Chemical-Reactivity Properties, Drug Likeness, and Bioactivity Scores of Seragamides A–F Anticancer Marine Peptides: Conceptual Density Functional Theory Viewpoint




Abstract
1. Introduction
2. Theoretical Background and Computational Methodology
3. Results and Discussion
| Electronegativity | |
| Global Hardness | |
| Electrophilicity | = |
| Electrodonating power | = |
| Electroaccepting power | = |
| Net electrophilicity |
> Seragamide C > Seragamide F > Tenilsetam > Seragamide A >
> Pyridoxamine > Seragamide D > Pioglitazone > Seragamide E
| Nucleophilic Fukui Function | |
| Electrophilic Fukui Function | , |
| Dual Descriptor | = - |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- La Barre, S.; Kornprobst, J.M. Outstanding Marine Molecules; Wiley-Blackwell: Weinheim, Germany, 2014. [Google Scholar]
- Kim, S.K. Marine Proteins and Peptides—Biological Activities and Applications; Wiley-Blackwell: Chichester, UK, 2013. [Google Scholar]
- Rekka, E.; Kourounakis, P. Chemistry and Molecular Aspects of Drug Design and Action; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Náray-Szabó, G.; Warshel, A. Computational Approaches to Biochemical Reactivity; Kluwer Academic Publishers: New York, NY, USA, 2002. [Google Scholar]
- Parr, R.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Chermette, H. Chemical Reactivity Indexes in Density Functional Theory. J. Comput. Chem. 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Ayers, P.; Parr, R. The Variational Principles for Describing Chemical Reactions: The Fukui Function and Chemical Hardness Revisited. J. Am. Chem. Soc. 2000, 122, 2010–2018. [Google Scholar] [CrossRef]
- Poater, A.; Saliner, A.G.; Carbó-Dorca, R.; Poater, J.; Solà, M.; Cavallo, L.; Worth, A.P. Modeling the Structure-Property Relationships of Nanoneedles: A Journey Toward Nanomedicine. J. Comput. Chem. 2009, 30, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Saliner, A.G.; Solá, M.; Cavallo, L.; Worth, A.P. Computational Methods to Predict the Reactivity of Nanoparticles Through Structure-Property Relationships. Expert Opin. Drug Deliv. 2010, 7, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Munro, M.H.G. (Eds.) Dictionary of Natural Marine Products; Chapman & Hall: Boca Raton, FL, USA, 2008. [Google Scholar]
- Frau, J.; Hernández-Haro, N.; Glossman-Mitnik, D. Computational Prediction of the pKas of Small Peptides through Conceptual DFT Descriptors. Chem. Phys. Lett. 2017, 671, 138–141. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Chemical Reactivity Theory Study of Advanced Glycation Endproduct Inhibitors. Molecules 2017, 22, 226. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Kumar, V. Chemical Drug Design; Walter de Gruyter GmbH: Berlin, Germany, 2016. [Google Scholar]
- Gore, M.; Jagtap, U.B. Computational Drug Discovery and Design; Springer Science+Business Media, LLC: New York, NY, USA, 2018. [Google Scholar]
- Frau, J.; Flores-Holguín, N.; Glossman-Mitnik, D. Chemical Reactivity Properties, pKa Values, AGEs Inhibitor Abilities and Bioactivity Scores of the Mirabamides A–H Peptides of Marine Origin Studied by Means of Conceptual DFT. Mar. Drugs 2018, 16, 302. [Google Scholar] [CrossRef]
- Lewars, E. Computational Chemistry—Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003. [Google Scholar]
- Young, D. Computational Chemistry—A Practical Guide for Applying Techniques to Real-World Problems; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Cramer, C. Essentials of Computational Chemistry—Theories and Models, 2nd ed.; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A Long-Range Correction Scheme for Generalized-Gradient- Approximation Exchange Functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Chai, J.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb- Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G. Efficient Hybrid Density Functional Calculations in Solids: Assessment of the Heyd-Scuseria-Ernzerhof Screened Coulomb Hybrid Functional. J. Chem. Phys. 2004, 121, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Kronik, L.; Baer, R. Reliable Prediction of Charge Transfer Excitations in Molecular Complexes Using Time-Dependent Density Functional Theory. J. Am. Chem. Soc. 2009, 131, 2818–2820. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Kronik, L.; Baer, R. Prediction of Charge-Transfer Excitations in Coumarin-Based Dyes Using a Range-Separated Functional Tuned From First Principles. J. Chem. Phys. 2009, 131, 244119. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Eisenberg, H.; Kronik, L.; Baer, R. Fundamental Gaps in Finite Systems from Eigenvalues of a Generalized Kohn-Sham Method. Phys. Rev. Lett. 2010, 105, 266802–266804. [Google Scholar] [CrossRef]
- Karolewski, A.; Stein, T.; Baer, R.; Kümmel, S. Communication: Tailoring the Optical Gap in Light-Harvesting Molecules. J. Chem. Phys. 2011, 134, 151101–151105. [Google Scholar] [CrossRef] [PubMed]
- Kuritz, N.; Stein, T.; Baer, R.; Kronik, L. Charge-Transfer-Like π→π* Excitations in Time-Dependent Density Functional Theory: A Conundrum and Its Solution. J. Chem. Theory Comput. 2011, 7, 2408–2415. [Google Scholar] [CrossRef]
- Ansbacher, T.; Srivastava, H.K.; Stein, T.; Baer, R.; Merkx, M.; Shurki, A. Calculation of Transition Dipole Moment in Fluorescent Proteins-Towards Efficient Energy Transfer. Phys. Chem. Chem. Phys. 2012, 14, 4109–4117. [Google Scholar] [CrossRef]
- Kronik, L.; Stein, T.; Refaely-Abramson, S.; Baer, R. Excitation Gaps of Finite-Sized Systems from Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2012, 8, 1515–1531. [Google Scholar] [CrossRef]
- Stein, T.; Autschbach, J.; Govind, N.; Kronik, L.; Baer, R. Curvature and Frontier Orbital Energies in Density Functional Theory. J. Phys. Chem. Lett. 2012, 3, 3740–3744. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT. Molecules 2018, 23, 559. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Glossman-Mitnik, D. Conceptual DFT Study of the Local Chemical Reactivity of the Dilysyldipyrrolones A and B Intermediate Melanoidins. Theor. Chem. Accounts 2018, 137, 1210. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Conceptual DFT Study of the Local Chemical Reactivity of the Colored BISARG Melanoidin and Its Protonated Derivative. Front. Chem. 2018, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Glossman-Mitnik, D. Molecular Reactivity of some Maillard Reaction Products Studied through Conceptual DFT. Contemp. Chem. 2018, 1, 1–14. [Google Scholar]
- Frau, J.; Glossman-Mitnik, D. Computational Study of the Chemical Reactivity of the Blue-M1 Intermediate Melanoidin. Comput. Theor. Chem. 2018, 1134, 22–29. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Chemical Reactivity Theory Applied to the Calculation of the Local Reactivity Descriptors of a Colored Maillard Reaction Product. Chem. Sci. Int. J. 2018, 22, 1–14. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Blue M2: An Intermediate Melanoidin Studied via Conceptual DFT. J. Mol. Model. 2018, 24, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. II. MMFF94 van der Waals and Electrostatic Parameters for Intermolecular Interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s Option for Energy Minimization Studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting Basis Sets for H to R. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.; Cramer, C.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. [Google Scholar] [CrossRef]
- Becke, A.D. Vertical Excitation Energies From the Adiabatic Connection. J. Chem. Phys. 2016, 145, 194107. [Google Scholar] [CrossRef]
- Baerends, E.J.; Gritsenko, O.V.; van Meer, R. The Kohn-Sham Gap, the Fundamental Gap and the Optical Gap: The Physical Meaning of Occupied and Virtual Kohn-Sham Orbital Energies. Phys. Chem. Chem. Phys. 2013, 15, 16408–16425. [Google Scholar] [CrossRef]
- Van Meer, R.; Gritsenko, O.V.; Baerends, E.J. Physical Meaning of Virtual Kohn-Sham Orbitals and Orbital Energies: An Ideal Basis for the Description of Molecular Excitations. J. Chem. Theory Comput. 2014, 10, 4432–4441. [Google Scholar] [CrossRef]
- Gázquez, J.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Chattaraj, P.; Chakraborty, A.; Giri, S. Net Electrophilicity. J. Phys. Chem. A 2009, 113, 10068–10074. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N. Advanced Glycation Endproducts–Role in Pathology of Diabetic Complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, S.; Figarola, J.L. Novel Inhibitors of Advanced Glycation Endproducts. Arch. Biochem. Biophys. 2003, 419, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Peyroux, J.; Sternberg, M. Advanced Glycation Endproducts (AGEs): Pharmacological Inhibition in Diabetes. Pathol. Biol. 2006, 54, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Toro-Labbé, A. (Ed.) Theoretical Aspects of Chemical Reactivity; Elsevier Science: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Grand, A.; Toro-Labbé, A. Theoretical Support for Using the Δf(r) Descriptor. Chem. Phys. Lett. 2006, 425, 342–346. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Revisiting Caffeate’s Capabilities as a Complexation Agent to Silver Cation in Mining Processes by means of the Dual Descriptor—A Conceptual DFT Approach. J. Mol. Model. 2012, 18, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Araya, J.I. Explaining Reaction Mechanisms Using the Dual Descriptor: A Complementary Tool to the Molecular Electrostatic Potential. J. Mol. Model. 2012, 19, 2715–2722. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Araya, J.I. Why is the Dual Descriptor a More Accurate Local Reactivity Descriptor than Fukui Functions? J. Math. Chem. 2015, 53, 451–465. [Google Scholar] [CrossRef]
- Santos, G.B.; Ganesan, A.; Emery, F.S. Oral Administration of Peptide-Based Drugs: Beyond Lipinski’s Rule. ChemMedChem 2016, 11, 2245–2251. [Google Scholar] [CrossRef]
- Capecchi, A.; Awale, M.; Probst, D.; Reymond, J.L. PubChem and ChEMBL beyond Lipinski. Mol. Inform. 2019, 38, 1900016. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]




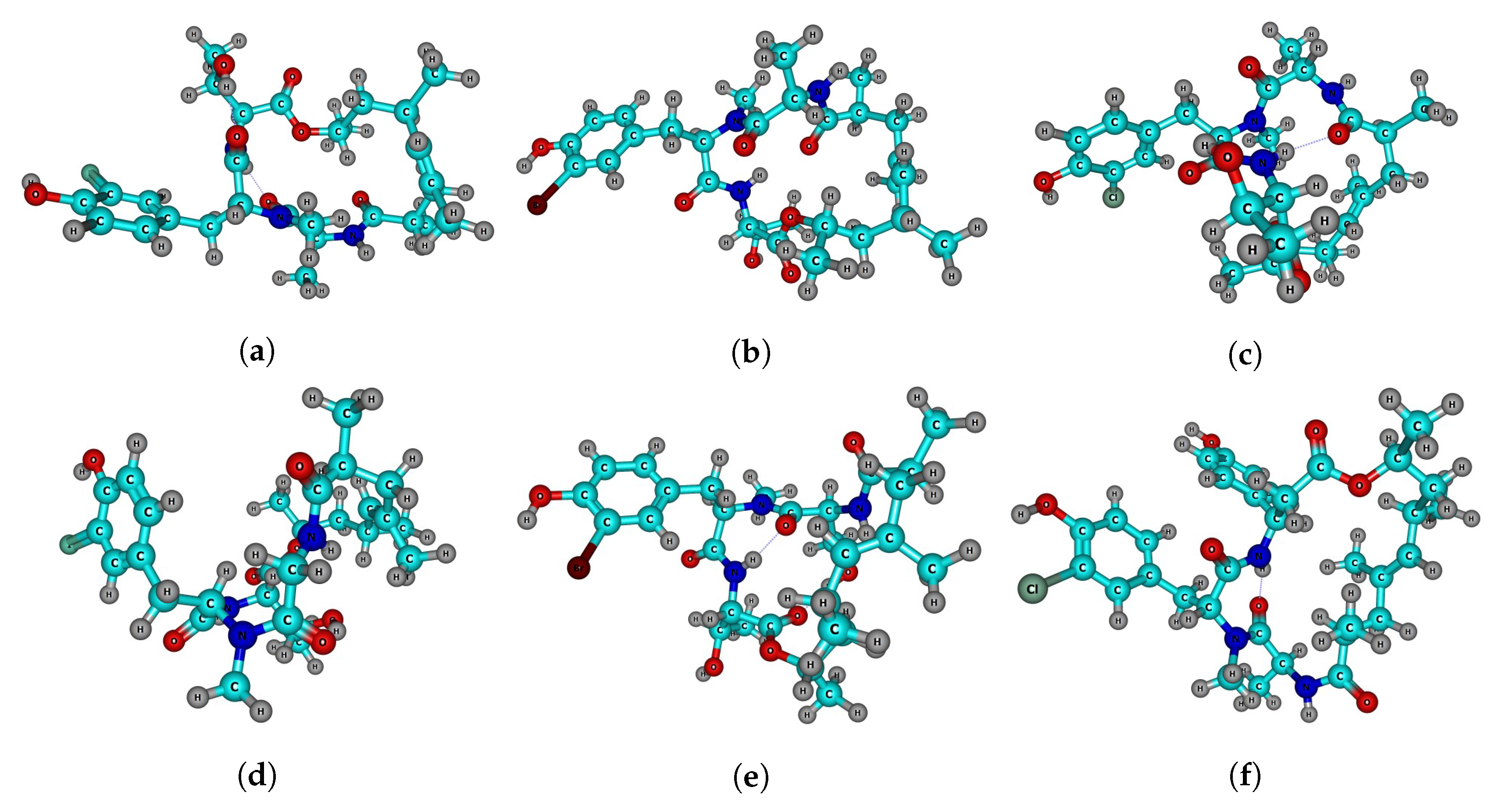{kind=link}
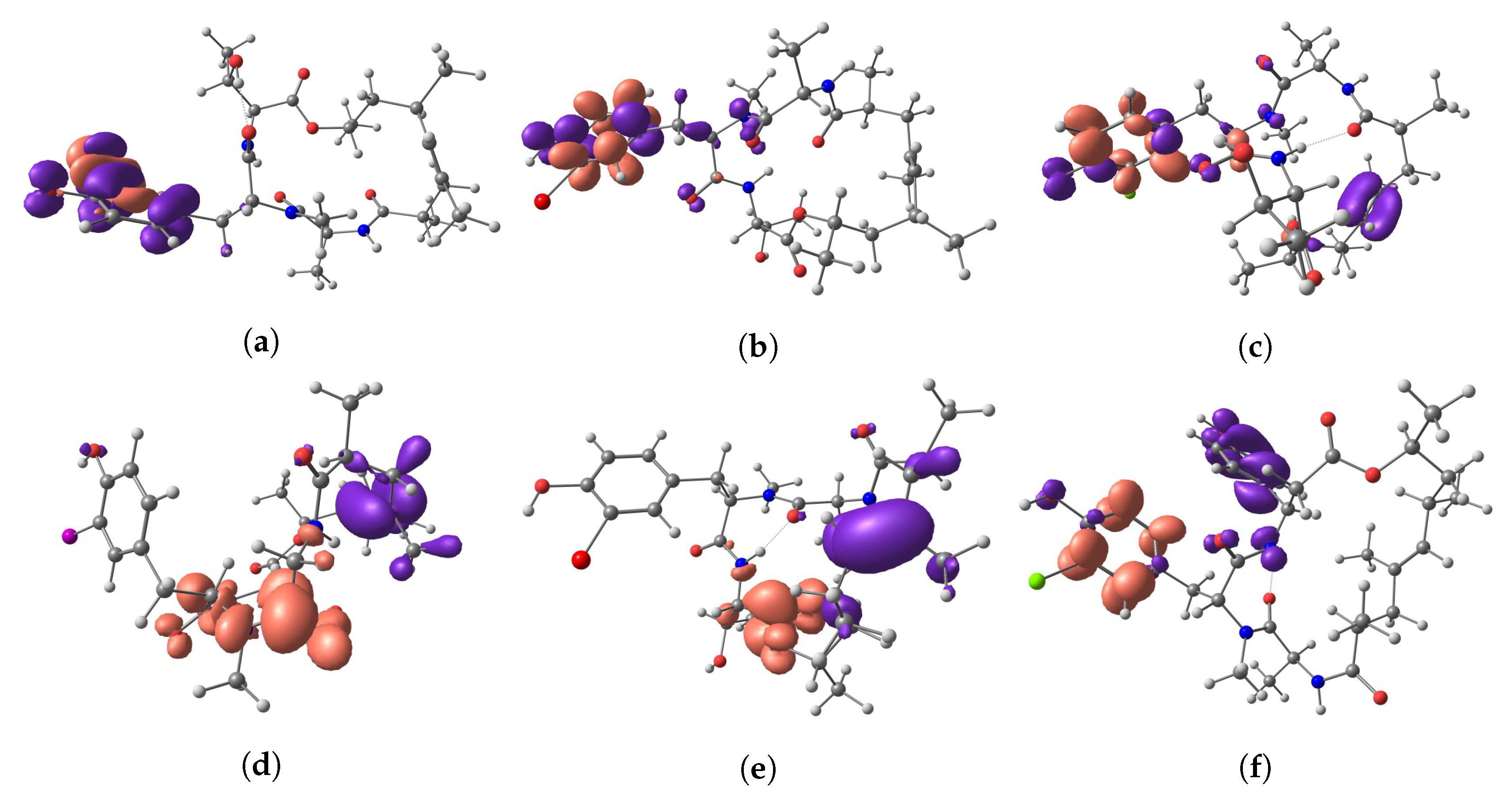{kind=link}
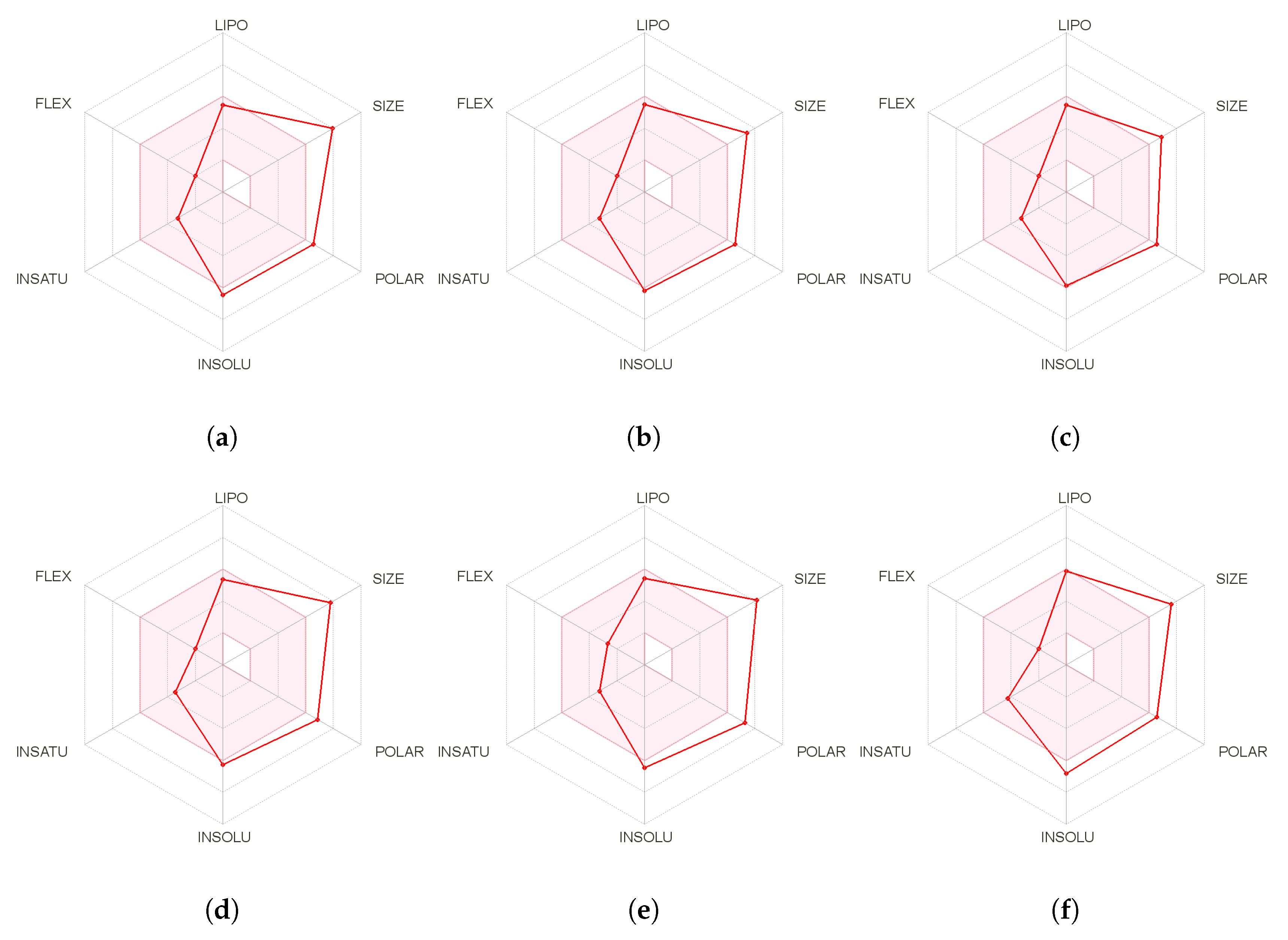{kind=link}
| Molecule | HOMO | LUMO | HOMO-LUMO Gap | |
|---|---|---|---|---|
| Seragamide A | −6.2902 | −0.9587 | 5.3315 | 233 |
| Seragamide B | −6.3789 | −0.7535 | 5.6254 | 220 |
| Seragamide C | −6.3247 | −0.7799 | 5.5449 | 224 |
| Seragamide D | −6.2567 | −1.3241 | 4.9326 | 251 |
| Seragamide E | −6.3422 | −1.0841 | 5.2581 | 236 |
| Seragamide F | −6.3038 | −0.8504 | 5.4534 | 227 |
| Molecule | Electronegativity | Global Hardness | Electrophilicity |
|---|---|---|---|
| Seragamide A | 3.6244 | 5.3315 | 1.2320 |
| Seragamide B | 3.5662 | 5.6254 | 1.1304 |
| Seragamide C | 3.5523 | 5.5449 | 1.1379 |
| Seragamide D | 3.7904 | 4.9326 | 1.4564 |
| Seragamide E | 3.7131 | 5.2581 | 1.3111 |
| Seragamide F | 3.5771 | 5.4534 | 1.1732 |
| Molecule | Electrodonating Power | Electroaccepting Power | Net Electrophilicity |
| Seragamide A | 4.6093 | 0.9849 | 5.5943 |
| Seragamide B | 4.3954 | 0.8293 | 5.2247 |
| Seragamide C | 4.3985 | 0.8462 | 5.2447 |
| Seragamide D | 5.1162 | 1.3528 | 6.4420 |
| Seragamide E | 4.8073 | 1.0942 | 5.9015 |
| Seragamide F | 4.4757 | 0.8986 | 5.3743 |
| Molecule | pKa |
|---|---|
| Seragamide A | 11.90 |
| Seragamide B | 11.66 |
| Seragamide C | 11.72 |
| Seragamide D | 12.23 |
| Seragamide E | 11.96 |
| Seragamide F | 11.80 |
| Molecule | LogP | TPSA | nAtoms | nON | NOHNH |
|---|---|---|---|---|---|
| Seragamide A | 3.69 | 145.27 | 40 | 10 | 4 |
| Seragamide B | 3.42 | 145.27 | 40 | 10 | 4 |
| Seragamide C | 3.29 | 145.27 | 40 | 10 | 4 |
| Seragamide D | 3.36 | 145.27 | 39 | 10 | 4 |
| Seragamide E | 2.44 | 165.49 | 41 | 11 | 5 |
| Seragamide F | 3.73 | 145.27 | 45 | 10 | 4 |
| Molecule | Nviol | Nrotb | Volume | MW | |
| Seragamide A | 1 | 3 | 547.28 | 671.57 | |
| Seragamide B | 1 | 3 | 541.17 | 624.57 | |
| Seragamide C | 1 | 3 | 536.82 | 580.12 | |
| Seragamide D | 1 | 3 | 530.69 | 657.55 | |
| Seragamide E | 2 | 4 | 549.43 | 640.57 | |
| Seragamide F | 1 | 3 | 591.64 | 642.19 |
| Molecule | GPCR Ligand | Ion Channel Modulator | Kinase Inhibitor |
|---|---|---|---|
| Seragamide A | 0.23 | −0.22 | −0.40 |
| Seragamide B | 0.17 | −0.36 | −0.39 |
| Seragamide C | 0.26 | −0.27 | −0.31 |
| Seragamide D | 0.22 | −0.20 | −0.43 |
| Seragamide E | 0.20 | −0.39 | −0.39 |
| Seragamide F | 0.04 | −0.73 | −0.66 |
| Molecule | Nuclear Receptor Ligand | Protease Inhibitor | Enzyme Inhibitor |
| Seragamide A | 0.01 | 0.30 | 0.19 |
| Seragamide B | −0.09 | 0.27 | 0.18 |
| Seragamide C | −0.04 | 0.32 | 0.21 |
| Seragamide D | 0.03 | 0.28 | 0.22 |
| Seragamide E | −0.06 | 0.31 | 0.21 |
| Seragamide F | −0.50 | 0.14 | −0.24 |
| ADME | Seragamide A | Seragamide B | Seragamide C | Seragamide D | Seragamide E | Seragamide F |
|---|---|---|---|---|---|---|
| GI absorption | Low | Low | Low | Low | Low | Low |
| BBB permeant | No | No | No | No | No | No |
| P-gp substrate | Yes | Yes | Yes | Yes | Yes | Yes |
| CYP1A2 inhibitor | No | No | No | No | No | No |
| CYP2C19 inhibitor | No | No | No | No | No | No |
| CYP2C9 inhibitor | No | No | No | No | No | No |
| CYP2D6 inhibitor | No | No | No | No | No | No |
| CYP3A4 inhibitor | Yes | Yes | Yes | Yes | Yes | Yes |
| Log K | −7.52 cm/s | −7.21 cm/s | −6.98 cm/s | −7.56 cm/s | −7.65 cm/s | −6.83 cm/s |
| (skin permeation) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Chemical-Reactivity Properties, Drug Likeness, and Bioactivity Scores of Seragamides A–F Anticancer Marine Peptides: Conceptual Density Functional Theory Viewpoint. Computation 2019, 7, 52. https://doi.org/10.3390/computation7030052
Flores-Holguín N, Frau J, Glossman-Mitnik D. Chemical-Reactivity Properties, Drug Likeness, and Bioactivity Scores of Seragamides A–F Anticancer Marine Peptides: Conceptual Density Functional Theory Viewpoint. Computation. 2019; 7(3):52. https://doi.org/10.3390/computation7030052
Chicago/Turabian StyleFlores-Holguín, Norma, Juan Frau, and Daniel Glossman-Mitnik. 2019. "Chemical-Reactivity Properties, Drug Likeness, and Bioactivity Scores of Seragamides A–F Anticancer Marine Peptides: Conceptual Density Functional Theory Viewpoint" Computation 7, no. 3: 52. https://doi.org/10.3390/computation7030052
APA StyleFlores-Holguín, N., Frau, J., & Glossman-Mitnik, D. (2019). Chemical-Reactivity Properties, Drug Likeness, and Bioactivity Scores of Seragamides A–F Anticancer Marine Peptides: Conceptual Density Functional Theory Viewpoint. Computation, 7(3), 52. https://doi.org/10.3390/computation7030052