | 97.11 | 3 | 3 | 6 | 13 | 5 |
| 5i3y-68K | 617.55 | 7.18 ** 8.59 ++ | 110 | 3 | 4 | 7 | 14 | 5 |
| 5i3v-68M | 404.548 | 4.96 ** 5.89 ++ | 80.9 | 2 | 3 | 5 | 8 | 3 |
| 5i3w-68L | 488.902 | 2.77 ** 4.43 ++ | 122.56 | 1 | 3 | 9 | 4 | 6 |
| 5ie1-6BS | 389.533 | 5.42 ** 6.25 ++ | 68.01 | 2 | 2 | 4 | 8 | 3 |
| 4lc7-1WP | 328.122 | 3.88 ** 4.23 ++ | 62.11 | 1 | 0 | 4 | 2 | 4 |
| 3tpp-5HA | 597.730 | 3.6 ** 3.86 ++ | 140.8 | 4 | 5 | 9 | 16 | 4 |
| Proteins in Figures S5 Pockets Found by SPDV Software | Hydrophobic Pocket Area Å2, Volume Å3 | Hydrophobic E = −25 × S.A (Å2) kcal/mol | PSA (Å2) | Estimated Hydrophobic Energy −25 × PSA kcal/mol |
|---|---|---|---|---|
| 5i3x Bound CR3 | 106, 61 | |||
| 105, 75 | −2.63 | 97.11 | −2.42 | |
| 90, 72 | ||||
| 71, 45 | ||||
| 5i3y | 93, 64 | |||
| Bound t CR3 | 87, 57 | −2.18 | 110 | −2.75 |
| 74, 48 | ||||
| 5ie1 | ||||
| CR3, Hexane ring | 96, 71 | −2.42 | 68.1 | −1.7 |
| 82,55 | ||||
| 67, 33 | ||||
| 5i3v | 126, 107 | |||
| Bound CR3 | 61, 37 | −1.54 | 80.9 | −2.03 |
| 58, 33 | ||||
| 55, 31 | ||||
| 3TPP no hyd | 115, 71 | 140.8 | ||
| No hyd | 74, 47 | 0.0 | ||
| No hyd | 59, 35 | |||
| 4lc7 | 165, 101 | |||
| Hexane ring | 100, 61 | −2.52 | 62.11 | |
| 89, 60 | ||||
| 5i3w | 80,39 | |||
| Close to ring | 61,35 | −1.54 | 122.56 | −3.06 |
| 61, 36 | ||||
| 56,33 |
| PDB ID-Inhibitor Number (from Figure 2) | NnH | LE = −∆G/NnH kcal/mol/Heavy Atom | ∆Gbind Calculated | ΔG Exp |
|---|---|---|---|---|
| 5i3X-(1) | 44 | 0.41 | −19.3 | −11.34 |
| 5i3Y-(2) | 47 | 0.27 | −12.4 (7) | −13.16 |
| 5iE1-(3) | 29 | 0.26 | −7.5 (6) | −9.60 |
| 5i3V-(4) | 30 | 0.36 | −10.66 (4) | −10.92 |
| 5i3W-(5) | 35 | 0.24 | −8.15 (4) | −12.9 |
| 3TPP-(6) | 41 | 0.23 | −9.4 (4) | −9.28 |
| 4LC7-(7) | 23 | 0.09 | −2.15 (5) | −6.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamed, M.Y. Prediction of Drug Potencies of BACE1 Inhibitors: A Molecular Dynamics Simulation and MM_GB(PB)SA Scoring. Computation 2020, 8, 106. https://doi.org/10.3390/computation8040106
Hamed MY. Prediction of Drug Potencies of BACE1 Inhibitors: A Molecular Dynamics Simulation and MM_GB(PB)SA Scoring. Computation. 2020; 8(4):106. https://doi.org/10.3390/computation8040106
Chicago/Turabian StyleHamed, Mazen Y. 2020. "Prediction of Drug Potencies of BACE1 Inhibitors: A Molecular Dynamics Simulation and MM_GB(PB)SA Scoring" Computation 8, no. 4: 106. https://doi.org/10.3390/computation8040106
APA StyleHamed, M. Y. (2020). Prediction of Drug Potencies of BACE1 Inhibitors: A Molecular Dynamics Simulation and MM_GB(PB)SA Scoring. Computation, 8(4), 106. https://doi.org/10.3390/computation8040106