
Towards TDDFT for Strongly Correlated Materials

Abstract
:1. Introduction
2. The TDDFT + DMFT Formalism
2.1. The Linear Response TDDFT
2.2. Dynamical Mean-Field Theory and the XC Kernel
2.2.1. Single-Electron Green’s Functions
2.2.2. Two-Electron Green’s Functions
3. The DMFT XC Kernel for the One-Band Hubbard Model
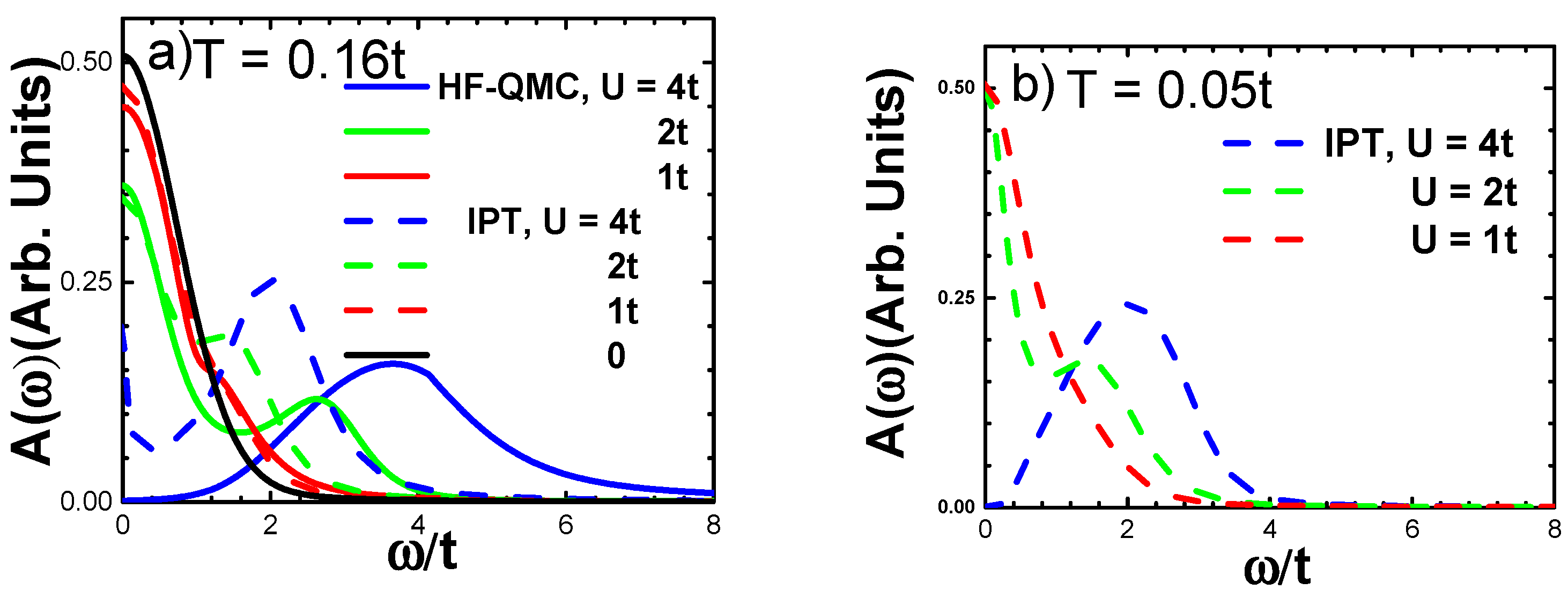
3.1. Density of States
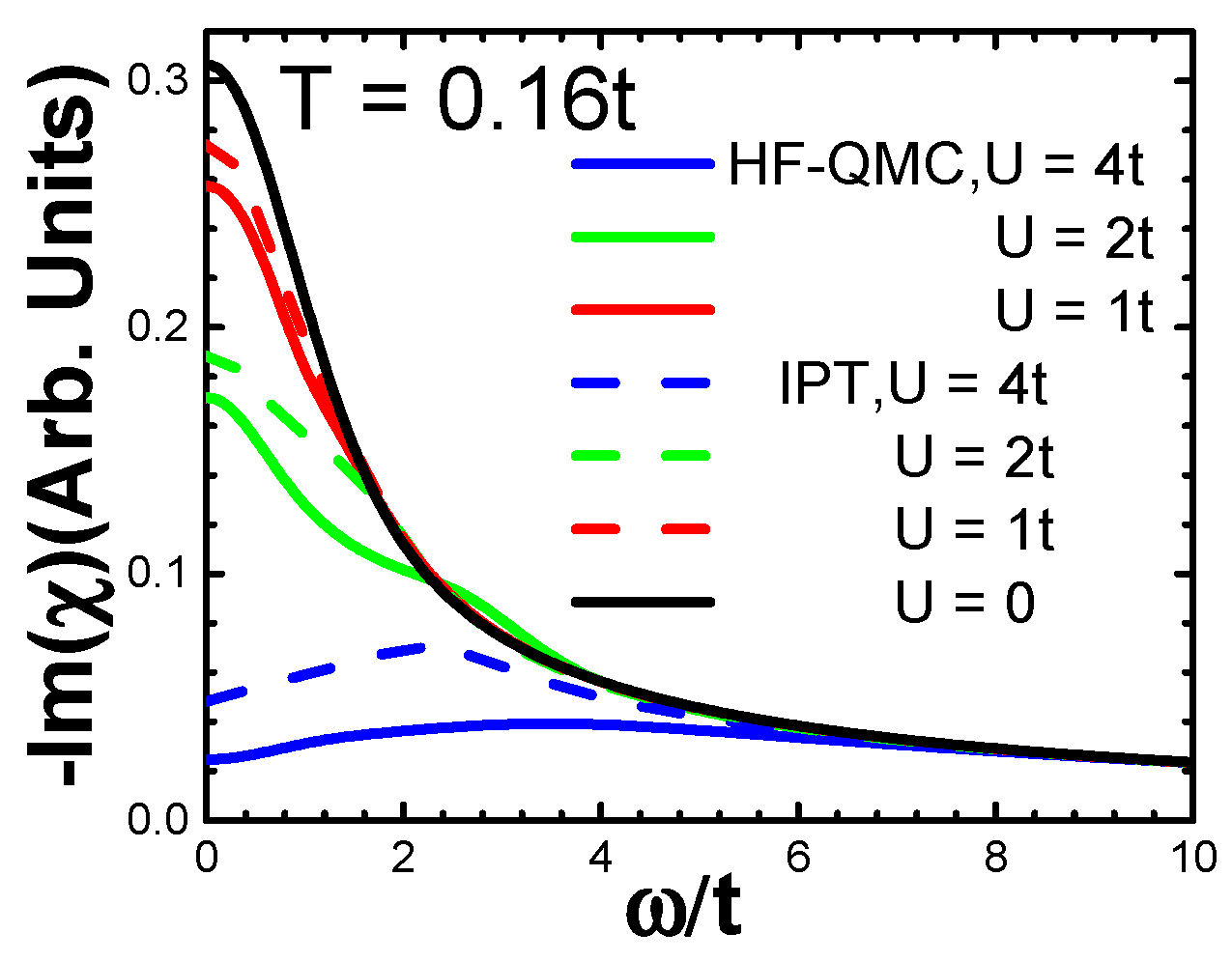
3.2. Charge Susceptibility
3.3. The XC Kernel
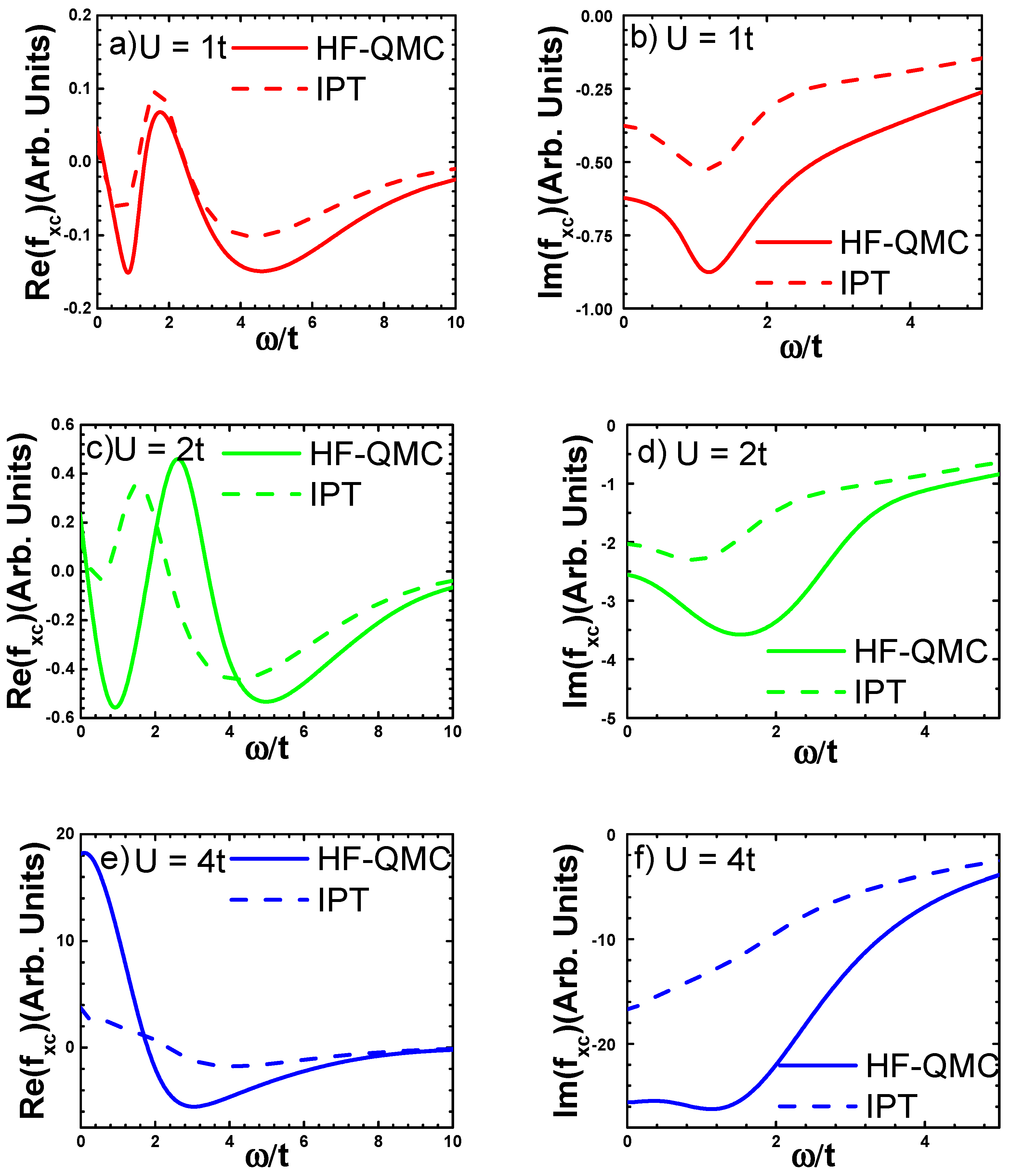
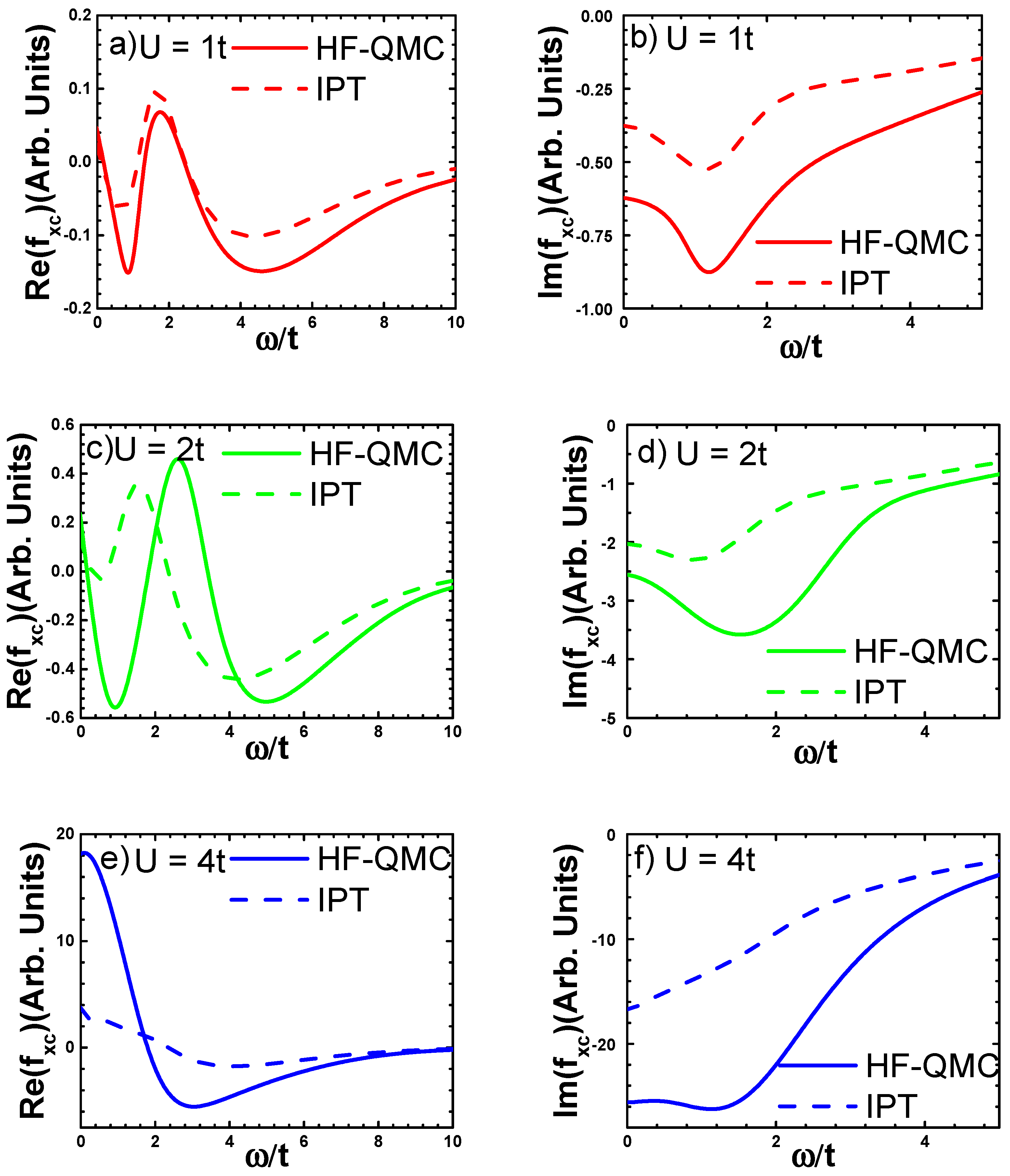
3.3.1. Numerical Results
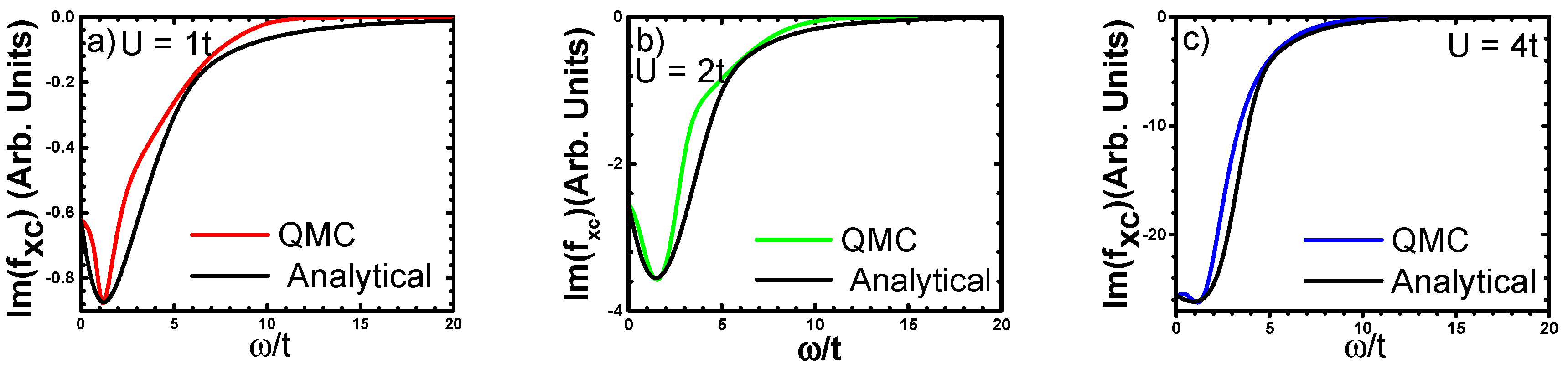
3.3.2. Analytical Fitting
4. Applications: Ultrafast Charge Response
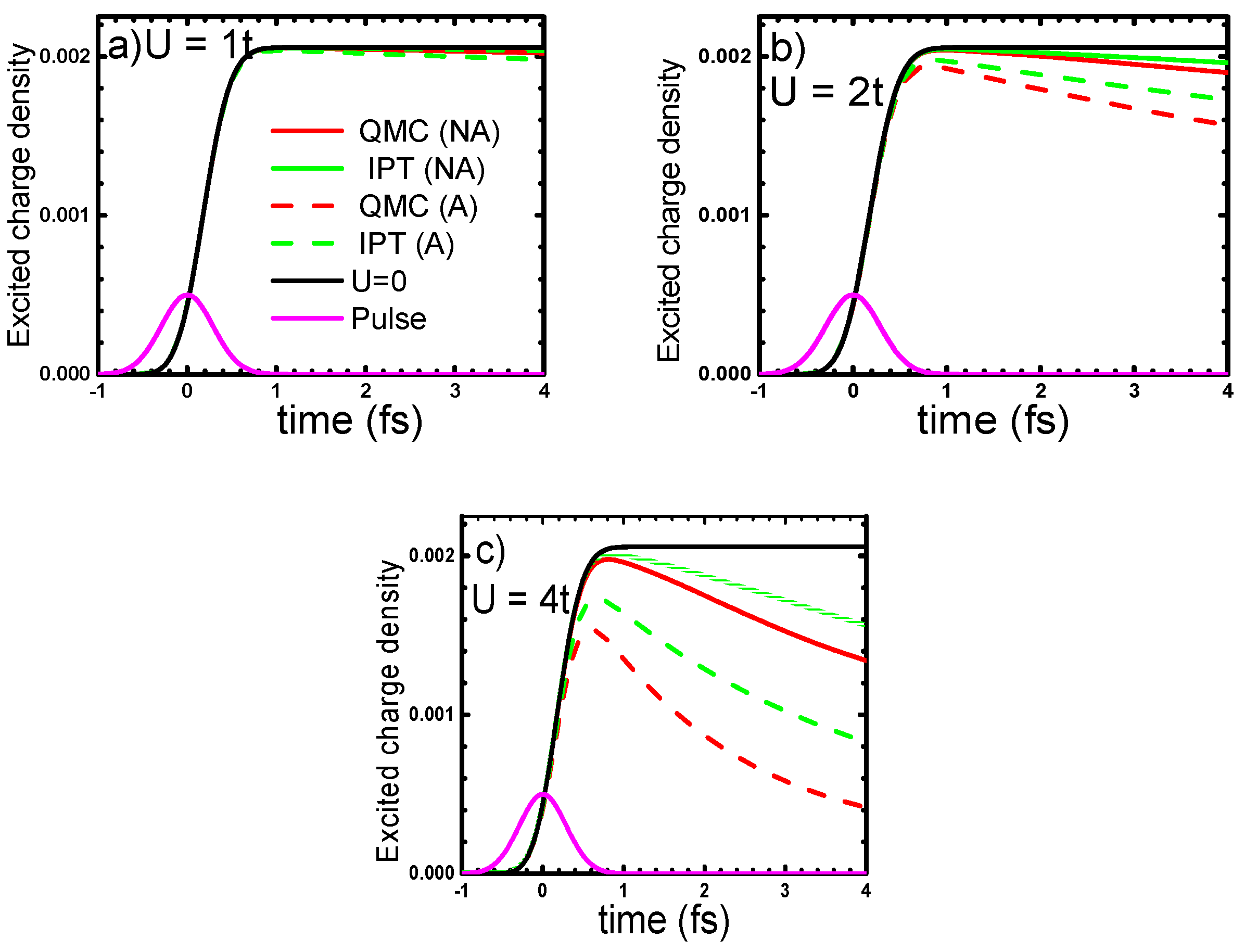
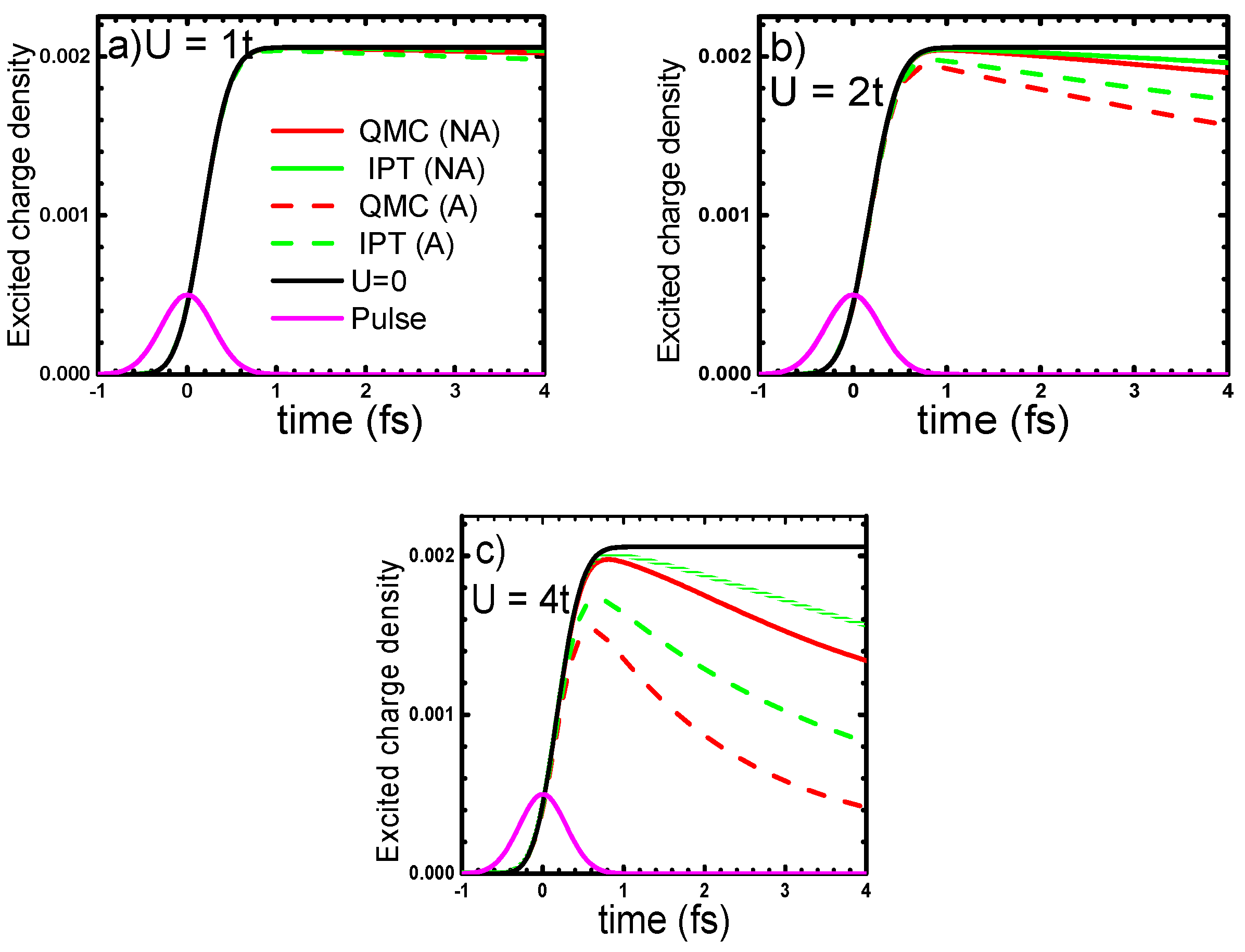
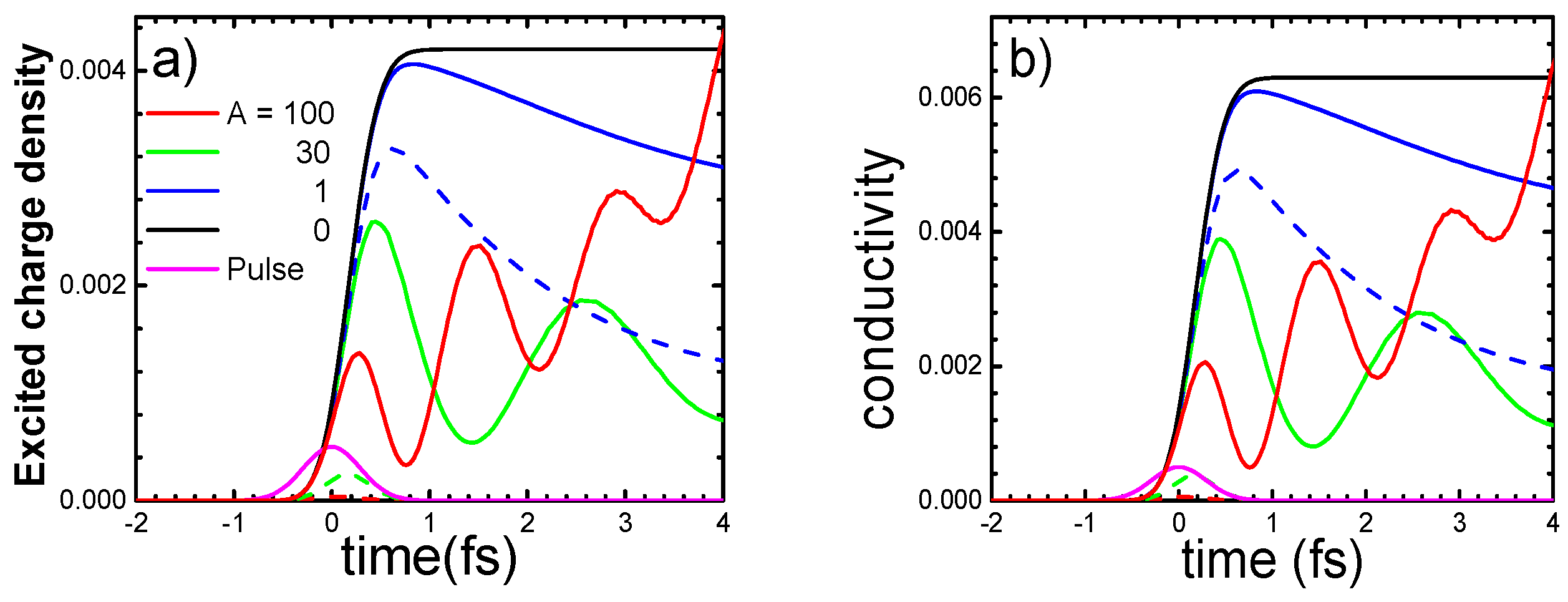
4.1. One-Band Hubbard Model
4.2. Mott Insulator YTiO3
5. The Non-Linear Response: A Possible Extension of the Formalism
6. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| DA | Dynamical Vertex Approximation |
| DMFT | Dynamical Mean-Field Theory |
| DOE | Department of Energy |
| DOS | Density Of States |
| HEG | Homogeneous Electron Gas |
| HF-QMC | Hirsch–Fye Quantum Monte Carlo |
| IPT | Iterative Perturbation Theory |
| KS | Kohn–Sham |
| LDA | Local Density Approximation |
| MEMS | Microelectromechanical Systems |
| RPA | Random Phase Approximation |
| SCMs | Strongly-Correlated Materials |
| TDDFT | Time-Dependent Density-Functional Theory |
| XC | Exchange-Correlation |
Appendix A. Iterative Perturbation Theory Approximation
Appendix B. The Hirsch–Fye Quantum Monte Carlo Scheme
Appendix C. The Density-Matrix TDDFT Formalism
References
- Anisimov, V.; Izyumov, Y. Electronic Structure of Strongly Correlated Materials; Springer: Berlin, Germany; London, UK, 2010. [Google Scholar]
- Loth, S.; Baumann, S.; Lutz, C.P.; Eigler, D.; Heinrich, A.J. Bistability in atomic-scale antiferromagnets. Science 2012, 335, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Shibuya, K.; Okuyama, D.; Hatano, T.; Ono, S.; Kawasaki, M.; Iwasa, Y.; Tokura, Y. Collective bulk carrier delocalization driven by electrostatic surface charge accumulation. Nature 2012, 487, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Tsunekawa, S.; Ishikawa, K.; Li, Z.-Q.; Kawazoe, Y.; Kasuya, A. Origin of anomalous lattice expansion in oxide nanoparticles. Phys. Rev. Lett. 2000, 85, 3440. [Google Scholar] [CrossRef] [PubMed]
- Hailstone, R.; DiFrancesco, A.; Leong, J.; Allston, T.; Reed, K. A study of lattice expansion in CeO2 nanoparticles by transmission electron microscopy. J. Phys. Chem. C 2009, 113, 15155–15159. [Google Scholar] [CrossRef]
- Sundaresan, A.; Bhargavi, R.; Rangarajan, N.; Siddesh, U.; Rao, C. Ferromagnetism as a universal feature of nanoparticles of the otherwise nonmagnetic oxides. Phys. Rev. B 2006, 74, 161306. [Google Scholar] [CrossRef]
- Ao, T.; Ping, Y.; Widmann, K.; Price, D.F.; Lee, E.; Tam, H.; Springer, P.T.; Ng, A. Optical properties in nonequilibrium phase transitions. Phys. Rev. Lett. 2006, 96, 055001. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.; Marvel, R.E.; Conley, H.J.; Miller, K.J.; Haglund Jr, R.F.; Weiss, S.M. Optically monitored electrical switching in VO2. ACS Photonics 2015, 2, 1175–1182. [Google Scholar] [CrossRef]
- Merced, E.; Torres, D.; Tan, X.; Sepúlveda, N. An Electrothermally Actuated VO2-Based MEMS Using Self-Sensing Feedback Control. J. Microelectromec. Syst. 2015, 24, 100–107. [Google Scholar] [CrossRef]
- Nie, G.; Zhang, L.; Lei, J.; Yang, L.; Zhang, Z.; Lu, X.; Wang, C. Monocrystalline VO2 (B) nanobelts: Large-scale synthesis, intrinsic peroxidase-like activity and application in biosensing. J. Mater. Chem. A 2014, 2, 2910–2914. [Google Scholar] [CrossRef]
- Yang, S.; Gong, Y.; Liu, Z.; Zhan, L.; Hashim, D.P.; Ma, L.; Vajtai, R.; Ajayan, P.M. Bottom-up approach toward single-crystalline VO2-graphene ribbons as cathodes for ultrafast lithium storage. Nano Lett. 2013, 13, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Runge, E.; Gross, E.K. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 1984, 52, 997. [Google Scholar] [CrossRef]
- Lieb, E.H.; Wu, F.Y. Absence of Mott transition in an exact solution of the short-range, one-band model in one dimension. Phys. Rev. Lett. 1968, 20, 1445. [Google Scholar] [CrossRef]
- Aryasetiawan, F.; Gunnarsson, O. Exchange-correlation kernel in time-dependent density functional theory. Phys. Rev. B 2002, 66, 165119. [Google Scholar] [CrossRef]
- López-Sandoval, R.; Pastor, G. Properties of the exact correlation-energy functional in Hubbard models. Phase Transit. 2005, 78, 839–850. [Google Scholar] [CrossRef]
- Carrascal, D.; Ferrer, J. Exact Kohn–Sham eigenstates versus quasiparticles in simple models of strongly correlated electrons. Phys. Rev. B 2012, 85, 045110. [Google Scholar] [CrossRef]
- Turkowski, V.; Rahman, T.S. Nonadiabatic time-dependent spin-density functional theory for strongly correlated systems. J. Phys. Condens. Matter 2014, 26, 022201. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fuks, J.I.; Maitra, N.T. Challenging adiabatic time-dependent density functional theory with a Hubbard dimer: The case of time-resolved long-range charge transfer. Phys. Chem. Chem. Phys. 2014, 16, 14504–14513. [Google Scholar] [CrossRef] [PubMed]
- Fuks, J.; Maitra, N. Charge transfer in time-dependent density-functional theory: Insights from the asymmetric Hubbard dimer. Phys. Rev. A 2014, 89, 062502. [Google Scholar] [CrossRef]
- Lani, G.; Di Marino, S.; Gerolin, A.; van Leeuwen, R.; Gori-Giorgi, P. The adiabatic strictly-correlated-electrons functional: Kernel and exact properties. Phys. Chem. Chem. Phys. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Chen, A.-H.; Tokatly, I.V.; Kurth, S. Lattice density functional theory at finite temperature with strongly density-dependent exchange-correlation potentials. Phys. Rev. B 2012, 86, 235139. [Google Scholar]
- Stefanucci, G.; Kurth, S. Towards a description of the Kondo effect using time-dependent density-functional theory. Phys. Rev. Lett. 2011, 107, 216401. [Google Scholar] [CrossRef] [PubMed]
- Uimonen, A.-M.; Khosravi, E.; Stan, A.; Stefanucci, G.; Kurth, S.; van Leeuwen, R.; Gross, E. Comparative study of many-body perturbation theory and time-dependent density functional theory in the out-of-equilibrium Anderson model. Phys. Rev. B 2011, 84, 115103. [Google Scholar] [CrossRef]
- Liu, Z.-F.; Bergfield, J.P.; Burke, K.; Stafford, C.A. Accuracy of density functionals for molecular electronics: The Anderson junction. Phys. Rev. B 2012, 85, 155117. [Google Scholar] [CrossRef]
- Liu, Z.-F.; Burke, K. Density functional description of Coulomb blockade: Adiabatic versus dynamic exchange correlation. Phys. Rev. B 2015, 91, 245158. [Google Scholar] [CrossRef]
- Schönhammer, K.; Gunnarsson, O.; Noack, R. Density-functional theory on a lattice: Comparison with exact numerical results for a model with strongly correlated electrons. Phys. Rev. B 1995, 52, 2504. [Google Scholar] [CrossRef]
- Verdozzi, C. Time-dependent density-functional theory and strongly correlated systems: Insight from numerical studies. Phys. Rev. Lett. 2008, 101, 166401. [Google Scholar] [CrossRef] [PubMed]
- Mancini, L.; Ramsden, J.; Hodgson, M.; Godby, R. Adiabatic and local approximations for the Kohn–Sham potential in time-dependent Hubbard chains. Phys. Rev. B 2014, 89, 195114. [Google Scholar] [CrossRef]
- Capelle, K.; Campo, V.L., Jr. Density functionals and model Hamiltonians: Pillars of many-particle physics. Phys. Rep. 2013, 528. [Google Scholar] [CrossRef]
- Carrascal, D.; Ferrer, J.; Smith, J.C.; Burke, K. The Hubbard dimer: A density functional case study of a many-body problem. J. Phys. Condens. Matter 2015, 27, 393001. [Google Scholar] [CrossRef] [PubMed]
- Lieb, E.H.; Seiringer, R.; Solovej, J.P. Ground-state energy of the low-density Fermi gas. Phys. Rev. A 2005, 71, 053605. [Google Scholar] [CrossRef]
- Giuliani, A. Ground state energy of the low density Hubbard model: An upper bound. J. Math. Phys. 2007, 48, 023302. [Google Scholar] [CrossRef]
- Metzner, W.; Vollhardt, D. Correlated lattice fermions in d=∞ dimensions. Phys. Rev. Lett. 1989, 62, 324. [Google Scholar] [CrossRef] [PubMed]
- Georges, A.; Kotliar, G.; Krauth, W.; Rozenberg, M.J. Dynamical mean-field theory of strongly correlated fermion systems and the limit of infinite dimensions. Rev. Mod. Phys. 1996, 68, 13. [Google Scholar] [CrossRef]
- Anisimov, V.; Poteryaev, A.; Korotin, M.; Anokhin, A.; Kotliar, G. First-principles calculations of the electronic structure and spectra of strongly correlated systems: Dynamical mean-field theory. J. Phys. Condens. Matter 1997, 9, 7359. [Google Scholar] [CrossRef]
- Lichtenstein, A.; Katsnelson, M. Ab initio calculations of quasiparticle band structure in correlated systems: LDA++ approach. Phys. Rev. B 1998, 57, 6884. [Google Scholar] [CrossRef]
- Kotliar, G.; Savrasov, S.Y.; Haule, K.; Oudovenko, V.S.; Parcollet, O.; Marianetti, C. Electronic structure calculations with dynamical mean-field theory. Rev. Mod. Phys. 2006, 78, 865. [Google Scholar] [CrossRef]
- Held, K.; Nekrasov, I.; Keller, G.; Eyert, V.; Blümer, N.; McMahan, A.; Scalettar, R.; Pruschke, T.; Anisimov, V.; Vollhardt, D. Realistic investigations of correlated electron systems with LDA+ DMFT. Phys. Status Solidi (b) 2006, 243, 2599–2631. [Google Scholar] [CrossRef]
- Turkowski, V.; Kabir, A.; Nayyar, N.; Rahman, T.S. A DFT+ DMFT approach for nanosystems. J. Phys.: Condens. Matter 2010, 22, 462202. [Google Scholar] [CrossRef] [PubMed]
- Turkowski, V.; Kabir, A.; Nayyar, N.; Rahman, T.S. Dynamical mean-field theory for molecules and nanostructures. J. Chem. Phys. 2012, 136, 114108. [Google Scholar] [CrossRef] [PubMed]
- Freericks, J.; Turkowski, V.; Zlatić, V. Nonequilibrium dynamical mean-field theory. Phys. Rev. Lett. 2006, 97, 266408. [Google Scholar] [CrossRef] [PubMed]
- Turkowski, V.; Freericks, J.K. Strongly Correlated Systems, Coherence and Entanglement; Carmelo, J.M.P., Lopes dos Santos, J.M.B., Vieira, V.R., Sacramento, P.D., Eds.; World Scientific: Singapore, 2007; p. 187. [Google Scholar]
- Aoki, H.; Tsuji, N.; Eckstein, M.; Kollar, M.; Oka, T.; Werner, P. Nonequilibrium dynamical mean-field theory and its applications. Rev. Mod. Phys. 2014, 86, 779. [Google Scholar] [CrossRef]
- Karlsson, D.; Privitera, A.; Verdozzi, C. Time-dependent density-functional theory meets dynamical mean-field theory: Real-time dynamics for the 3d Hubbard model. Phys. Rev. Lett. 2011, 106, 116401. [Google Scholar] [CrossRef] [PubMed]
- Turkowski, V.; Rahman, T.S. Nonadiabatic exchange-correlation kernel for strongly correlated materials. ArXiv Preprint 2014. [Google Scholar]
- Ullrich, C.A. Time-Dependent Density-Functional Theory: Concepts and Applications; Oxford University Press: Oxford, NY, USA, 2012. [Google Scholar]
- Toschi, A.; Katanin, A.; Held, K. Dynamical vertex approximation: A step beyond dynamical mean-field theory. Phys. Rev. B 2007, 75, 045118. [Google Scholar] [CrossRef]
- Rohringer, G.; Valli, A.; Toschi, A. Local electronic correlation at the two-particle level. Phys. Rev. B 2012, 86, 125114. [Google Scholar] [CrossRef]
- Gross, E.; Kohn, W. Local density-functional theory of frequency-dependent linear response. Phys. Rev. Lett. 1985, 55, 2850. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, N.; Gross, E. Correlation effects on the third-frequency-moment sum rule of electron liquids. Phys. Rev. B 1987, 35, 3003. [Google Scholar] [CrossRef]
- Wegkamp, D.; Stähler, J. Ultrafast dynamics during the photoinduced phase transition in VO2. Prog. Surf. Sci. 2015, 90, 464–502. [Google Scholar] [CrossRef]
- He, Z.; Millis, A.J. Photoinduced phase transitions in narrow-gap Mott insulators: The case of VO2. Phys. Rev. B 2016, 93, 115126. [Google Scholar] [CrossRef]
- Freericks, J.K.; Turkowski, V.M.; Zlatić, V. F-electron spectral function of the Falicov-Kimball model in infinite dimensions: the half-filled case. Phys. Rev. B 2005, 71, 115111. [Google Scholar] [CrossRef]
- Pavarini, E.; Yamasaki, A.; Nuss, J.; Andersen, O. How chemistry controls electron localization in 3d1 perovskites: A Wannier-function study. New J. Phys. 2005, 7, 188. [Google Scholar] [CrossRef]
- Pavarini, E.; Biermann, S.; Poteryaev, A.; Lichtenstein, A.; Georges, A.; Andersen, O. Mott transition and suppression of orbital fluctuations in orthorhombic 3d1 perovskites. Phys. Rev. Lett. 2004, 92, 176403. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Sato, H.; Higashi, M.; Yoshikawa, K.; Shimada, K.; Nakatake, M.; Ueda, Y.; Namatame, H.; Taniguchi, M.; Tsubota, M. Unoccupied electronic structure of Y1−xCaxTiO3 investigated by inverse photoemission spectroscopy. Phys. Rev. B 2007, 75, 205124. [Google Scholar] [CrossRef]
- Bruggemann, D.A.G. Berechnung verschiedener physikalischer Konstanten von heterogenen Substanzen. I. Dielektrizitätskonstanten und Leitfähigkeiten der Mischkörper aus isotropen Substanzen. Ann. Phys. (Leipzig) 1935, 24, 636. [Google Scholar] [CrossRef]
- Van Leeuwen, R. The Sham-Schlüter equation in time-dependent density-functional theory. Phys. Rev. Lett. 1996, 76, 3610. [Google Scholar] [CrossRef] [PubMed]
- Sham, L.; Schlüter, M. Density-functional theory of the energy gap. Phys. Rev. Lett. 1983, 51, 1888. [Google Scholar] [CrossRef]
- Sham, L. Exchange and correlation in density-functional theory. Phys. Rev. B 1985, 32, 3876. [Google Scholar] [CrossRef]
- Turkowski, V.M.; Freericks, J.K. Spectral moment sum rules for strongly correlated electrons in time-dependent electric fields. Phys. Rev. B 2006, 73, 075108, Erratum: Phys. Rev. B 2006, 73, 209902. [Google Scholar] [CrossRef]
- Turkowski, V.M.; Freericks, J.K. Nonequilibrium sum rules for the retarded self-energy of strongly correlated electrons. Phys. Rev. B 2008, 77, 205102, Erratum: Phys. Rev. B 2010, 82, 119904. [Google Scholar] [CrossRef]
- Craco, L.; Laad, M.; Müller-Hartmann, E. Orbital Kondo Effect in CrO2: A Combined Local-Spin-Density-Approximation Dynamical-Mean-Field-Theory Study. Phys. Rev. Lett. 2003, 90, 237203. [Google Scholar] [CrossRef] [PubMed]
- Wernsdorfer, J.; Harder, G.; Schollwoeck, U.; Hofstetter, W. Signatures of delocalization in the fermionic 1d Hubbard model with box disorder: Comparative study with DMRG and R-DMFT. ArXiv Preprint 2011. [Google Scholar]
- Semmler, D.; Byczuk, K.; Hofstetter, W. Anderson-Hubbard model with box disorder: Statistical dynamical mean-field theory investigation. Phys. Rev. B 2011, 84, 115113. [Google Scholar] [CrossRef]
- Hirsch, J.E.; Fye, R.M. Monte Carlo method for magnetic impurities in metals. Phys. Rev. Lett. 1986, 56, 2521. [Google Scholar] [CrossRef] [PubMed]
- Turkowski, V.; Ullrich, C.A. Time-dependent density-functional theory for ultrafast interband excitations. Phys. Rev. B 2008, 77, 075204. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| U/t | a | Imfxc(0) | b |
|---|---|---|---|
| 1 | −0.40 | 1.56 | 0.14 |
| 2 | −1.32 | 1.93 | 0.09 |
| 4 | −1.14 | 22.42 | 0.01 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acharya, S.R.; Turkowski, V.; Rahman, T.S. Towards TDDFT for Strongly Correlated Materials. Computation 2016, 4, 34. https://doi.org/10.3390/computation4030034
Acharya SR, Turkowski V, Rahman TS. Towards TDDFT for Strongly Correlated Materials. Computation. 2016; 4(3):34. https://doi.org/10.3390/computation4030034
Chicago/Turabian StyleAcharya, Shree Ram, Volodymyr Turkowski, and Talat S. Rahman. 2016. "Towards TDDFT for Strongly Correlated Materials" Computation 4, no. 3: 34. https://doi.org/10.3390/computation4030034
APA StyleAcharya, S. R., Turkowski, V., & Rahman, T. S. (2016). Towards TDDFT for Strongly Correlated Materials. Computation, 4(3), 34. https://doi.org/10.3390/computation4030034