
Excitons in Solids from Time-Dependent Density-Functional Theory: Assessing the Tamm-Dancoff Approximation


Abstract
:1. Introduction
2. Theoretical Background
2.1. Dyson Equation
2.2. Casida Equation
2.3. Local-Field Effect
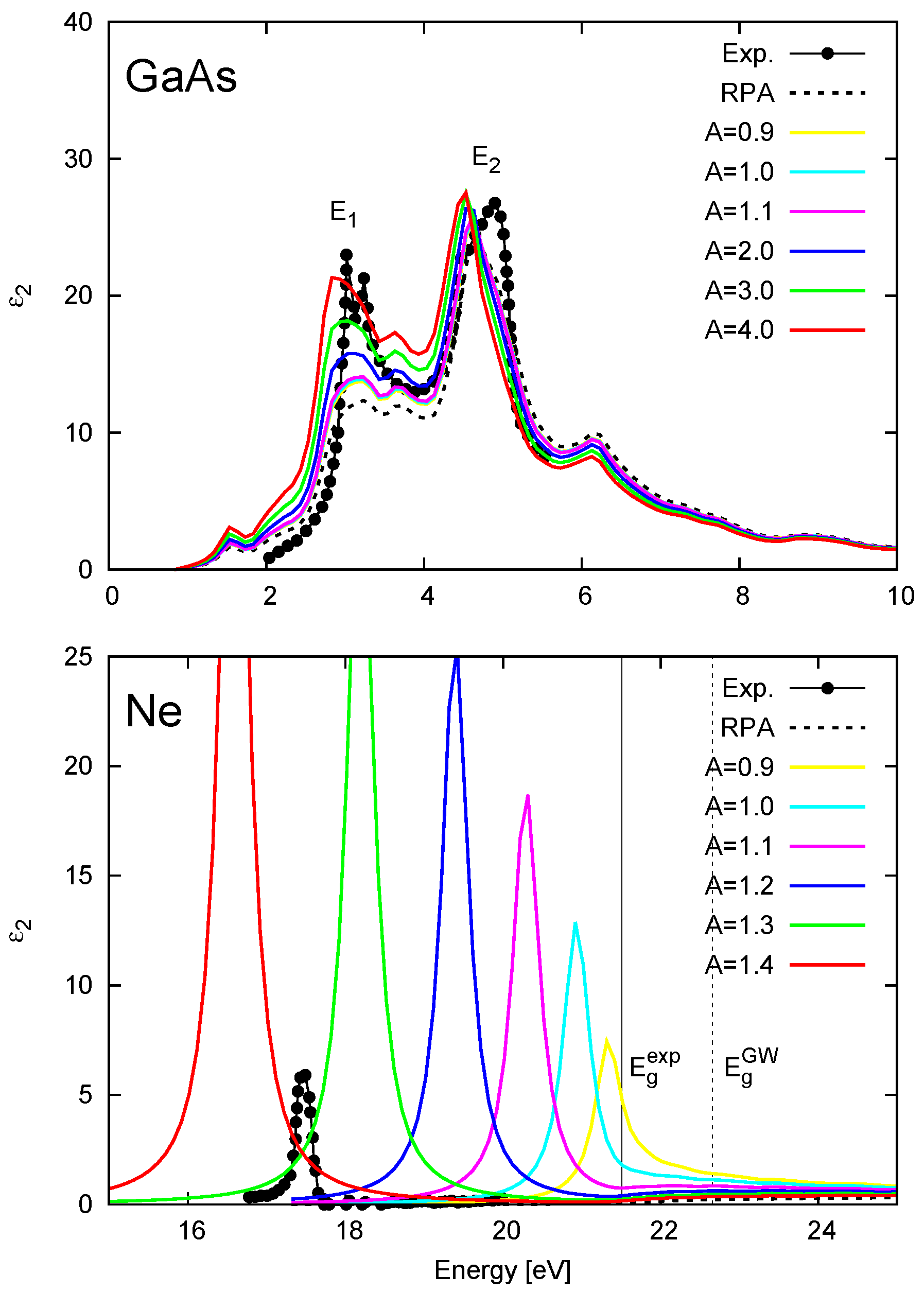
2.4. LRC Kernel: Head-Only vs. Diagonal
2.5. Tamm-Dancoff Approximation
2.6. Band-Gap Corrections: LDA vs. Scissors Shift
3. Computational Details
4. Results and Discussion
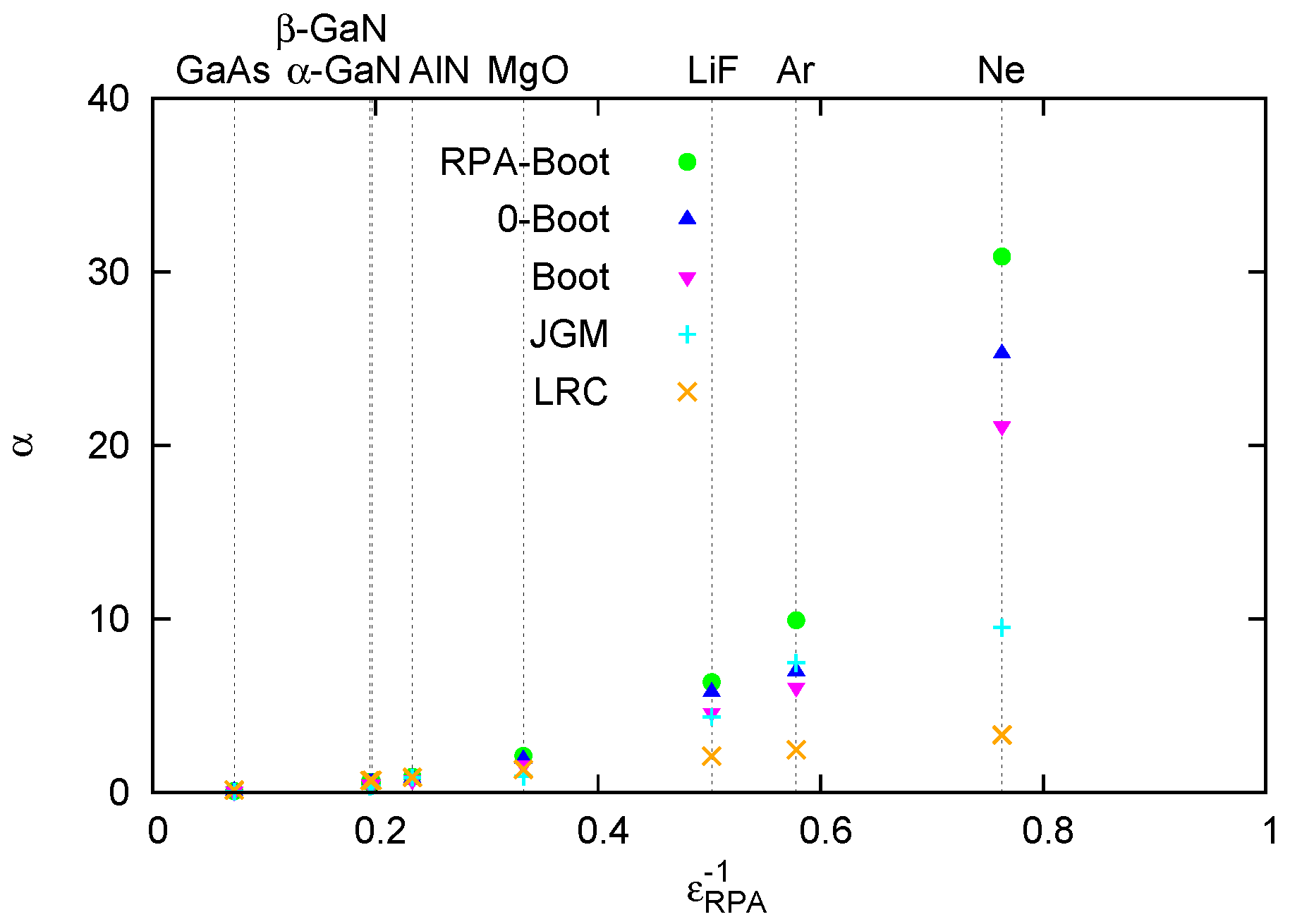
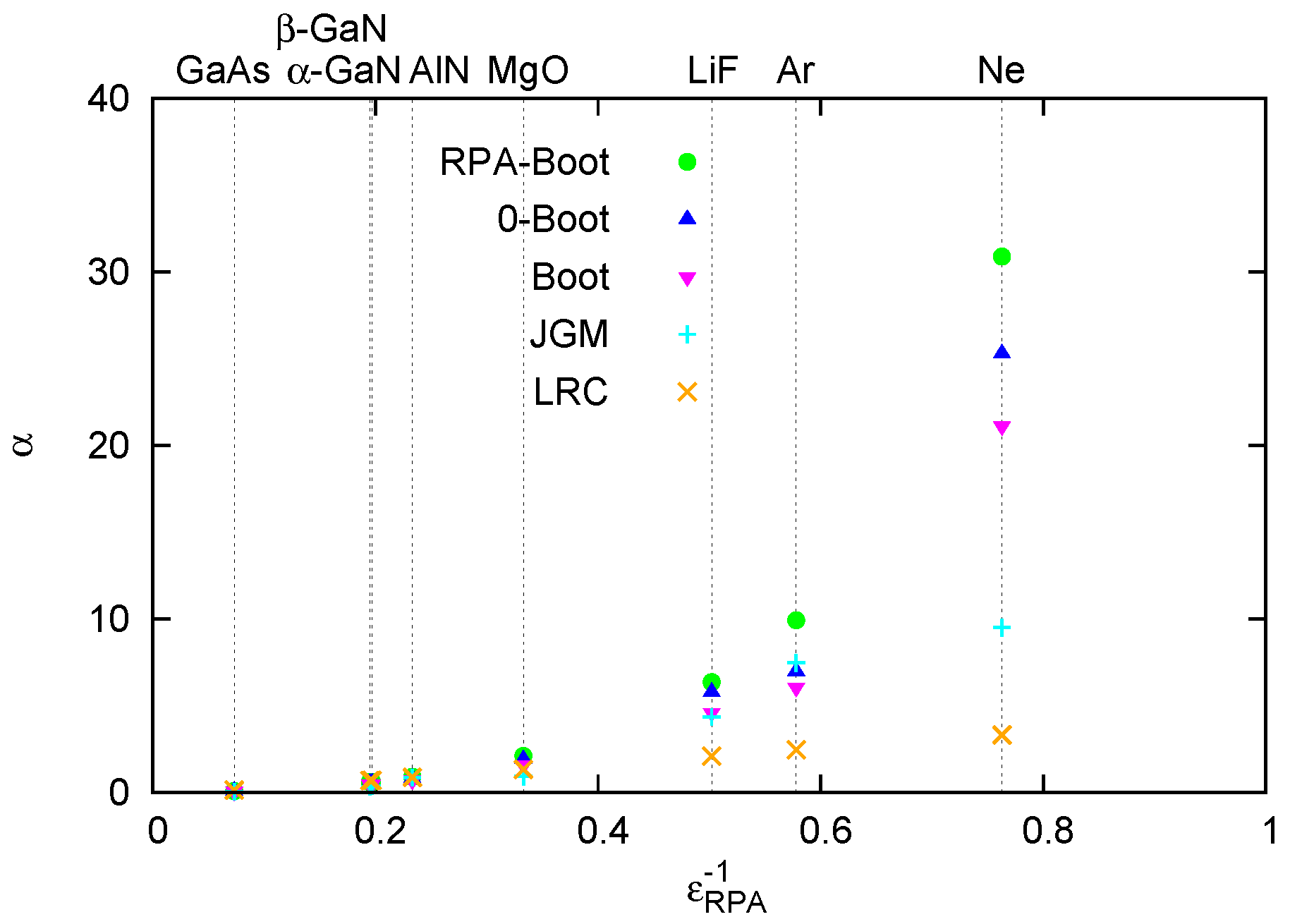
4.1. Overview of LRC-Type Kernels
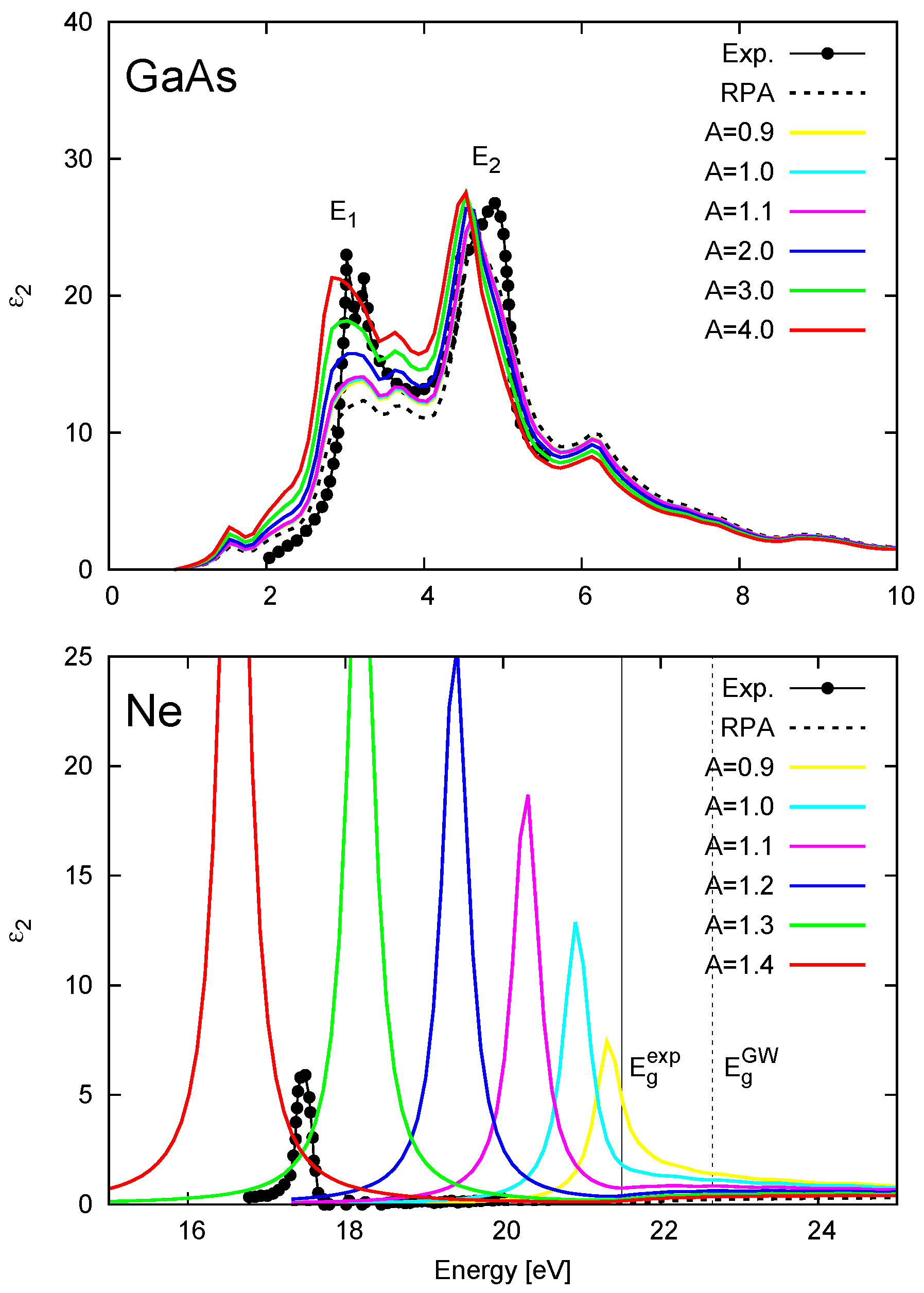
4.2. Effect of the LRC Kernel on Optical Spectra
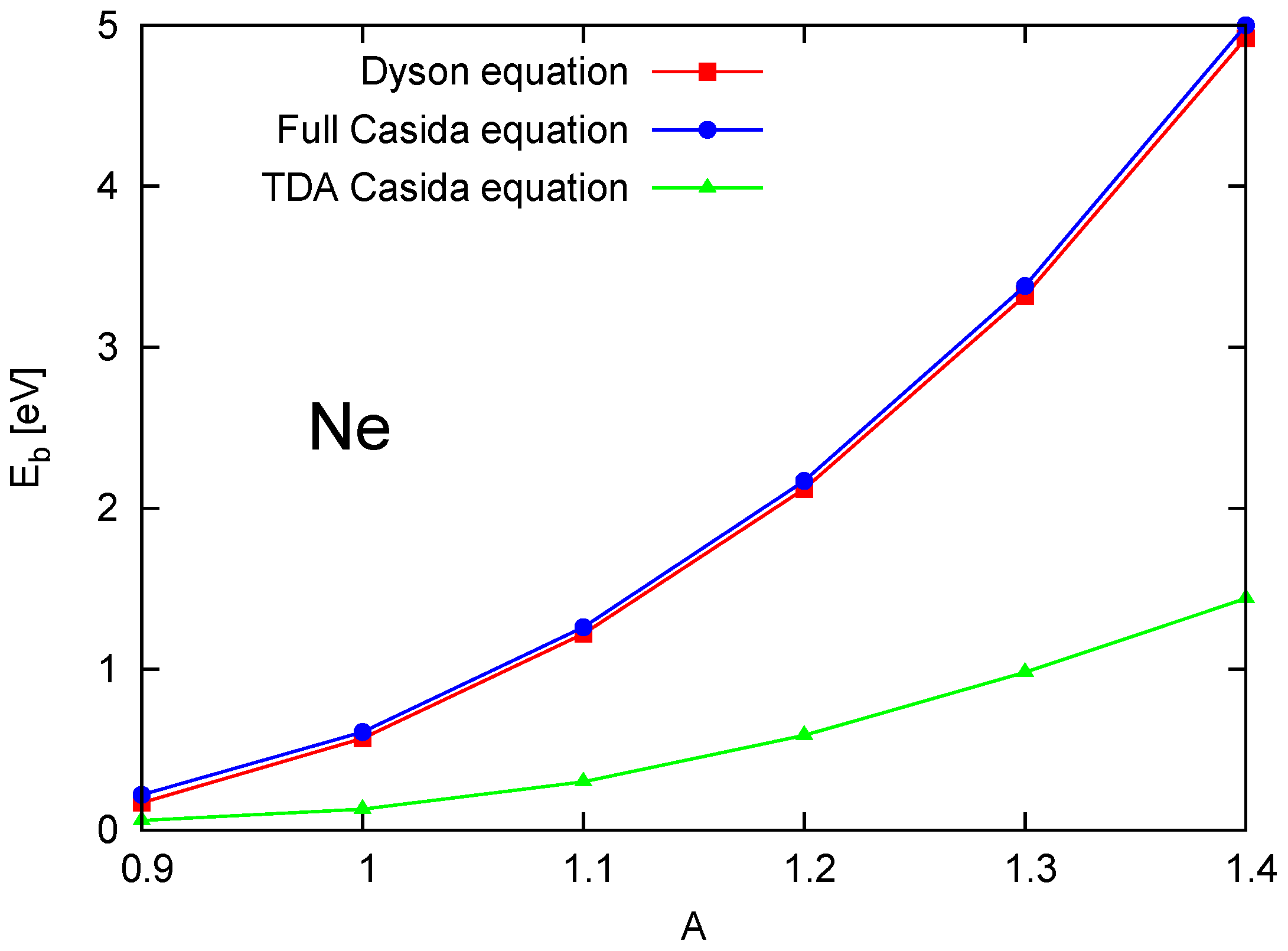
4.3. TDA and Exciton Binding Energies
4.4. Comparison of Dyson and Full Casida Equations
4.5. Limitations of Our Findings
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miyata, A.; Mitioglu, A.; Plochocka, P.; Portugall, O.; Wang, J.T.W.; Stranks, S.D.; Snaith, H.J.; Nicholas, R.J. Direct measurement of the exciton binding energy and effective masses for charge carriers in organic–inorganic tri-halide perovskites. Nat. Phys. 2015, 11, 582–587. [Google Scholar] [CrossRef]
- Onida, G.; Reining, L.; Rubio, A. Electronic excitations: Density-functional versus many-body Green’s-function approaches. Rev. Mod. Phys. 2002, 74, 601. [Google Scholar] [CrossRef]
- Ullrich, C.A.; Yang, Z.H. Excitons in time-dependent density-functional theory. In Density-Functional Methods for Excited States; Ferré, N., Filatov, M., Huix-Rotllant, M., Eds.; Springer: Berlin, Germany, 2015; Volume 368. [Google Scholar]
- Reining, L.; Olevano, V.; Rubio, A.; Onida, G. Excitonic effects in solids described by time-dependent density-functional theory. Phys. Rev. Lett. 2002, 88, 066404. [Google Scholar] [CrossRef] [PubMed]
- Botti, S.; Sottile, F.; Vast, N.; Olevano, V.; Reining, L.; Weissker, H.C.; Rubio, A.; Onida, G.; del Sole, R.; Godby, R.W. Long-range contribution to the exchange-correlation kernel of time-dependent density functional theory. Phys. Rev. B 2004, 69, 155112–155114. [Google Scholar] [CrossRef]
- Sharma, S.; Dewhurst, J.K.; Sanna, A.; Gross, E.K.U. Bootstrap approximation for the exchange-correlation kernel of time-dependent density-functional theory. Phys. Rev. Lett. 2011, 107, 186401. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, S.; Botti, S.; Veniard, V.; Draxl, C.; Reining, L.; Sottile, F. Estimating excitonic effects in the absorption spectra of solids: Problems and insight from a guided iteration scheme. Phys. Rev. Lett. 2015, 114, 146402. [Google Scholar] [CrossRef]
- Berger, J.A. Fully parameter-free calculation of optical spectra for insulators, semiconductors, and metals from a simple polarization functional. Phys. Rev. Lett. 2015, 115, 137402. [Google Scholar] [CrossRef] [PubMed]
- Trevisanutto, P.E.; Terentjevs, A.; Constantin, L.A.; Olevano, V.; Sala, F.D. Optical spectra of solids obtained by time-dependent density functional theory with the jellium-with-gap-model exchange-correlation kernel. Phys. Rev. B 2013, 87, 205143. [Google Scholar] [CrossRef]
- Casida, M.E. Time-dependent density functional response theory for molecules. In Recent Advances in Density Functional Methods; Chong, D.E., Ed.; World Scientific: Singapore, 1995; Volume 1, pp. 155–192. [Google Scholar]
- Yang, Z.H.; Ullrich, C.A. Direct calculation of exciton binding energies with time-dependent density-functional theory. Phys. Rev. B 2013, 87, 195204. [Google Scholar] [CrossRef]
- Yang, Z.H.; Sottile, F.; Ullrich, C.A. A simple screened exact-exchange approach for excitonic properties in solids. Phys. Rev. B 2015, 92, 035202. [Google Scholar] [CrossRef]
- Sander, T.; Maggio, E.; Kresse, G. Beyond the Tamm-Dancoff approximation for extended systems using exact diagonalization. Phys. Rev. B 2015, 92, 045209. [Google Scholar] [CrossRef]
- Shao, M.; da Jornada, F.H.; Yang, C.; Deslippe, J.; Louie, S.G. Structure preserving parallel algorithms for solving the Bethe–Salpeter eigenvalue problem. Linear Algebra Its Appl. 2016, 488, 148–167. [Google Scholar] [CrossRef]
- Dreuw, A.; Head-Gordon, M. Single-reference ab initio methods for the calculation of excited states of large molecules. Chem. Rev. 2005, 105, 4009–4037. [Google Scholar] [CrossRef] [PubMed]
- Petersilka, M.; Gossmann, U.J.; Gross, E.K.U. Excitation energies from time-dependent density-functional theory. Phys. Rev. Lett. 1996, 76, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Casida, M.E.; Gutierrez, F.; Guan, J.; Cadea, F.X.; Salahub, D.; Daudey, J.P. Charge-transfer correction for improved time-dependent local density approximation excited-state potential energy curves: Analysis within the two-level model with illustration for H2 and LiH. J. Chem. Phys. 2000, 113, 7062–7071. [Google Scholar] [CrossRef]
- Baroni, S.; Resta, R. Ab initio calculation of the macroscopic dielectric constant in silicon. Phys. Rev. B 1986, 33, 7017. [Google Scholar] [CrossRef]
- Yang, Z.H.; Li, Y.; Ullrich, C.A. A minimal model for excitons within time-dependent density-functional theory. J. Chem. Phys. 2012, 137, 014513. [Google Scholar] [CrossRef] [PubMed]
- Byun, Y.M.; Ullrich, C.A. Systematic assessment of long-range-corrected exchange-correlation kernels for solids: Accurate excitonic properties via a nonuniformly scaled Bootstrap kernel. Phys. Rev. B 2017. In preparation. [Google Scholar]
- Levine, Z.H.; Allan, D.C. Linear optical response in silicon and germanium including self-energy effects. Phys. Rev. Lett. 1989, 63, 1719. [Google Scholar] [CrossRef] [PubMed]
- Gonze, X.; Amadon, B.; Anglade, P.M.; Beuken, J.M.; Bottin, F.; Boulanger, P.; Bruneval, F.; Caliste, D.; Caracas, R.; Côté, M.; et al. ABINIT: First-principles approach to material and nanosystem properties. Comput. Phys. Commun. 2009, 180, 2582–2615. [Google Scholar] [CrossRef]
- Olevano, V.; Reining, L.; Sottile, F. The Dp Code 1997. Available online: http://www.dp-code.org/ (accessed on 25 January 2017).
- Lautenschlager, P.; Garriga, M.; Logothetidis, S.; Cardona, M. Interband critical points of GaAs and their temperature dependence. Phys. Rev. B 1987, 35, 9174. [Google Scholar] [CrossRef]
- Sonntag, B. Dielectric and optical properties. In Rare Gas Solids; Klein, M.L., Venables, J.A., Eds.; Academic Press: London, UK, 1976; Volume II, p. 1021. [Google Scholar]
- Parenteau, M.; Carlone, C.; Khanna, S.M. Damage coefficient associated with free exciton lifetime in GaAs irradiated with neutrons and electrons. J. Appl. Phys. 1992, 71, 3747. [Google Scholar] [CrossRef]
- As, D.J.; Schmilgus, F.; Wang, C.; Schöttker, B.; Schikora, D.; Lischka, K. The near band edge photoluminescence of cubic GaN epilayers. Appl. Phys. Lett. 1997, 70, 1311. [Google Scholar] [CrossRef]
- Muth, J.F.; Lee, J.H.; Shmagin, I.K.; Kolbas, R.M.; Casey, H.C.; Keller, B.P.; Mishra, U.K.; DenBaars, S.P. Absorption coefficient, energy gap, exciton binding energy, and recombination lifetime of GaN obtained from transmission measurements. Appl. Phys. Lett. 1997, 71, 2572. [Google Scholar] [CrossRef]
- Haensel, R.; Keitel, G.; Koch, E.E.; Skibowski, M.; Schreiber, P. Reflection spectrum of solid argon in the vacuum ultraviolet. Phys. Rev. Lett. 1969, 23, 1160. [Google Scholar] [CrossRef]
- Roessler, D.M.; Walker, W.C. Optical constants of magnesium oxide and lithium fluoride in the far ultraviolet. J. Opt. Soc. Am. 1967, 57, 835–836. [Google Scholar] [CrossRef]
- Saile, V.; Koch, E.E. Bulk and surface excitons in solid neon. Phys. Rev. B 1979, 20, 784. [Google Scholar] [CrossRef]
- Leute, R.A.R.; Feneberg, M.; Sauer, R.; Thonke, K.; Thapa, S.B.; Scholz, F.; Taniyasu, Y.; Kasu, M. Photoluminescence of highly excited AlN: Biexcitons and exciton-exciton scattering. Appl. Phys. Lett. 2009, 95, 031903. [Google Scholar] [CrossRef]
- Roessler, D.M.; Walker, W.C. Electronic Spectrum and Ultraviolet Optical Properties of Crystalline MgO. Phys. Rev. 1967, 159, 733. [Google Scholar] [CrossRef]
- Nazarov, V.U.; Vignale, G. Optics of semiconductors from meta-generalized-gradient approximation based time-dependent density-functional theory. Phys. Rev. Lett. 2011, 107, 216402. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623. [Google Scholar] [CrossRef]
- Bernasconi, L.; Tomić, S.; Ferrero, M.; Rérat, M.; Orlando, R.; Dovesi, R.; Harrison, N.M. First-principles optical response of semiconductors and oxide materials. Phys. Rev. B 2011, 83, 195325. [Google Scholar] [CrossRef]
- Tomić, S.; Bernasconi, L.; Searle, B.G.; Harrison, N.M. Electronic and Optical Structure of Wurtzite CuInS2. J. Phys. Chem. C 2014, 118, 14478–14484. [Google Scholar] [CrossRef]
- Refaely-Abramson, S.; Jain, M.; Sharifzadeh, S.; Neaton, J.B.; Kronik, L. Solid-state optical absorption from optimally tuned time-dependent range-separated hybrid density functional theory. Phys. Rev. B 2015, 92, 081204. [Google Scholar] [CrossRef]
- Ullrich, C.A. Time-Dependent Density-Functional Theory: Concepts and Applications; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Grüning, M.; Marini, A.; Gonze, X. Exciton-plasmon states in nanoscale materials: Breakdown of the Tamm-Dancoff approximation. Nano Lett. 2009, 9, 2820–2824. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Casida Equation | GaAs | α-GaN | β-GaN | AlN | MgO | LiF | Ar | Ne | |
|---|---|---|---|---|---|---|---|---|---|
| Exp. | 3.27 | 20.4 | 26.0 | 48.0 | 80.0 | 1600 | 1900 | 4080 | |
| RPA-Boot | TDA | 0.334 | 0.927 | 0.875 | 0.00 | 1.72 | 33.3 | 37.7 | 666 |
| 0-Boot | TDA | 0.285 | 0.811 | 0.720 | 0.00 | 1.43 | 22.4 | 10.8 | 128 |
| Boot | TDA | 0.267 | 0.651 | 0.562 | 0.00 | 1.03 | 10.7 | 7.70 | 39.7 |
| JGM | TDA | 0.137 | 0.387 | 0.226 | 0.00 | 0.348 | 9.12 | 12.9 | 5.30 |
| LRC | TDA | 0.636 | 1.16 | 1.14 | 0.00 | 0.747 | 1.61 | 1.46 | 1.01 |
| RPA-Boot | Full | 0.344 | 1.06 | 1.01 | 0.00 | 2.12 | 94.7 | 96.0 | 2400 |
| 0-Boot | Full | 0.293 | 0.919 | 0.829 | 0.00 | 1.72 | 43.2 | 13.7 | 612 |
| Boot | Full | 0.278 | 0.735 | 0.649 | 0.00 | 1.20 | 14.8 | 9.14 | 101 |
| JGM | Full | 0.141 | 0.438 | 0.279 | 0.00 | 0.397 | 12.1 | 17.1 | 5.96 |
| LRC | Full | 0.670 | 1.33 | 1.32 | 0.00 | 0.855 | 1.89 | 1.54 | 1.06 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byun, Y.-M.; Ullrich, C.A. Excitons in Solids from Time-Dependent Density-Functional Theory: Assessing the Tamm-Dancoff Approximation. Computation 2017, 5, 9. https://doi.org/10.3390/computation5010009
Byun Y-M, Ullrich CA. Excitons in Solids from Time-Dependent Density-Functional Theory: Assessing the Tamm-Dancoff Approximation. Computation. 2017; 5(1):9. https://doi.org/10.3390/computation5010009
Chicago/Turabian StyleByun, Young-Moo, and Carsten A. Ullrich. 2017. "Excitons in Solids from Time-Dependent Density-Functional Theory: Assessing the Tamm-Dancoff Approximation" Computation 5, no. 1: 9. https://doi.org/10.3390/computation5010009
APA StyleByun, Y.-M., & Ullrich, C. A. (2017). Excitons in Solids from Time-Dependent Density-Functional Theory: Assessing the Tamm-Dancoff Approximation. Computation, 5(1), 9. https://doi.org/10.3390/computation5010009