Localized Polycentric Orbital Basis Set for Quantum Monte Carlo Calculations Derived from the Decomposition of Kohn-Sham Optimized Orbitals

Abstract
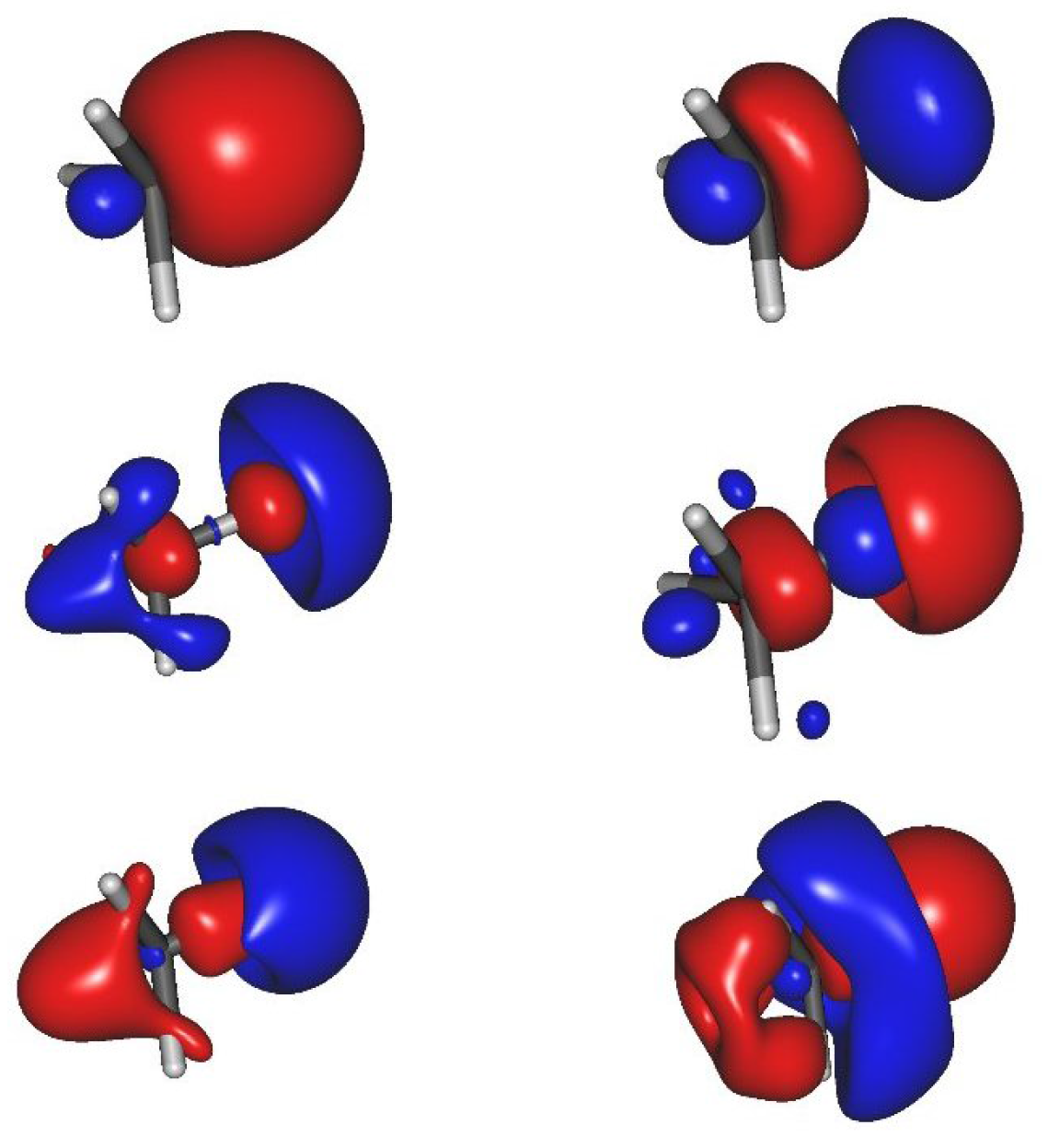
: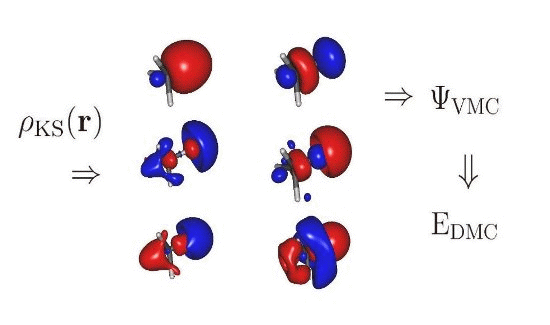
1. Introduction
2. Theory
3. Results and Discussion
3.1. Computational Details
3.2. Fragmentation of Hydrazine

| J-LGVBn | VMC [13] | VMC (This Work) | DMC [13] | DMC (This Work) |
|---|---|---|---|---|
| 0 | −22.2438 | −22.2406 | −22.2724 | –22.2721 |
| 1 | −22.2538 | −22.2509 | −22.2762 | –22.2755 |
| 2 | −22.2565 | −22.2534 | −22.2777 | –22.2771 |
| J-LGVBn | VMC [13] | DMC [13] | VMC (This Work) | DMC (This Work) | Exp [27] |
|---|---|---|---|---|---|
| 0 | 67.3 | 70.8 | 65.4 | 71.6 | – |
| 1 | 69.5 | 71.8 | 69.3 | 72.3 | – |
| 2 | 70.8 | 72.4 | 70.3 | 72.9 | – |
| 10 | 71.1 | 72.8 | – | – | – |
| exp | – | – | – | – | 73.39 |
3.3. Barrier Height in a Prototypical Hydrogen Transfer Reaction
| Forward Reaction Barrier Height | |||
|---|---|---|---|
| Method | DMC [31] | DMC (This Work) | Literature |
| J-LGVB0 | 15.3 | 14.8 | – |
| J-LGVB1 | 14.9 | 14.5 | – |
| J-LGVB2 | 14.6 | 14.3 | – |
| CCSD(T)/aug-cc-pvQZ [31] | – | – | 14.19 |
| DFT(MPWB1K) [30,32] | – | – | 13.15 |
| best reference [29,33] | – | – | 12.7 |
| Reverse Reaction Barrier Height | |||
| J-LGVB0 | 5.2 | 4.9 | – |
| J-LGVB1 | 4.7 | 4.7 | – |
| J-LGVB2 | 4.6 | 4.7 | – |
| CCSD(T)/aug-cc-pvQZ [31] | – | – | 3.86 |
| DFT(MPWB1K) [30,32] | – | – | 5.04 |
| reference [29,33] | – | – | 3.2 |
| Reaction Energy | |||
| J-LGVB0 | 10.1 | 9.9 | – |
| J-LGVB1 | 10.2 | 9.8 | – |
| J-LGVB2 | 10.0 | 9.6 | – |
| CCSD(T)/aug-cc-pvQZ [31] | – | – | 10.33 |
| DFT(MPWB1K) [30,32] | – | – | 8.12 |
| reference [29,33] | – | – | 9.5 |
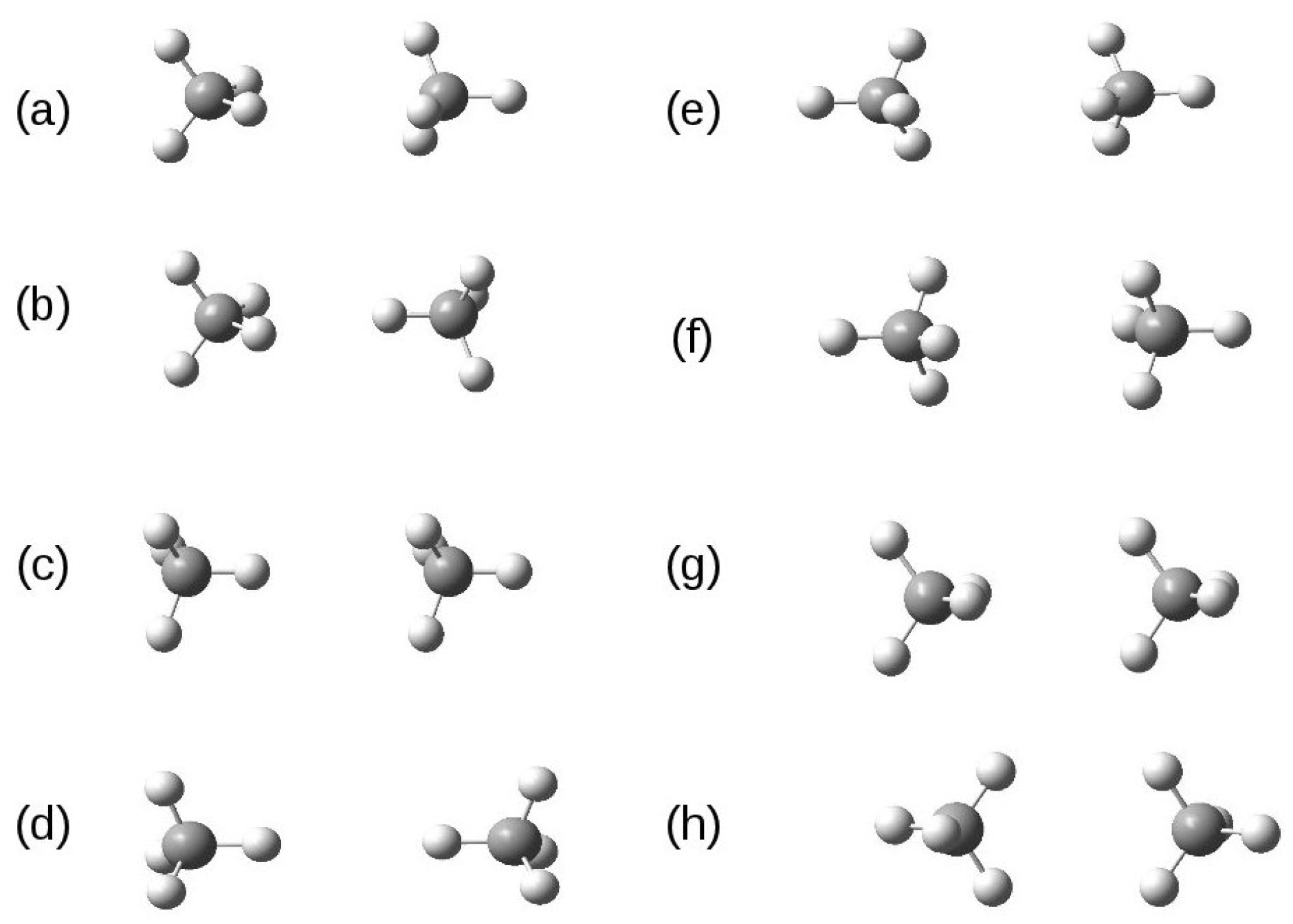
3.4. Methane Dimer Intermolecular Potential
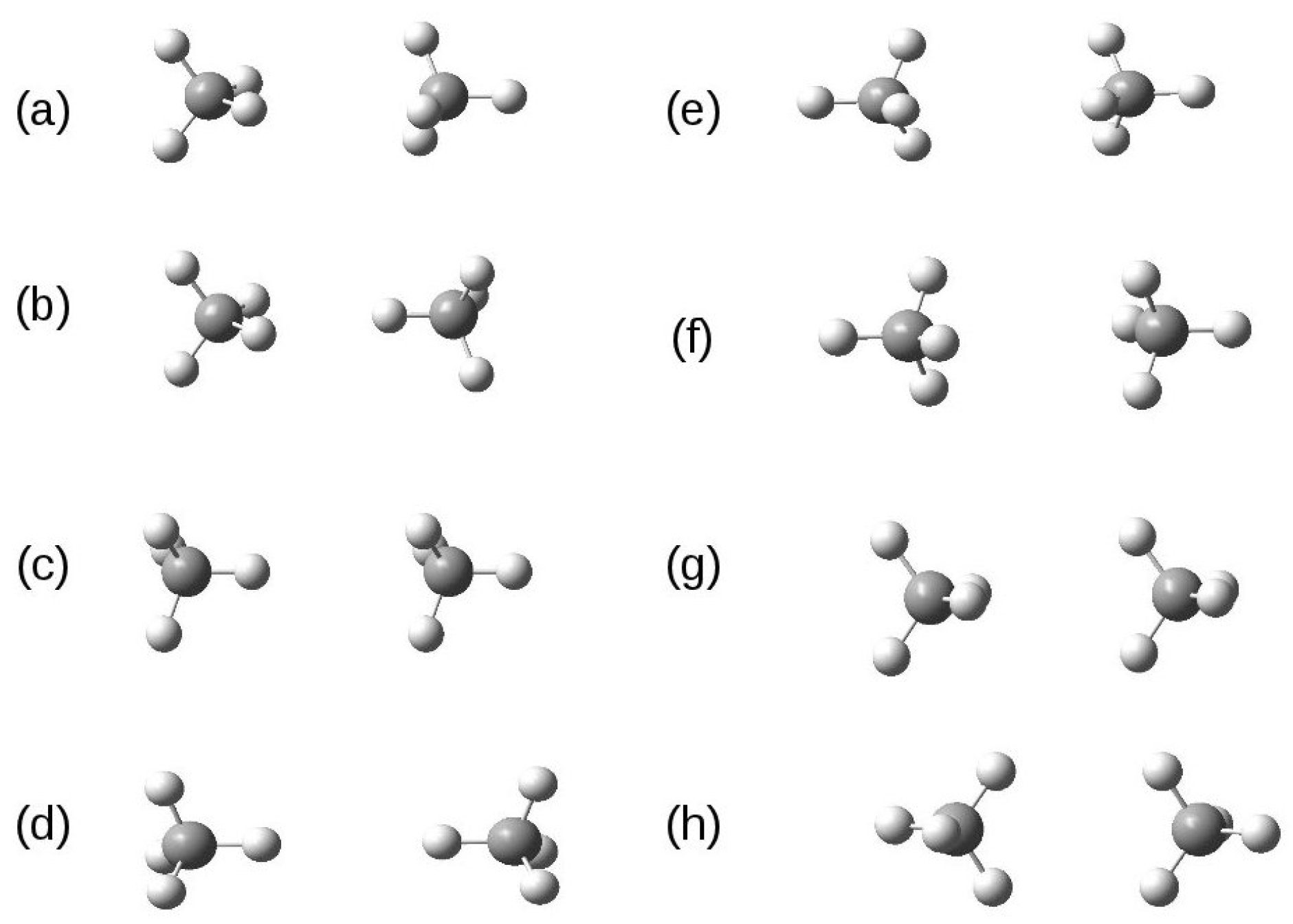
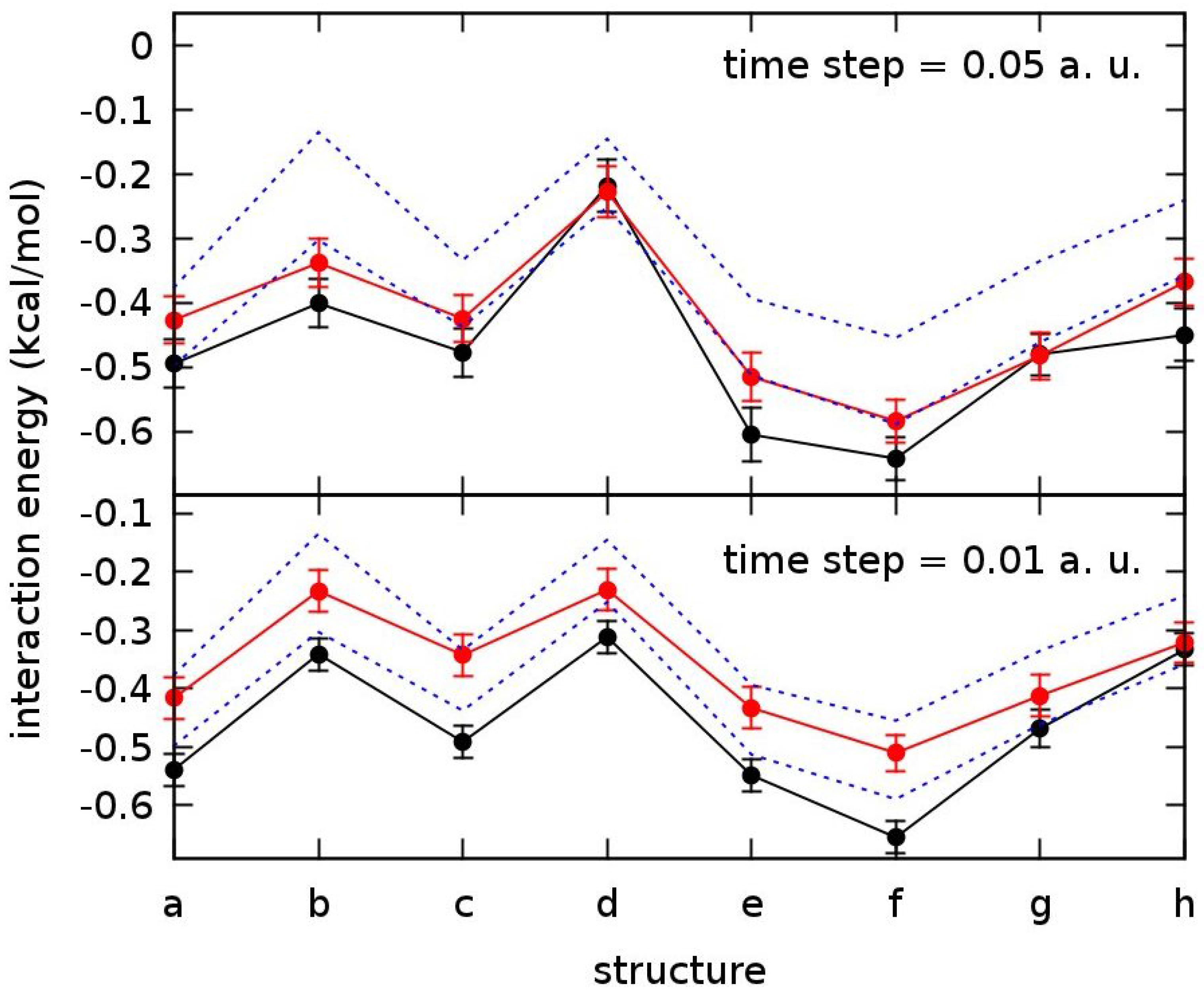
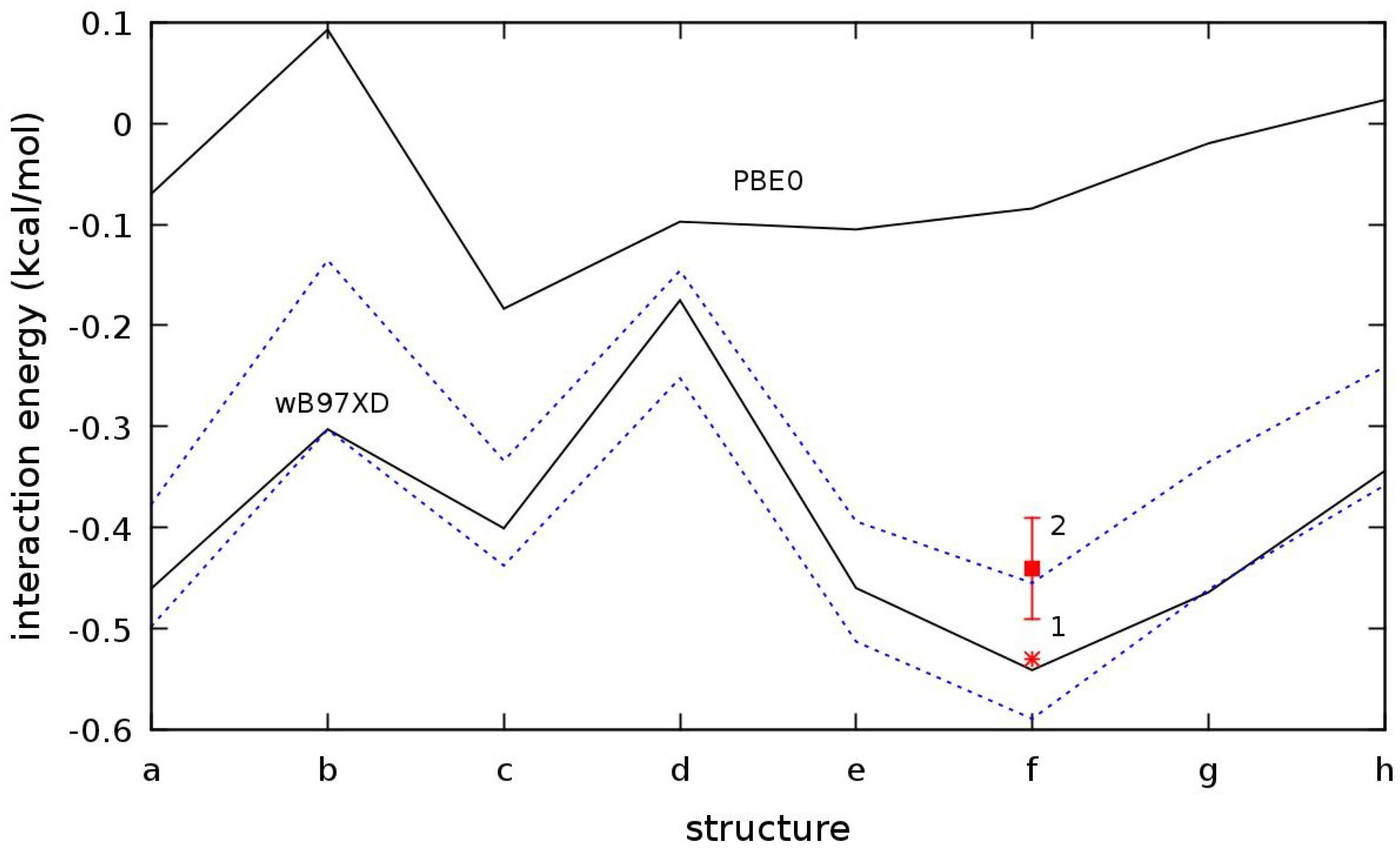
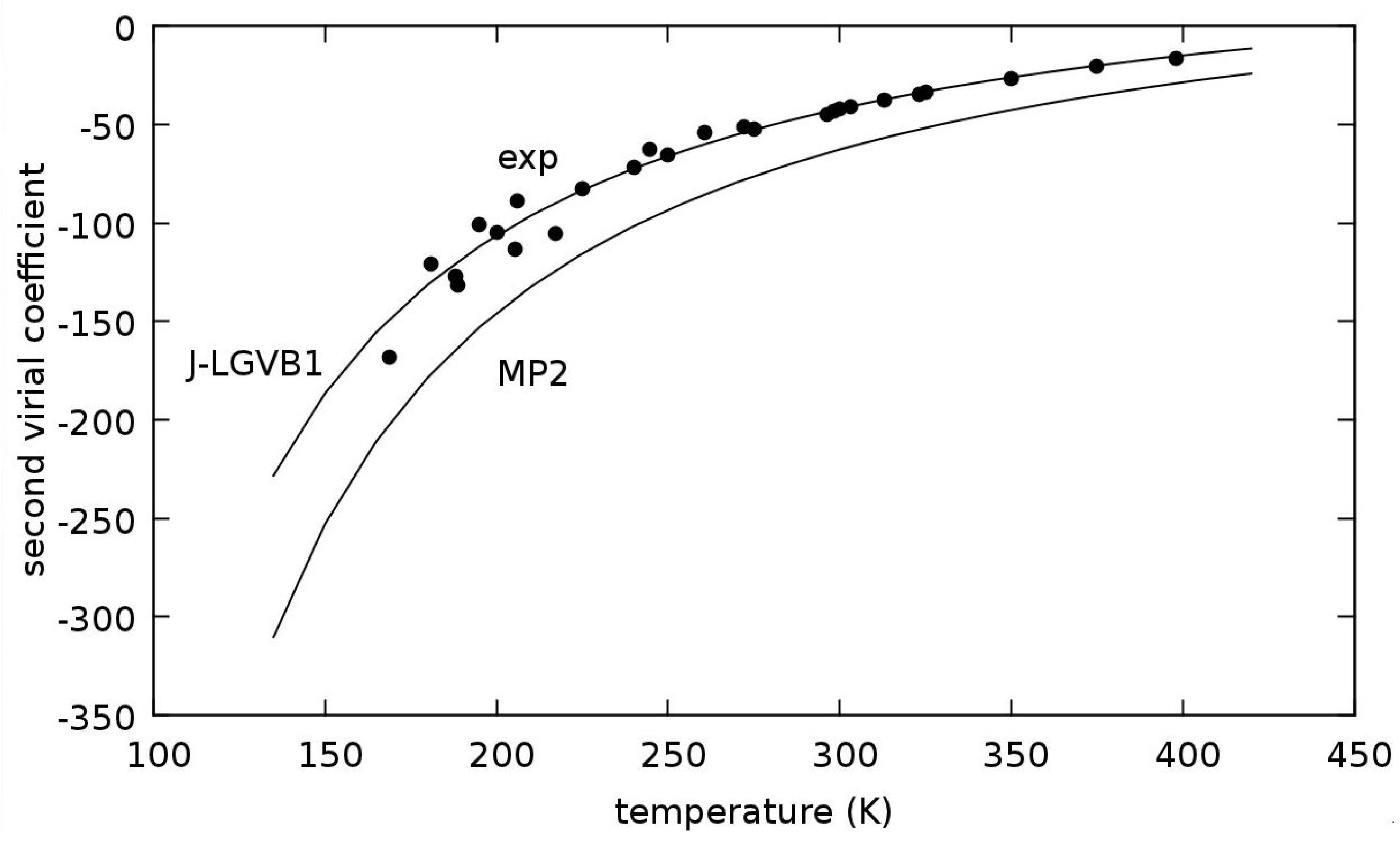
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fermi, E. Un metodo statistico per la determinazione di alcune proprietà dell’atomo. Rend. Accad. Lincei 1927, 6, 602–607. [Google Scholar]
- Thomas, L.H. The calculation of atomic fields. Proc. Camb. Philos. Soc. 1927, 23, 542–548. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Levy, M. Universal variational functionals of electron densities, first order density matrices and natural spin-orbitals and solution of the v-representability problem. Proc. Natl. Acad. Sci. USA 1979, 76, 6062–6065. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W.; MacDougall, P.J.; Lau, C.D.H. Bonded and nonbonded charge concentrations and their relation to molecular geometry and reactivity. J. Am. Chem. Soc. 1984, 106, 1594–1605. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Reynolds, P.J.; Ceperley, D.M.; Alder, B.J.; Lester, W.A., Jr. Fixed-node quantum Monte Carlo for molecules. J. Chem. Phys. 1982, 77, 5593–5603. [Google Scholar] [CrossRef]
- Foulkes, W.M.C.; Mitas, L.; Needs, R.J.; Rajagopal, G. Quantum Monte Carlo simulations of solids. Rev. Mod. Phys. 2001, 73, 33–83. [Google Scholar] [CrossRef]
- Lüchow, A. Quantum Monte Carlo methods. WIREs Comput. Mol. Sci. 2011, 1, 388–402. [Google Scholar] [CrossRef]
- Austin, B.M.; Zubarev, D.Y.; Lester, W.A., Jr. Quantum Monte Carlo and Related Approaches. Chem. Rev. 2012, 112, 263–288. [Google Scholar] [CrossRef] [PubMed]
- Benedek, N.A.; Snook, I.K.; Towler, M.D.; Needs, R.J. Quantum Monte Carlo calculations of the dissociation energy of the water dimer. J. Chem. Phys. 2006, 125, 104302. [Google Scholar] [CrossRef] [PubMed]
- Fracchia, F.; Filippi, C.; Amovilli, C. Size-Extensive Wave Functions for Quantum Monte Carlo: A Linear Scaling Generalized Valence Bond Approach. J. Chem. Theory Comput. 2012, 8, 1943–1951. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Amovilli, C.; March, N.H.; Bogar, F.; Gal, T. Use of ab initio methods to classify four existing energy density functionals according to their possible variational validity. Phys. Lett. A 2009, 373, 3158–3160. [Google Scholar] [CrossRef]
- Edmiston, C.; Ruedenberg, K. Localized Atomic and Molecular Orbitals. Rev. Mod. Phys. 1963, 35, 457–465. [Google Scholar] [CrossRef]
- CHAMP is a Quantum Monte Carlo Program Package. Available online: http://pages.physics.cornell.edu/cyrus/champ.html (accessed on 5 February 2016).
- Burkatzki, M.; Filippi, C.; Dolg, M. Energy-consistent pseudopotentials for quantum Monte Carlo calculations. J. Chem. Phys. 2007, 126, 234105. [Google Scholar] [CrossRef] [PubMed]
- Dolg, M.; Filippi, C.; (University of Twente, Enschede, The Netherlands). Private communication, 2012.
- Kendall, R.; Dunning, T., Jr.; Harrison, R. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Casula, M. Beyond the locality approximation in the standard diffusion Monte Carlo method. Phys. Rev. B 2006, 74, 161102. [Google Scholar] [CrossRef]
- Filippi, C.; Umrigar, C.J. Multiconfiguration wave functions for quantum Monte Carlo calculations of first-row diatomic molecules. J. Chem. Phys. 1996, 105, 213–226. [Google Scholar] [CrossRef]
- Umrigar, C.J.; Toulouse, J.; Filippi, C.; Sorella, S.; Hennig, R.G. Alleviation of the fermion-sign problem by optimization of many-body wave functions. Phys. Rev. Lett. 2007, 98, 110201. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.gaussian.com/ (accessed on 5 February 2016).
- Ruscic, B.; Pinzon, R.; Morton, M.; von Laszevski, G.; Bittner, S.; Nijsure, S.; Amin, K.; Minkoff, M.; Wagner, A. Introduction to Active Thermochemical Tables: Several “Key” Enthalpies of Formation Revisited. J. Phys. Chem. A 2004, 108, 9979–9997. [Google Scholar] [CrossRef]
- Karton, A.; Daon, S.; Martin, J.M. W4-11: A high-confidence benchmark dataset for computational thermochemistry derived from first-principles W4 data. Chem. Phys. Lett. 2011, 510, 165–178. [Google Scholar] [CrossRef]
- Zhao, Y.; Lynch, B.; Truhlar, D. Multi-coefficient extrapolated density functional theory for thermochemistry and thermochemical kinetics. Phys. Chem. Chem. Phys. 2005, 7, 43–52. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Hybrid Meta Density Functional Theory Methods for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions: The MPW1B95 and MPWB1K Models and Comparative Assessments for Hydrogen Bonding and van der Waals Interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Fracchia, F.; Filippi, C.; Amovilli, C. Barrier Heights in Quantum Monte Carlo with Linear-Scaling Generalized-Valence-Bond Wave Functions. J. Chem. Theory Comput. 2013, 9, 3453–3462. [Google Scholar] [CrossRef] [PubMed]
- Minnesota Database Collection. Available online: http://t1.chem.umn.edu/misc/database_group/database_therm_bh (accessed on 5 February 2016).
- Lynch, B.; Truhlar, D. What Are the Best Affordable Multi-Coefficient Strategies for Calculating Transition State Geometries and Barrier Heights? J. Phys. Chem. A 2002, 106, 842–846. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Hellmann, R.; Bich, E.; Vogel, E. Ab initio intermolecular potential energy surface and second pressure virial coefficients of methane. J. Chem. Phys. 2008, 128, 214303. [Google Scholar] [CrossRef] [PubMed]
- Dubecký, M.; Derian, R.; Jurečka, P.; Mitas, L.; Hobza, P.; Otyepka, M. Quantum Monte Carlo for Noncovalent Interactions: Analysis of Protocols and Simplified Scheme Attaining Benchmark Accuracy. Phys. Chem. Chem. Phys. 2014, 16, 20915–20923. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Kerl, K. Interferometric Determination of Mean Polarizabilities and Second Density Virial Coefficients of Methane Between 128 K and 890 K. Int. J. Phys. Chem. Ber. Bunsen-Ges. 1991, 95, 36–42. [Google Scholar]
- Trusler, J.P.M. The Speed of Sound in (0.8CH4 + 0.2C2H6) (G) at Temperatures between 200 K and 375 K and Amount-of-Substance Densities up to 5 Mol/dm3. J. Chem. Therm. 1994, 26, 751–763. [Google Scholar] [CrossRef]
- Ramazanova, A.E. Volumetric Properties and Virial Coefficients of (Water + Methane). J. Chem. Therm. 1993, 25, 249–259. [Google Scholar]
- Renner, C.A. Excess Second Virial Coefficients for Binary Mixtures of Carbon Dioxide with Methane, Ethane and Propane. J. Chem. Eng. Data 1990, 35, 314–317. [Google Scholar]
- Katayama, T. The Interaction Second Virial Coefficients for Seven Binary Systems Containing Carbon Dioxide, Methane, Ethylene, Ethane and Propylene at 25 °C. J. Chem. Eng. Jpn. 1981, 14, 71–72. [Google Scholar]
- Katayama, T. Interaction Second Virial Coefficients for Six Binary Systems Containing Carbon Dioxide, Methane, Ethylene and Propylene at 125 °C. J. Chem. Eng. Jpn. 1982, 15, 85–90. [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amovilli, C.; Floris, F.M.; Grisafi, A. Localized Polycentric Orbital Basis Set for Quantum Monte Carlo Calculations Derived from the Decomposition of Kohn-Sham Optimized Orbitals. Computation 2016, 4, 10. https://doi.org/10.3390/computation4010010
Amovilli C, Floris FM, Grisafi A. Localized Polycentric Orbital Basis Set for Quantum Monte Carlo Calculations Derived from the Decomposition of Kohn-Sham Optimized Orbitals. Computation. 2016; 4(1):10. https://doi.org/10.3390/computation4010010
Chicago/Turabian StyleAmovilli, Claudio, Franca Maria Floris, and Andrea Grisafi. 2016. "Localized Polycentric Orbital Basis Set for Quantum Monte Carlo Calculations Derived from the Decomposition of Kohn-Sham Optimized Orbitals" Computation 4, no. 1: 10. https://doi.org/10.3390/computation4010010
APA StyleAmovilli, C., Floris, F. M., & Grisafi, A. (2016). Localized Polycentric Orbital Basis Set for Quantum Monte Carlo Calculations Derived from the Decomposition of Kohn-Sham Optimized Orbitals. Computation, 4(1), 10. https://doi.org/10.3390/computation4010010