1. Introduction
Titanium dioxide (TiO
2) is widely recognized for its photocatalytic capabilities, which are harnessed in applications such as water splitting, pollutant degradation, and solar energy conversion [
1]. TiO
2 has a wide bandgap of about 3.2 eV for anatase, which limits its ability to absorb visible light [
2]. Most solar energy is in the visible range (approximately 400–700 nm), so TiO
2’s efficiency at utilizing sunlight is inherently limited by its bandgap. Through the doping of foreign atoms, the bandgap of TiO
2 can be optimized, leading to enhanced absorption of solar energy in the visible spectrum. Numerous experimental and theoretical studies have investigated this approach in depth [
3,
4,
5,
6]. Molybdenum (Mo) doping in TiO
2 can effectively shift the absorption edge toward the visible light region because the size of the Mo cation is comparable to that of the Ti cation. This can result in a stable doped system [
7]. Ohno et al. developed S-doped TiO
2 photocatalysts by substituting sulfur (S
4+) in place of certain titanium atoms within the lattice structure. These modified photocatalysts exhibit enhanced visible light absorption at wavelengths beyond 500 nm and demonstrate high photocatalytic activity. S-doped TiO
2 powder shows potential for various applications, including the oxidation of 2-propanol in aqueous solution, degradation of methylene blue, and partial oxidation of adamantane when exposed to wavelengths above 440 nm [
8]. A few studies have shown that S-doped anatase enhances visible light absorption [
9,
10,
11].
The experimental study investigated the effects of S, Se, and Te doping on the anatase-to-rutile phase transition and microbial disinfection properties of chalcogen-doped TiO
2 at high calcination temperatures. TiO
2 samples doped with 2 mol% S, Se, or Te were synthesized using a sol–gel method. The substitutional incorporation of chalcogens into the TiO
2 lattice improved visible light absorption. It was concluded that Te-doped TiO
2 displayed similar bactericidal efficiency to control anatase under visible light, indicating that Te maintains TiO
2’s photocatalytic activity even at temperatures up to 750 °C [
12]. Titanium dioxide samples doped with varying amounts of Se
4+ and Te
4+ ions were synthesized via homogeneous hydrolysis using amorphous Se and Te. The Se
4+- and Te
4+-doped titania samples with the highest photocatalytic activity under UV and visible light were identified as TiSe
3 (11.5 wt% Se) and TiTe
3 (8.0 wt% Te), respectively. This demonstrates that incorporating Se and Te into the anatase lattice positively influences photocatalytic activity in the visible light range [
13]. Another study investigated the Se (IV)-doped TiO
2 system using both experimental methods and density functional theory (DFT) to assess its optoelectronic and photocatalytic properties. Se-doped TiO
2 demonstrated photocatalytic activity when exposed to direct sunlight, with its bandgap extending from 420 to 650 nm. The 3
p orbitals of Se contribute to the formation of additional electronic states within the bandgap, which reduces the wide bandgap of pristine anatase and enhances its photocatalytic activity [
14]. It was found that substituting anions such as S, Se, and Te is more effective than cation doping, with the redshift becoming more pronounced as the atomic number of the chalcogen element increases [
15]. Doping TiO
2 by replacing O sites with chalcogen atoms has become a prominent research focus due to the unique electronic and optical properties of chalcogens. It is also of interest to explore Mo co-doping within the chalcogen element series, as this is a novel approach that has never been reported before.
As demonstrated in previous experimental studies, doping TiO2 with metal atoms such as molybdenum (Mo) and chalcogen elements like sulfur (S), selenium (Se), and tellurium (Te) can enhance its photocatalytic properties. In this work, we investigated the material characteristics of anatase TiO2 when co-doped with Mo and chalcogen atoms. Specifically, we studied the optoelectronic properties of anatase TiO2 doped with Mo at titanium sites and S, Se, or Te at oxygen sites, and we further explored how the spatial arrangement of dopants when the doped atoms are in the adjacent position or at a distant position in the anatase TiO2 structure affects these ground state properties. The electronic structure was calculated using the GGA+U method, while the HSE hybrid functional was employed for accurate optical property predictions. We examined the structural, electronic, and optical characteristics and compared them with the existing mono-doped chalcogen models. Additionally, formation energies were analyzed under oxygen-rich and titanium-rich conditions to assess the thermodynamic stability of the dopants within the anatase lattice. The photocatalytic potential of the doped systems was evaluated by analyzing their band edge positions relative to the normal hydrogen electrode (NHE) for water splitting.
2. Computational Methodology
The projector-augmented wave (PAW) pseudopotentials [
16] were used for all the DFT calculations in the VASP code [
17]. Initially, geometry optimization was performed for both the pure and doped models in the GGA method, which was parametrized at the Perdew−Burke−Ernzerhof (PBE) level [
18] with the Monkhorst−Pack k-point mesh [
19] of 4 × 4 × 4. Here, we used the plane wave basis set with a cutoff energy of 560 eV for our calculations [
1]. For the electronic property calculations, the spin-polarized density of states (DOS) of the doped structures were computed on the Monkhorst−Pack k-point grid on 7 × 7 × 5 along the x, y, and z directions of high symmetry in the first Brillouin zone [
1]. The DFT+U approach, as seen in Equation (1), incorporates an on-site correction to account for intra-atomic electron–electron interactions [
20,
21], which helps improve the description of systems with localized d and f electrons, often resulting in more accurate bandgap predictions than standard GGA.
Here,
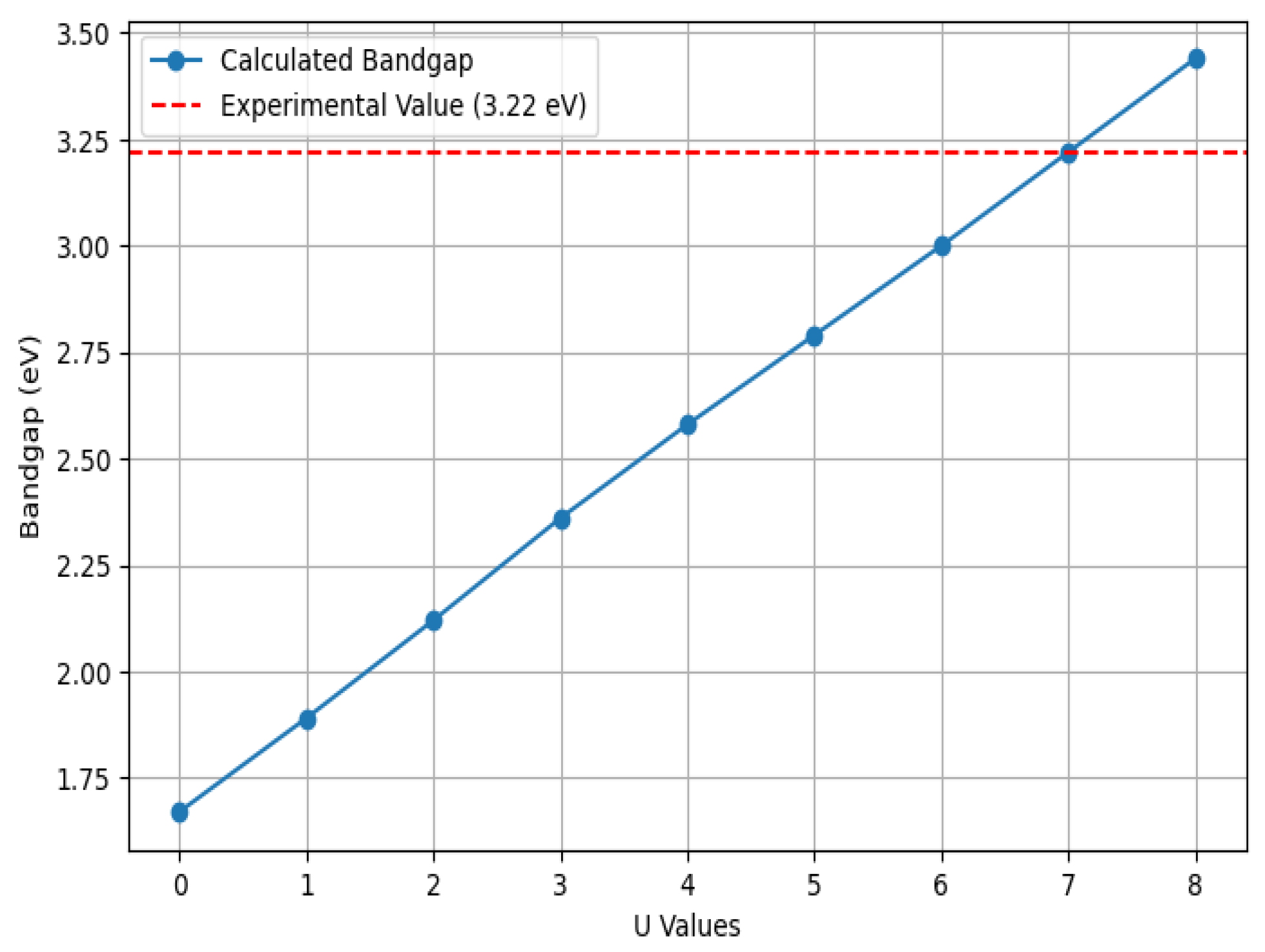
is the occupation matrix for spin σ, and Tr is the trace operator. The spherically averaged Hubbard parameter U quantifies the energy increase associated with adding an extra electron to the system. The parameter J represents (1 eV) as the screened exchange energy. In this case, the effective on-site Coulomb interactions were set to U = 7.0 eV for Ti 3
d and U = 5.38 eV for transition metal (TM) d electrons in the GGA+U approach. To determine the U value for pure anatase TiO
2, we computed the bandgap values by varying U from 0 to 8 eV and found that U = 7 eV gives a value closest to the experimental bandgap of 3.22 eV, as shown in
Figure 1. The U value for Mo was taken from the Materials Project [
22].
Simulating a system that has strong electron correlation and a need to correct the electronic structure, especially in transition metal oxides or correlated materials, GGA+U could be more effective, but it may not offer the same accuracy for optical properties as HSE06. Thus, optical property calculations were performed in the screened hybrid functional called (Heyd−Scuseria−Ernzerhof) HSE06 [
23]. VESTA is a three-dimensional visualization program that was used to visualize the equilibrium crystal lattices [
24].
The doped system consists of a 2 × 2 × 1 supercell containing a total of 48 atoms, which provides a realistic environment for introducing dopants at low concentrations while minimizing artificial interactions between periodic images of atoms [
1]. The supercell was generated using the Phonopy code to ensure proper structural setup for the simulations [
25]. S, Se, and Te doping in the anatase TiO
2 supercell was performed by replacing a single O atom with S, Se, or Te at its regular lattice site, while Mo-doped TiO
2 was obtained by replacing the Ti site with the Mo atom simultaneously. Here, Mo
4+ substitutes Ti
4+, and the valence band remains fully occupied, with no free carriers (electrons or holes). The concentration of each dopant element in this supercell is approximately 2.08%.
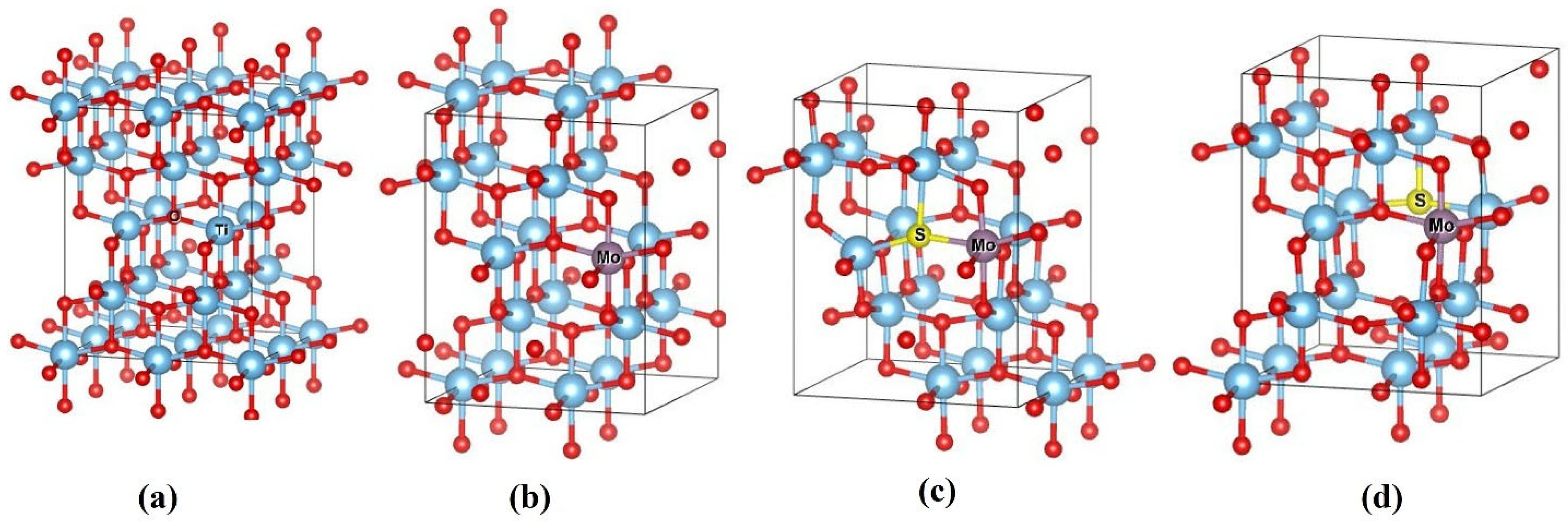
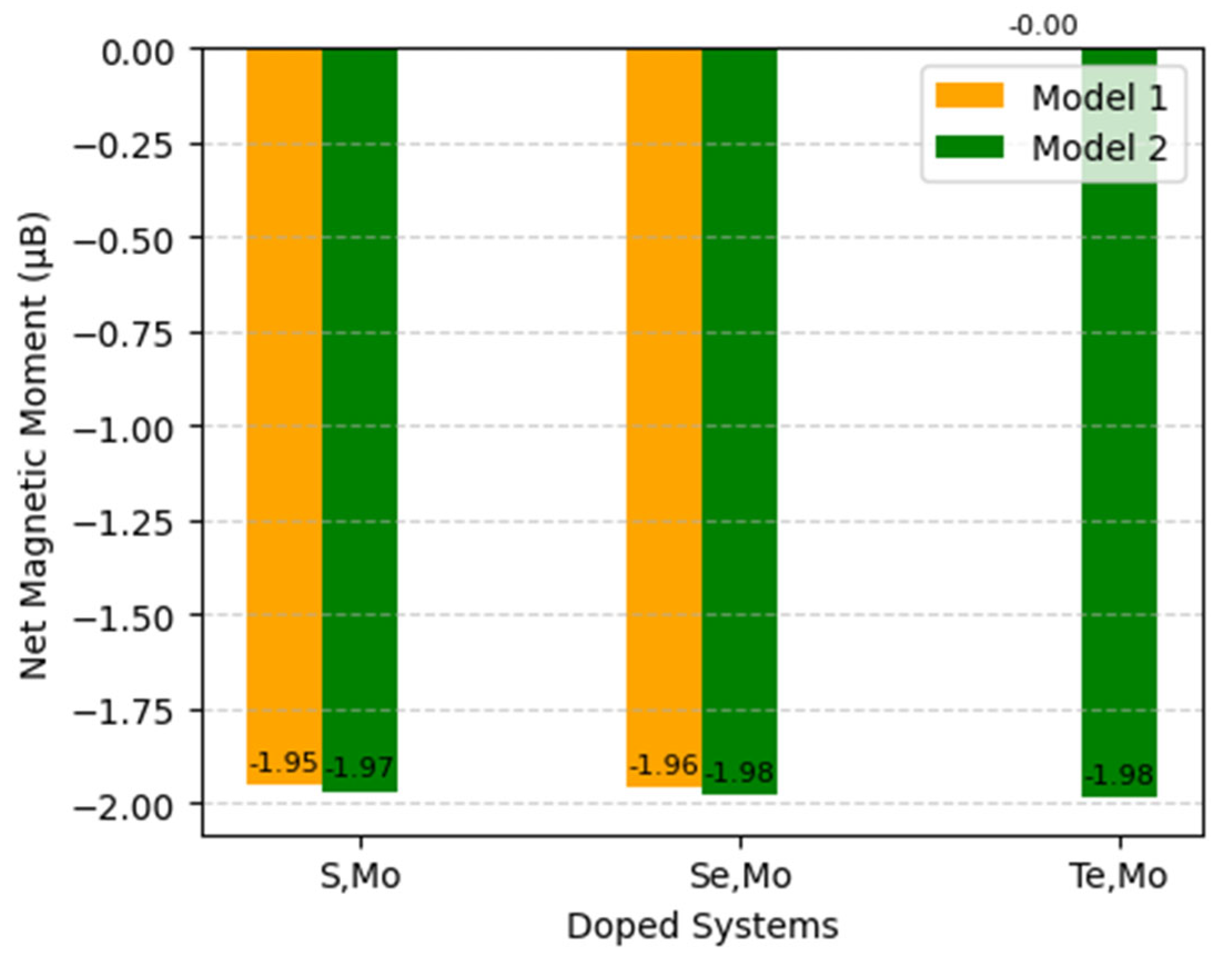
Two different models were constructed for the co-doping system to investigate the impact of dopant locations on the electronic and optical properties of these six configurations. In the first model (hereafter referred to as Model 1), S, Se, Te, and Mo were doped adjacently by replacing O and Ti atoms at a distance of 1.9576 Å. In the second model (hereafter referred to as Model 2), S, Se, Te, and Mo were placed at a distance of 4.3099 Å. Both doped models were geometrically optimized, and their electronic and optical properties were subsequently evaluated. The configurations of these models are illustrated in
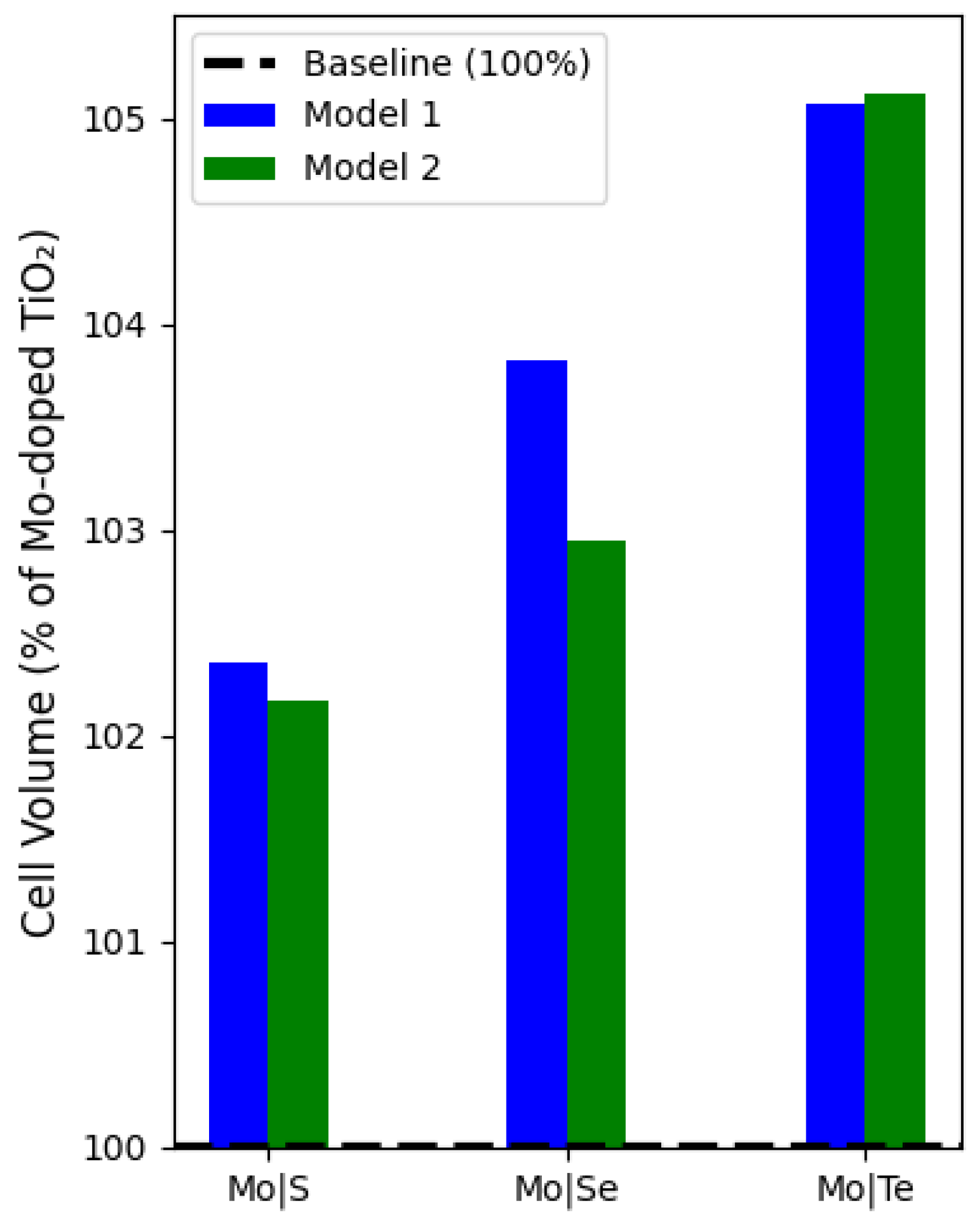
Figure 2. Energy versus volume data for these doped structures concluded that Model 1 exhibits a lower ground state energy than Model 2, indicating that Model 1 is more stable across all doped structures. The S-doped configuration of Model 1 has the most stable structure among the Se- and Te-doped compounds. Consequently, the adjacent positioning of the Mo and S dopants results in the most optimal structural stability relative to the other models, as seen in
Figures S1–S4. In adjacent co-doping, the introduced cation and anion can form a strong bond via direct charge transfer, which generally results in the system achieving the lowest total energy [
26].
4. Formation Energy
The formation energies for co-doped anatase models were calculated and analyzed under both oxygen-rich and titanium-rich conditions. The formation energy is given by Equation (2),
where
represents the total energy of the pure titanium dioxide supercell,
and
represent the chemical potentials of the transition metal and titanium atom, respectively, and
and
are used to represent the chemical potentials of the nonmetal and oxygen atoms, respectively.
The formation energy as in Equation (3) of TiO
2-based photocatalysts depends on the growth conditions and varies between Ti-rich (TRC) and O-rich (ORC) chemical environments. Here,
was computed by normalizing the energy value of the pure supercell.
Under the oxygen-rich growth conditions, the chemical potential of oxygen is the one calculated from the ground state energy of O2, . Then, is obtained using Equation (3). Conversely, under the titanium-rich conditions, is the energy of one titanium atom in bulk titanium, and is then computed.
The chemical potentials for the chalcogen elements S, Se, and Te are determined with Equation (4):
Here,
was calculated by Equations (3) and (4) through
. The chemical potentials (μ
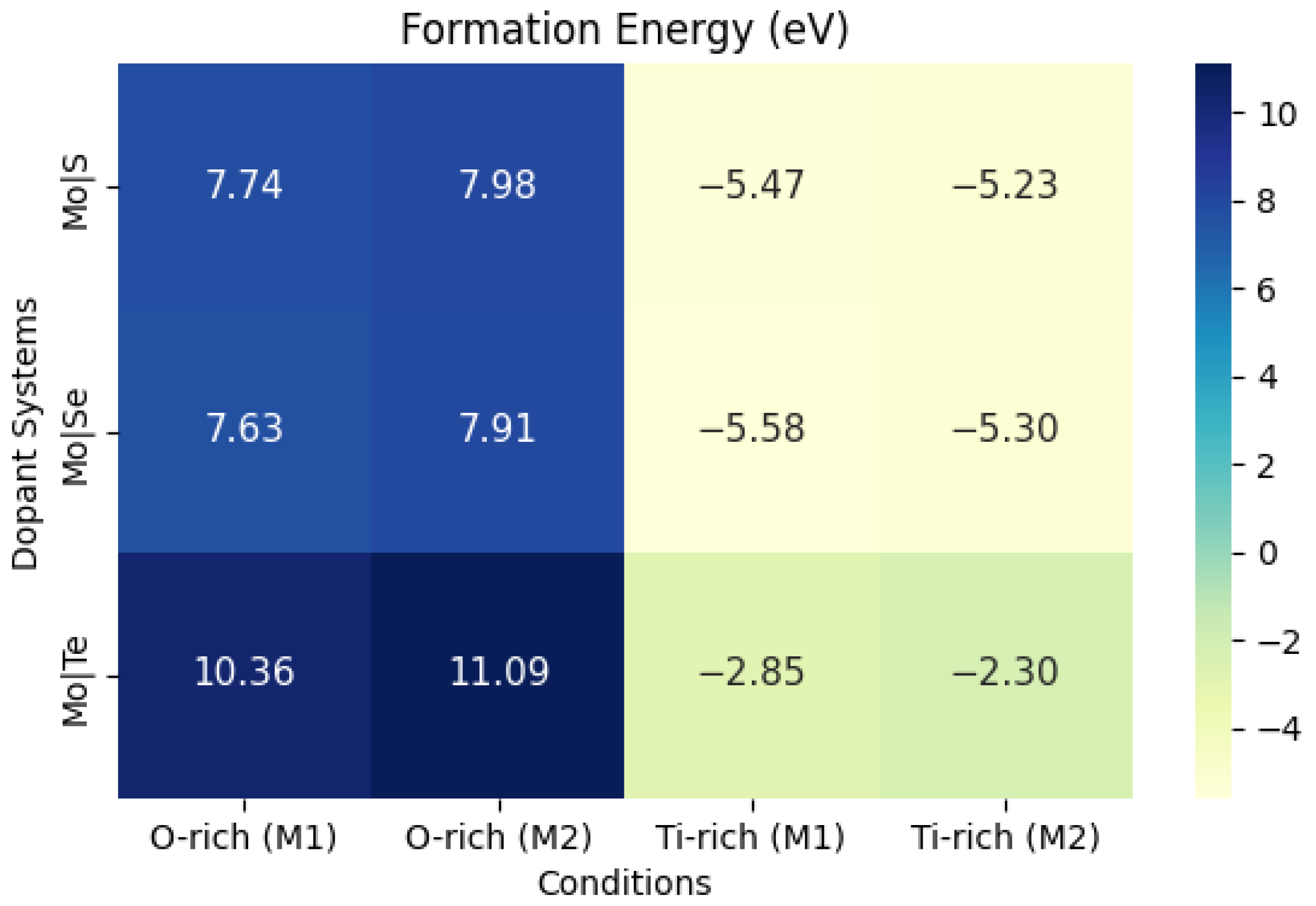
X), where X represents S, Se, and Te, were calculated under ORC, TRC, and in their respective bulk phases. Among these, the chemical potentials under O-rich conditions were found to be stable, and therefore, they were used for further calculations. The formation energies of the dopant elements are calculated using Equation (1) within the GGA approximation and are presented in a heatmap in
Figure 4 to convey the precise values. The computed values for E
tot [Ti
16O
32], μ
O, μ
Ti, and μ
Mo are −423.89, −4.94, −16.64 eV, and −10.95 eV, respectively [
1].
Overall, the results in
Figure 4 show that ORC obtained higher positive formation energy values, as under TRC it has negative formation energy values. Under ORC, the formation energy (E
f) values follow almost this order: Mo/Se doping < Mo/S doping < Mo/Te doping. These positive formation energies can be attributed to the significant electronic and structural disruptions caused by the dopants within the TiO
2 lattice, which adversely affect the overall stability of the system. Specifically, the incorporation of Mo/Te induces substantial distortion in the TiO
2 lattice, and additional energy is required to overcome unfavorable lattice interactions. As a result, higher formation energies are observed. These materials may still be synthesized under specific conditions (e.g., high temperatures or particular partial pressures of gases) that provide the necessary energy for their formation. However, the high formation energy values indicate that while these configurations are theoretically possible, they may not be easily achievable or stable in typical synthesis environments. On the other hand, under TRC conditions, all the formation energies are negative, indicating that the material has a thermodynamically spontaneous tendency to form.
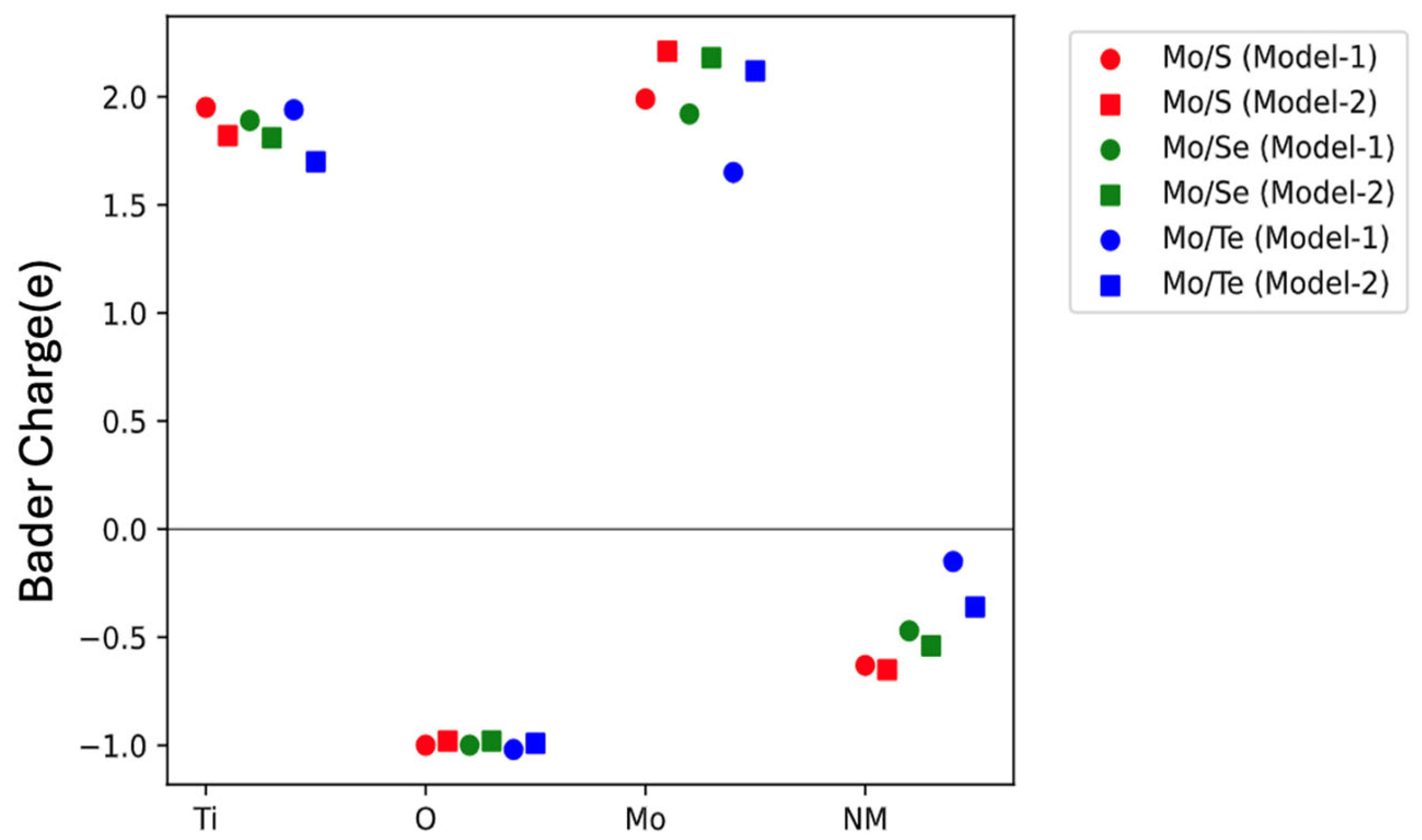
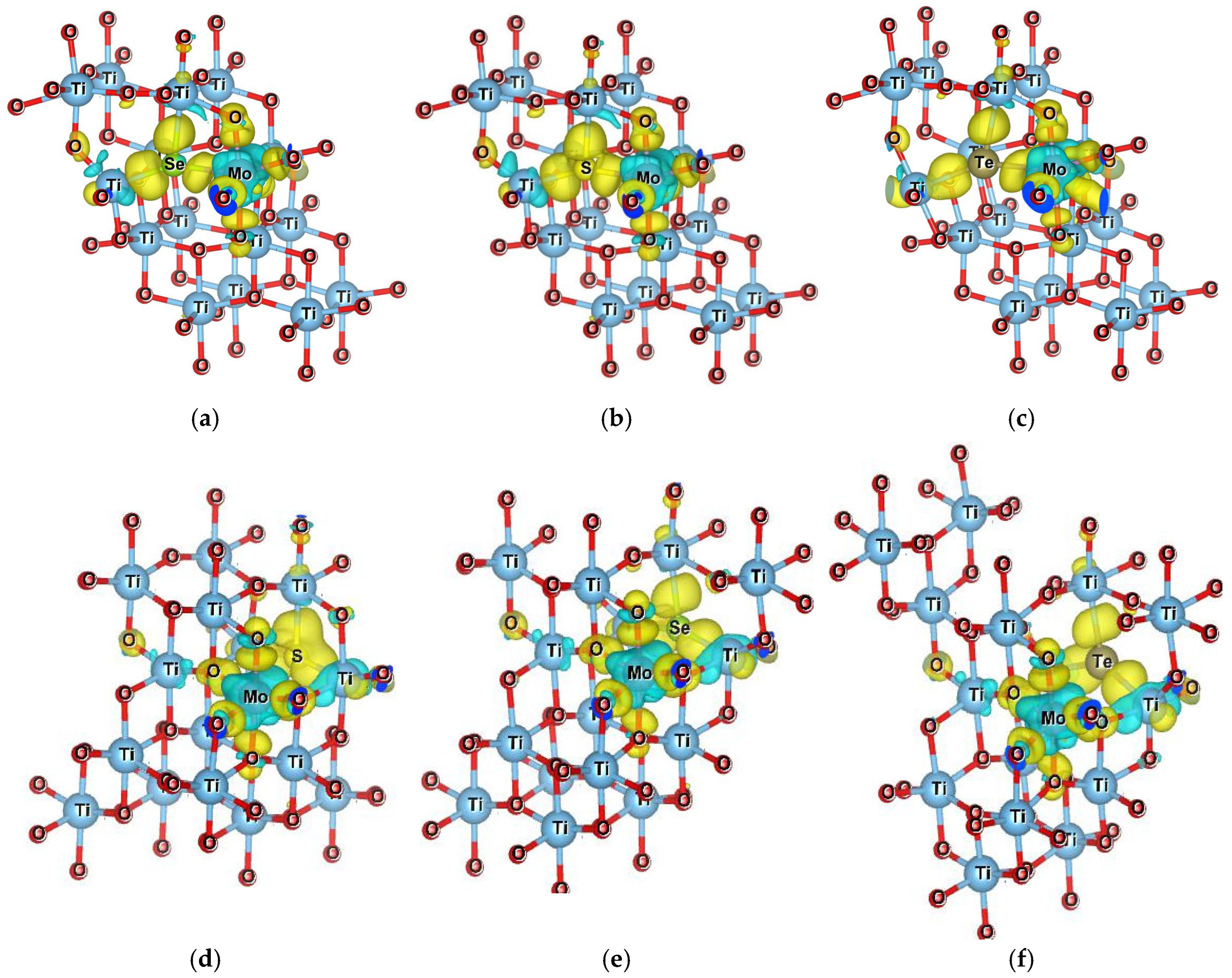
The impact of dopants on the stability of the host material can be understood through Bader charge analysis. As shown in
Figure 5, in
Section 7 dopants that exhibit more negative Bader charges tend to withdraw more electron density from their surroundings. This strong electron-withdrawing behavior may disrupt the local electronic environment and bonding structure within the host lattice. As a result, such dopants can destabilize the system, which is reflected in their higher formation energies. There are limited previous results available for comparing the formation energies of doped models. However, the co-doped models with other dopants (such as Cr/B and Co/B) exhibit higher positive formation energies rather than lower or negative values [
4]. This indicates that the co-doped models have higher formation energies, which are positive rather than negative, suggesting that additional effort is required to form these systems. Furthermore, the high formation energies suggest that these materials may be prone to decomposing back into their constituent phases or transforming into more stable forms under standard conditions. This indicates that the materials may not be practical for certain applications due to the significant energy required to maintain their structures.
5. Electronic Properties
The GGA + U calculation was introduced to obtain values closer to the experimental bandgaps. The pure anatase bandgap value was computed as 3.22 eV, which is almost equal to the experimentally found bandgap value of 3.23 eV [
28]. The energy values of the bands are shifted by subtracting the Fermi level from all energy values, effectively setting the Fermi level to 0 in the plot. The band structure in
Figure 6a along the high-symmetry directions of the Brillouin zone (BZ) and the density of states (DOS) for the valence band (VB) and conduction band (CB) of a perfect TiO
2 crystal are shown in
Figure 7. According to
Figure 7, the VB and CB are composed of contributions from both the Ti-3
d and O-2
p orbitals.
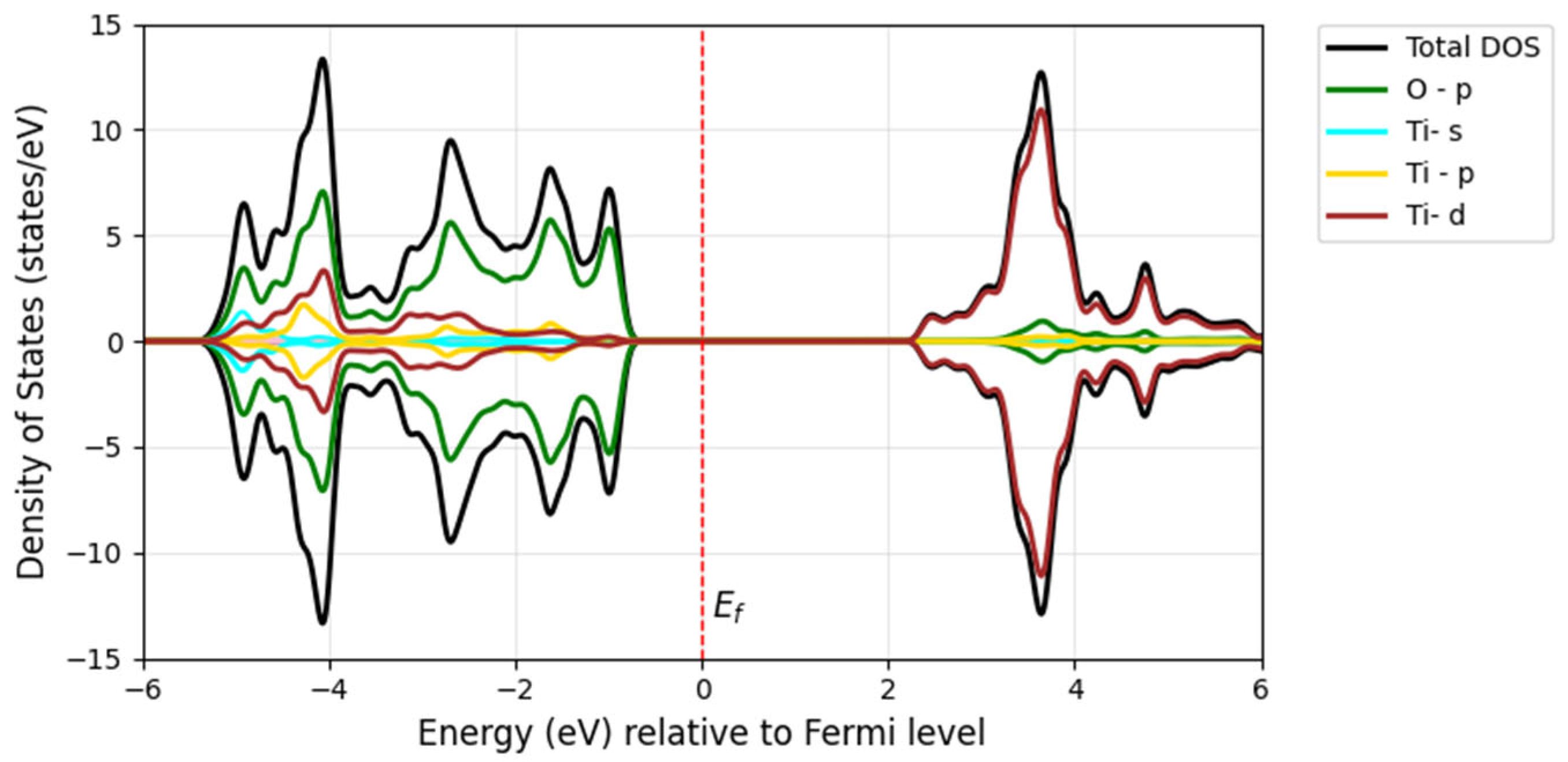
Figure 8 illustrates the decomposition of the TiO
2 DOS, where the Ti-3
d orbital is split into two components: the t
2g and e
g states.
In the anatase TiO
2 lattice, each titanium (Ti) atom is coordinated with six oxygen (O) atoms in an octahedral arrangement as depicted in
Figure 2a. The bonding between the Ti
d-orbitals and O
p-orbitals can be explained using molecular orbital theory (
Figure 9). The Ti e
g orbitals, with lobes pointing directly toward the oxygen atoms, form strong σ bonds with the O
p orbitals. These highly directional σ bonds play a crucial role in stabilizing the metal–ligand interactions within the lattice. According to
Figure 8, VBs are primarily composed of O
p and Ti e
g states, whereas the CB consists of Ti t
2g states. The Ti e
g orbitals, which include the d
z2 and d
x2−y2 orbitals, play a significant role in the CB. In contrast, the Ti t
2g orbitals, comprising the d
xy, d
xz, and d
yz orbitals, contribute largely to the CB. The energy bands of pure TiO
2 within the groups of bands where the bands are between 0 and −5 eV originate mainly from oxygen 2
p orbitals, and the bands above the Fermi energy are dominated by contributions from titanium 3
d orbitals. The bonding nature of the conduction and valence bands arises from the hybridization and interaction of these orbitals between the Ti and O atoms, which directly influence the material’s electronic and optical properties.
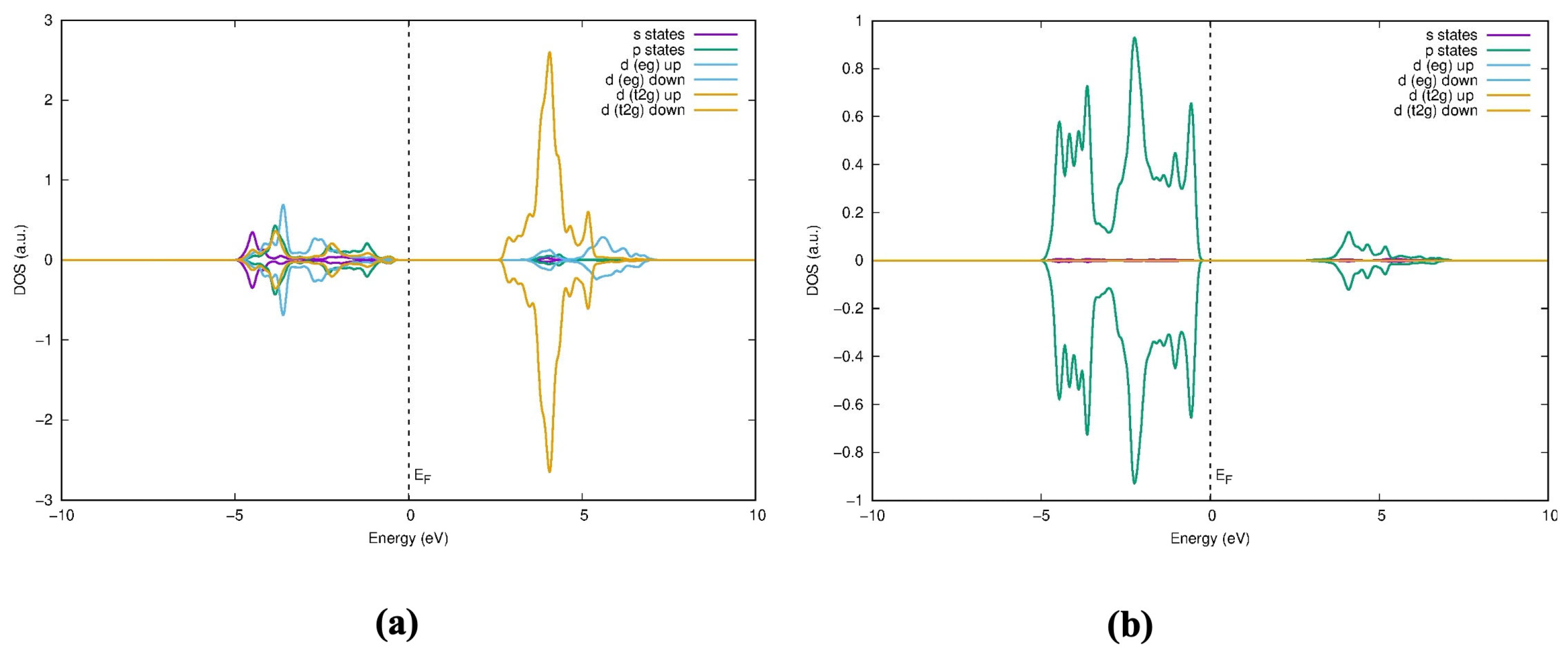
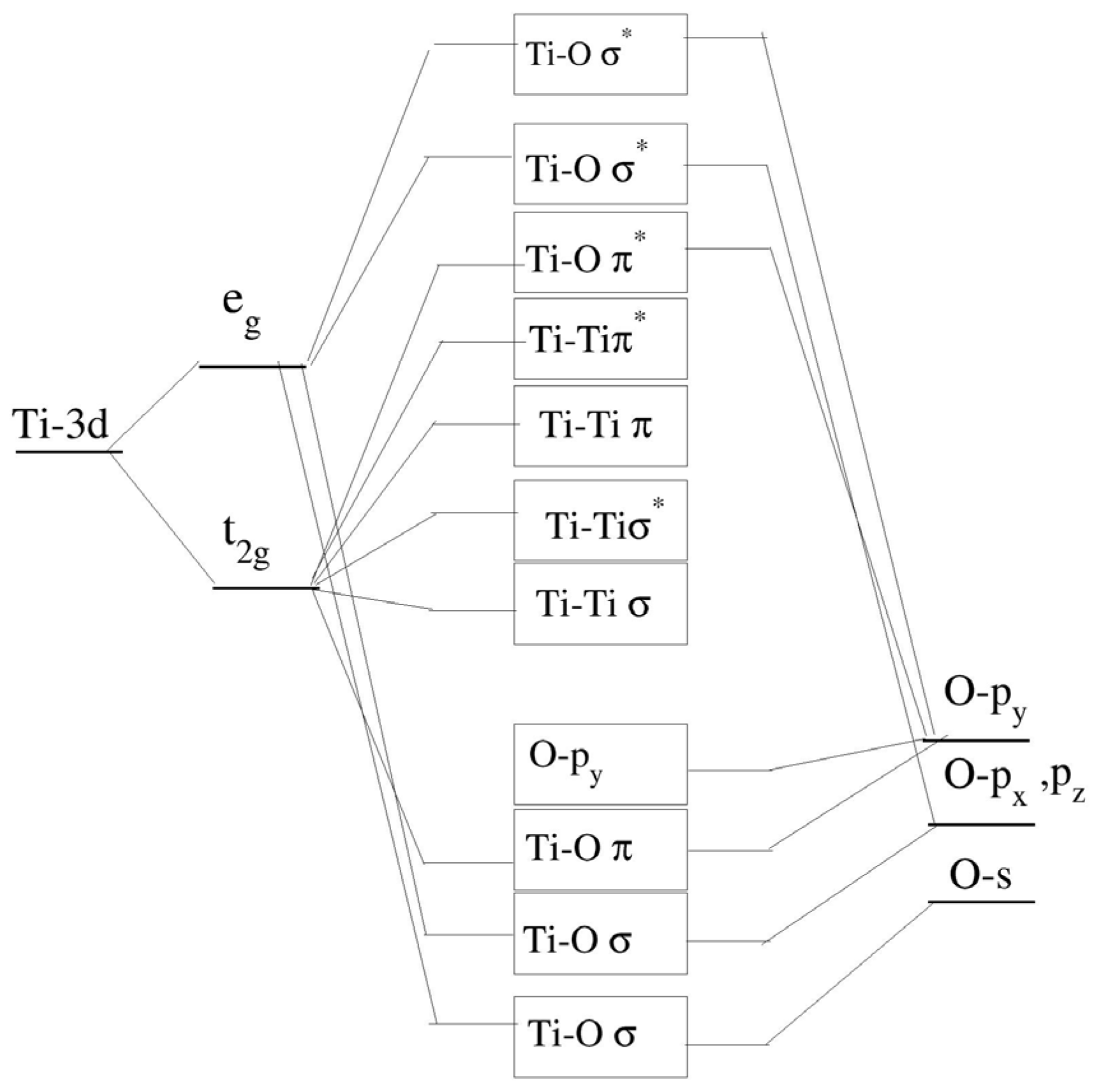
When examining the bonding nature of the conduction band and valence band between the atoms, Soratin and Schwarz provide a detailed description of the molecular orbital diagram [
29], where the lower conduction band (CB) consists of the sigma bonding and antibonding interactions of the Ti t
2g-Ti t
2g states. In the middle of the CB, the remaining Ti t
2g states participate in pi bonding and antibonding with Ti t
2g-Ti t
2g interactions, as well as antibonding interactions between Ti t
2g and O p
y orbitals. The upper CB consists of sigma antibonding interactions between O p
y and Ti e
g states. The valence band (VB) is divided into two regions: the lower-energy region is composed of sigma bonding interactions involving Ti e
g and O s, p
x, and p
z orbitals, while the upper region of the VB is primarily made up of O p
y orbitals and pi-bonding interactions between Ti t
2g and O p
y.
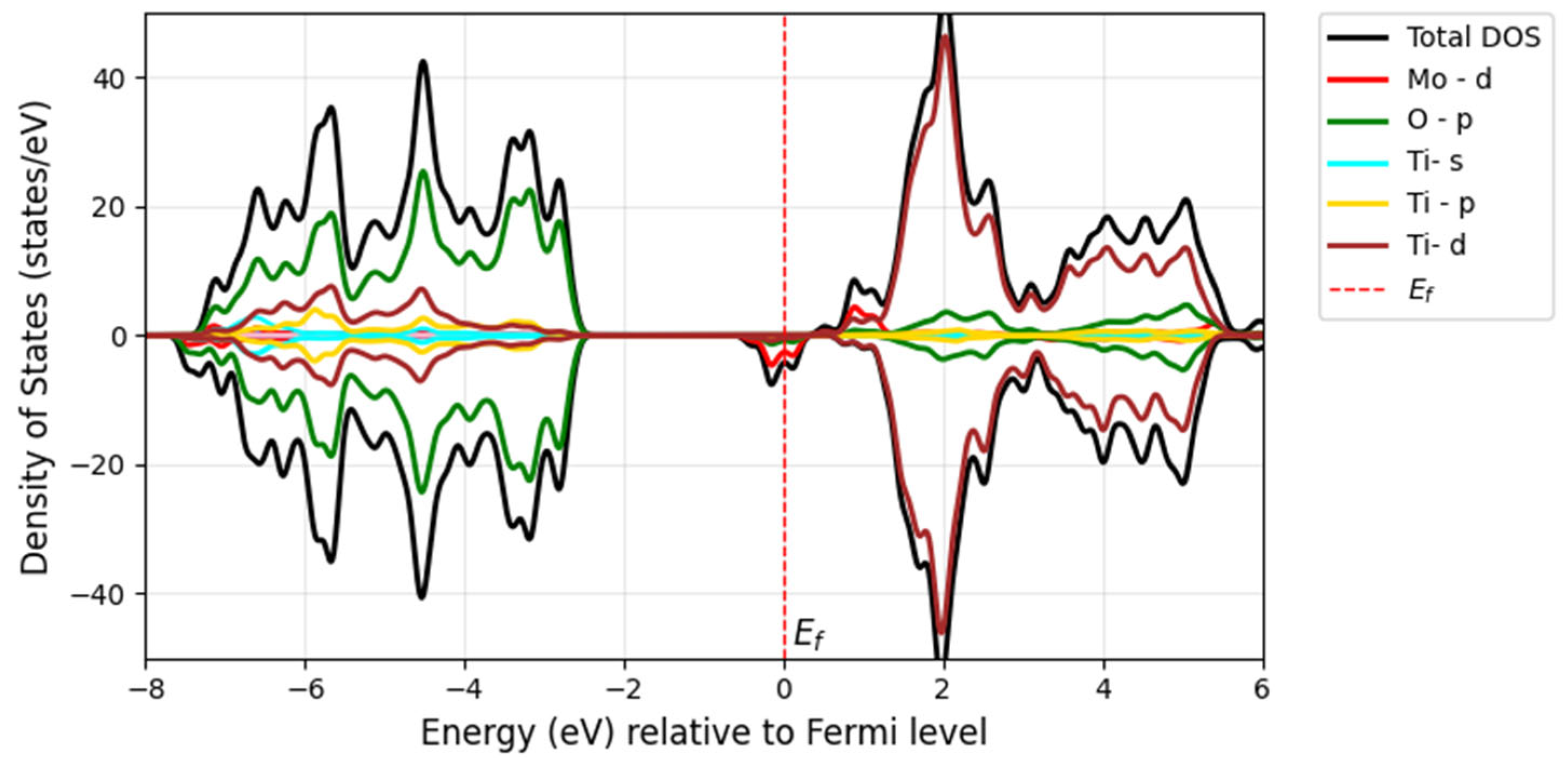
In molybdenum-doped TiO
2, molybdenum, which has more valence electrons than titanium, creates defect states within the bandgap, leading to the formation of intermediate bands (IBs). These intermediate states arise from Mo-4
d states and are localized at the conduction band, as shown in
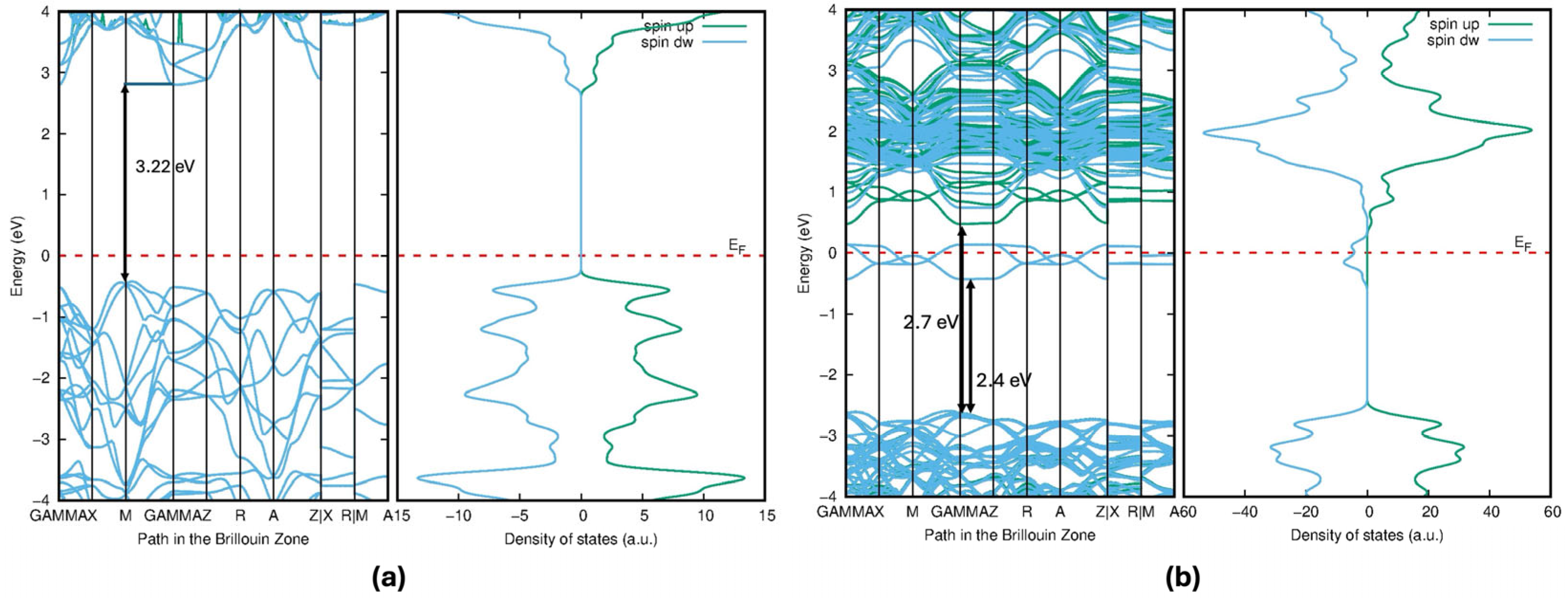
Figure 10. According to
Figure 6b, the impurity states act as shallow donor states at the Fermi level (indicated by the red dashed line), exhibiting half-metallic ferromagnetic behavior, metallic in the spin-down channel but semiconducting in the spin-up channel in the band diagram. In the Mo-doped model, the bandgap was reduced by 2.7 eV compared with the pure compound of 3.22 eV, and the reduction was about 0.52 eV. These states extend into the conduction band, contributing to the reduction in the bandgap in pristine TiO
2. This bandgap reduction occurs due to the hybridization of Mo-4
d and Ti-3
d states. Soussi et al. analyzed the electronic structure of TiO
2 with varying concentrations of Mo doping. Their findings revealed that, in all cases, the Fermi level shifted into the conduction band, indicating n-type metallic doping behavior [
30]. This reduction in the bandgap is able to shift the absorption edge to the visible light region, which is good for the optical absorption efficiency. Thus, it could enhance photocatalytic efficiency. Experimentally proved, Mo-doped TiO
2 nano powders were synthesized, significantly enhancing light absorption, even in the visible light range, and the photocatalytic activity of the synthesized TiO
2 nano powders was improved by Mo doping [
31].
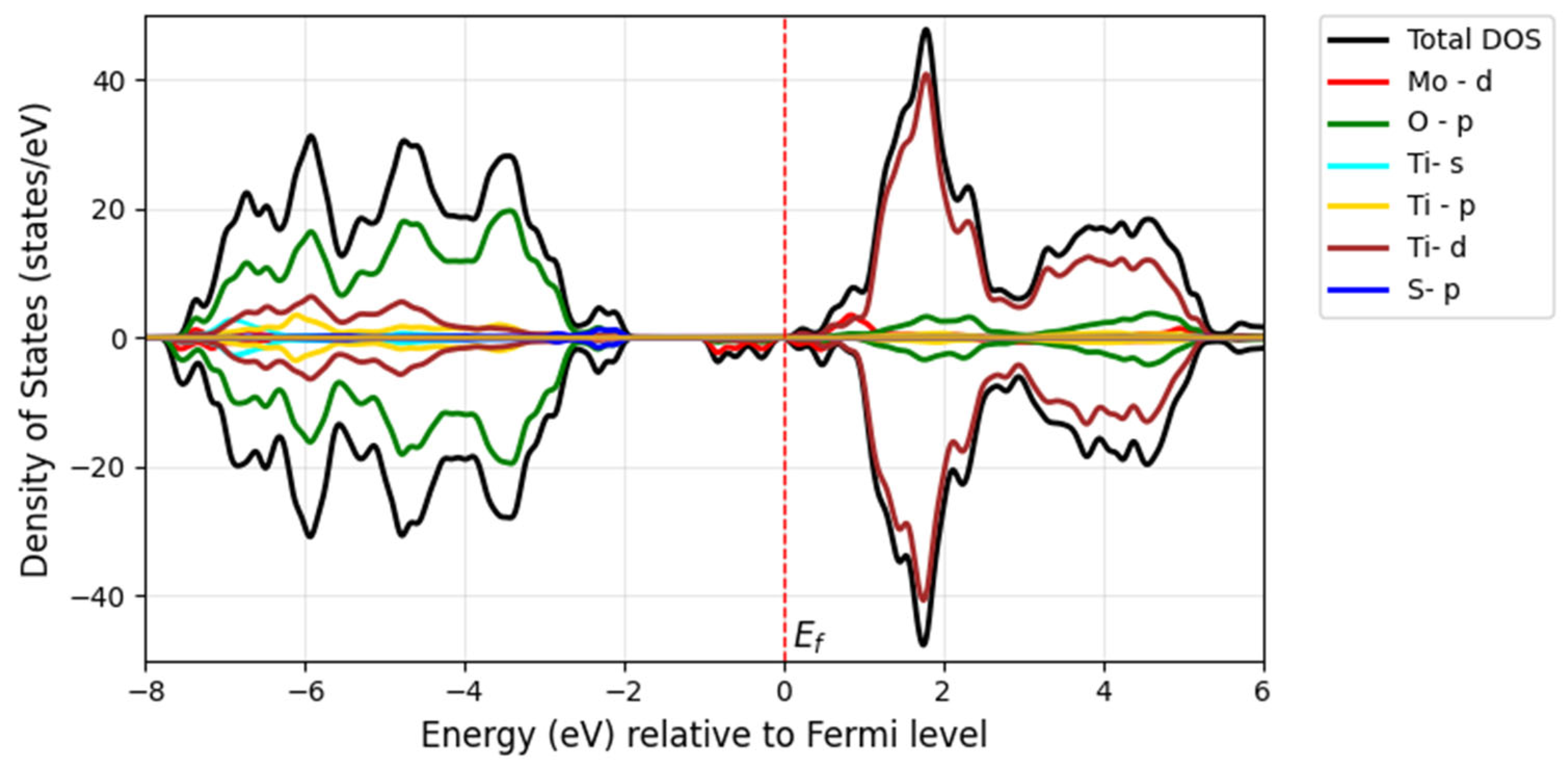
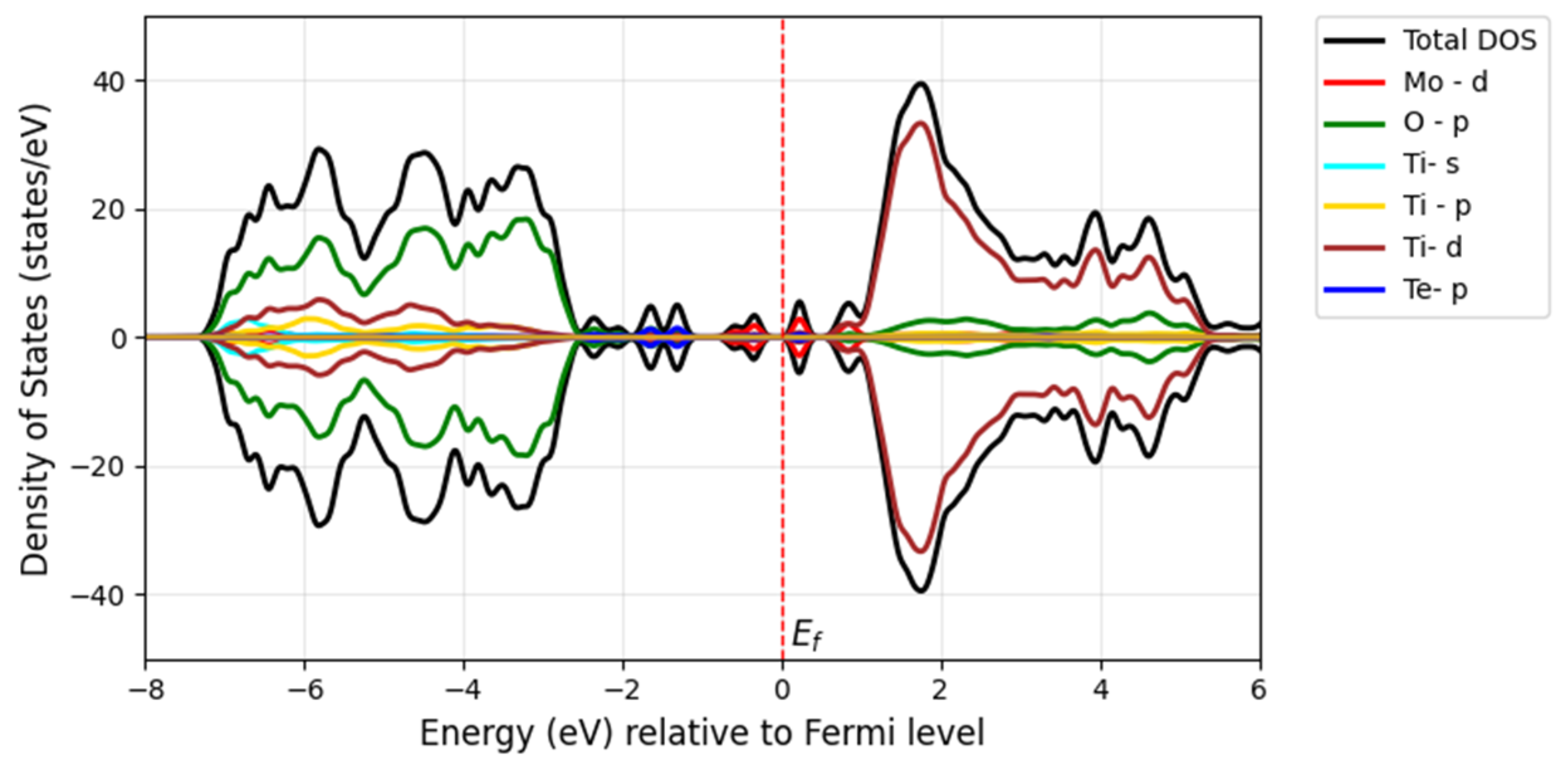
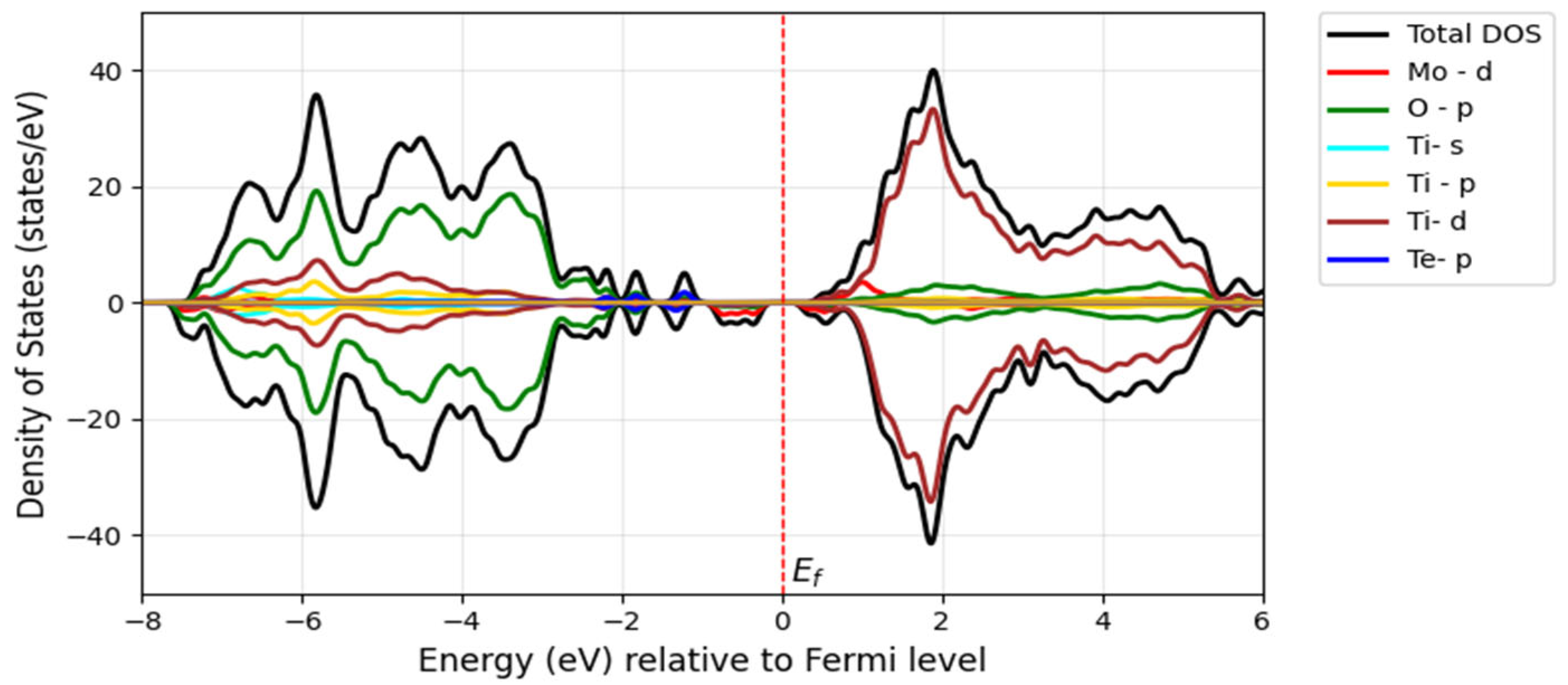
The incorporation of the chalcogen atom as a secondary dopant into Mo-doped TiO
2 further shifts the absorption edge of TiO
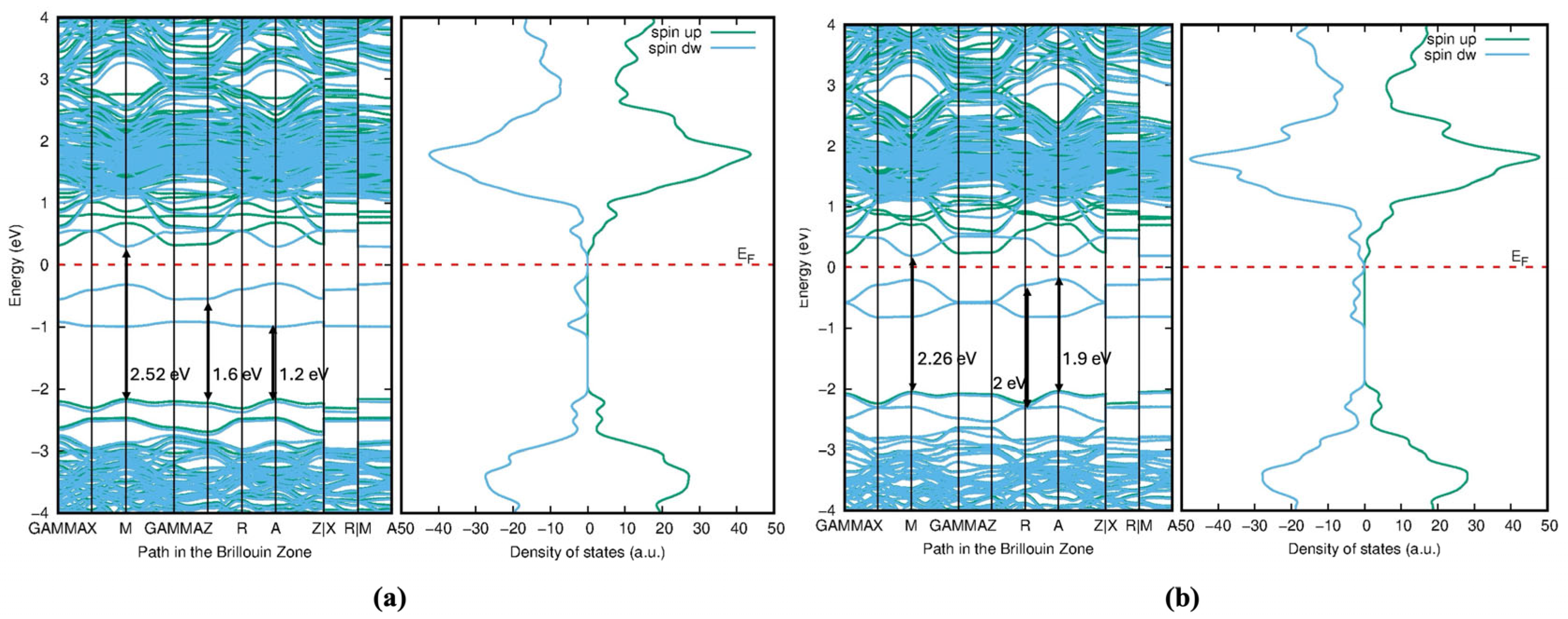
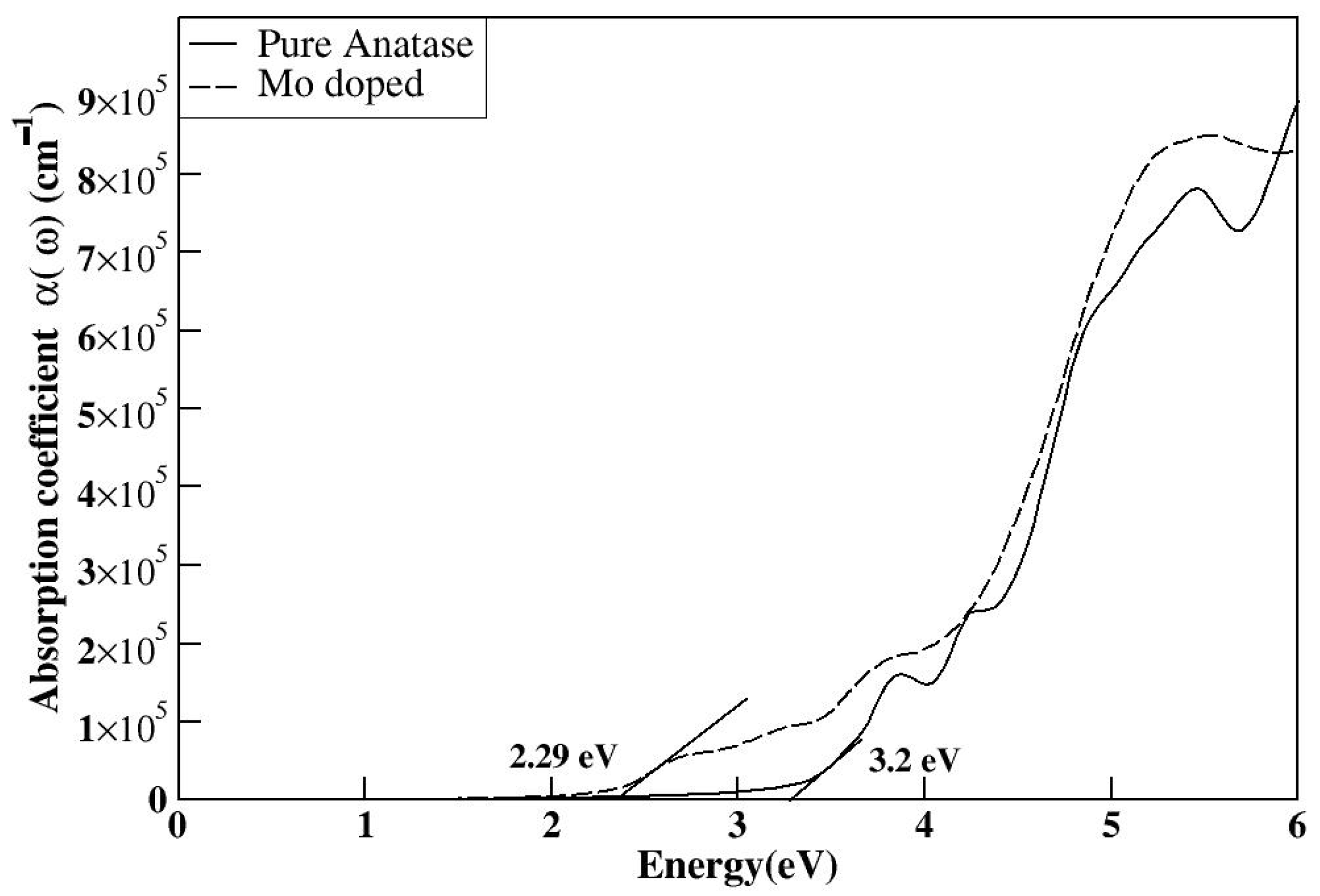
2 into the visible spectrum and even toward longer wavelengths. The redshift of the bandgap transition occurs further in the visible light region after doping with S, Se, and Te along with Mo. Additionally, the Fermi level shifts to the bottom of the conduction band minimum, indicating the absence of metallic nature. The bandgap of the Mo/S-doped system for Model 1 is 2.52 eV in
Figure 11a, and Model 2 shows the narrower bandgap of 2.26 eV in
Figure 11b. However, its interaction with Mo-
d may be stronger in Model 2, influencing the electronic properties.
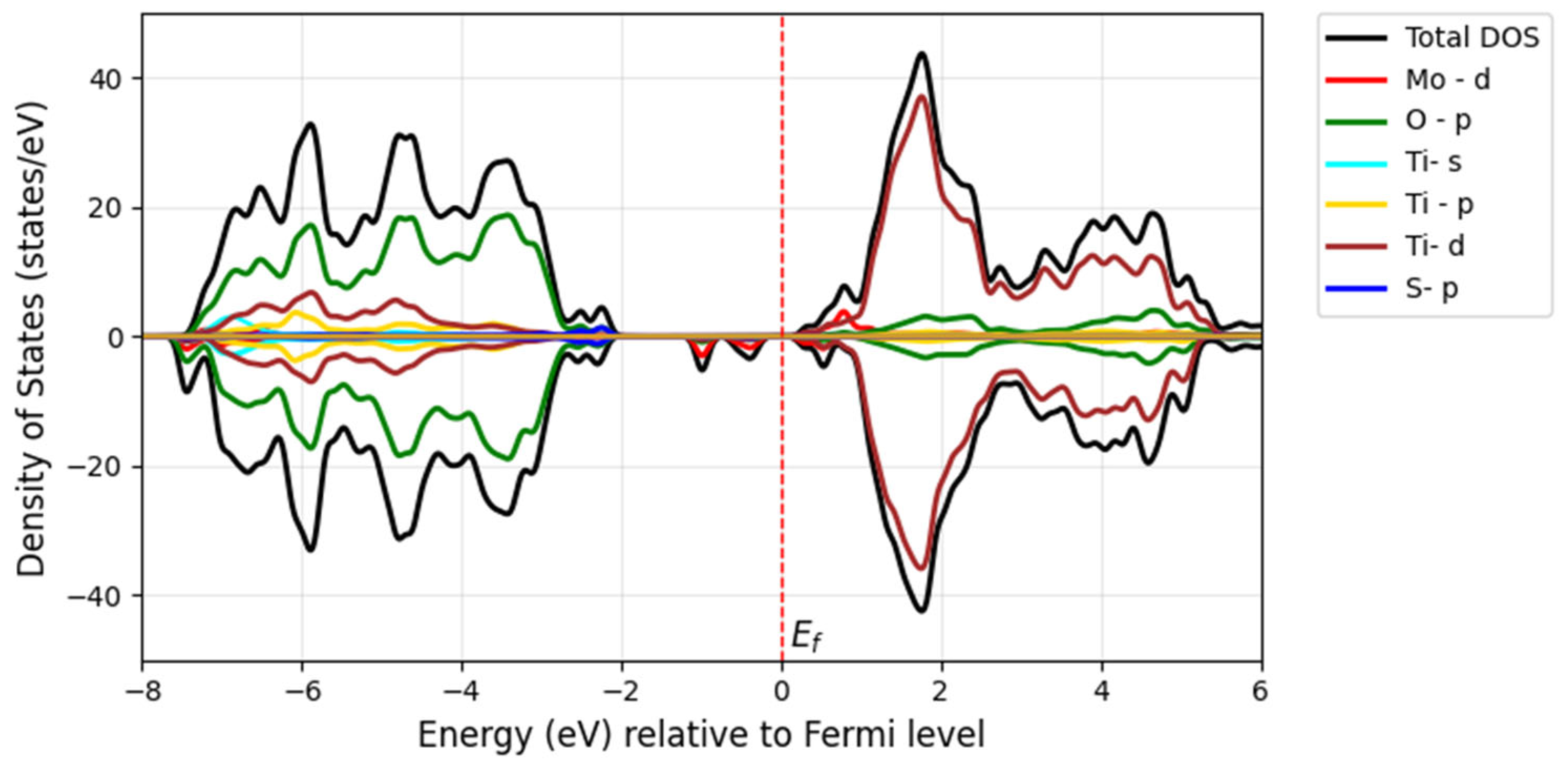
The S substitution at the oxygen site introduces S-
p states located at the edge of the valence band maximum, forming a tail of the valence band (blue) with a minor contribution, as seen in
Figure 12 and
Figure 13. This contributes to a further bandgap reduction compared with the mono-doped Mo model by hybridizing S-3
p states with O-2
p states, making it capable of absorbing visible light. The literature shows that doping with S can lower the bandgap and cause a redshift in the absorption spectra of TiO
2 as sulfur concentration increases. This effect is attributed to the presence of impurity states of S-3
p at the valence band maximum, which aligns with our findings [
32]. Experimentally, it has been proven that sulfur doping shifts the absorption edge of TiO
2 to a lower energy region within the wavelength range of 650 nm [
33].
Overall, our findings for the doped Mo, S/TiO2 system show that the decrease in the bandgap of pure TiO2 is related to the hybridization between the 3p S states and the 3d Ti and 4d Mo orbitals, which leads to the formation of new states within the bandgap.
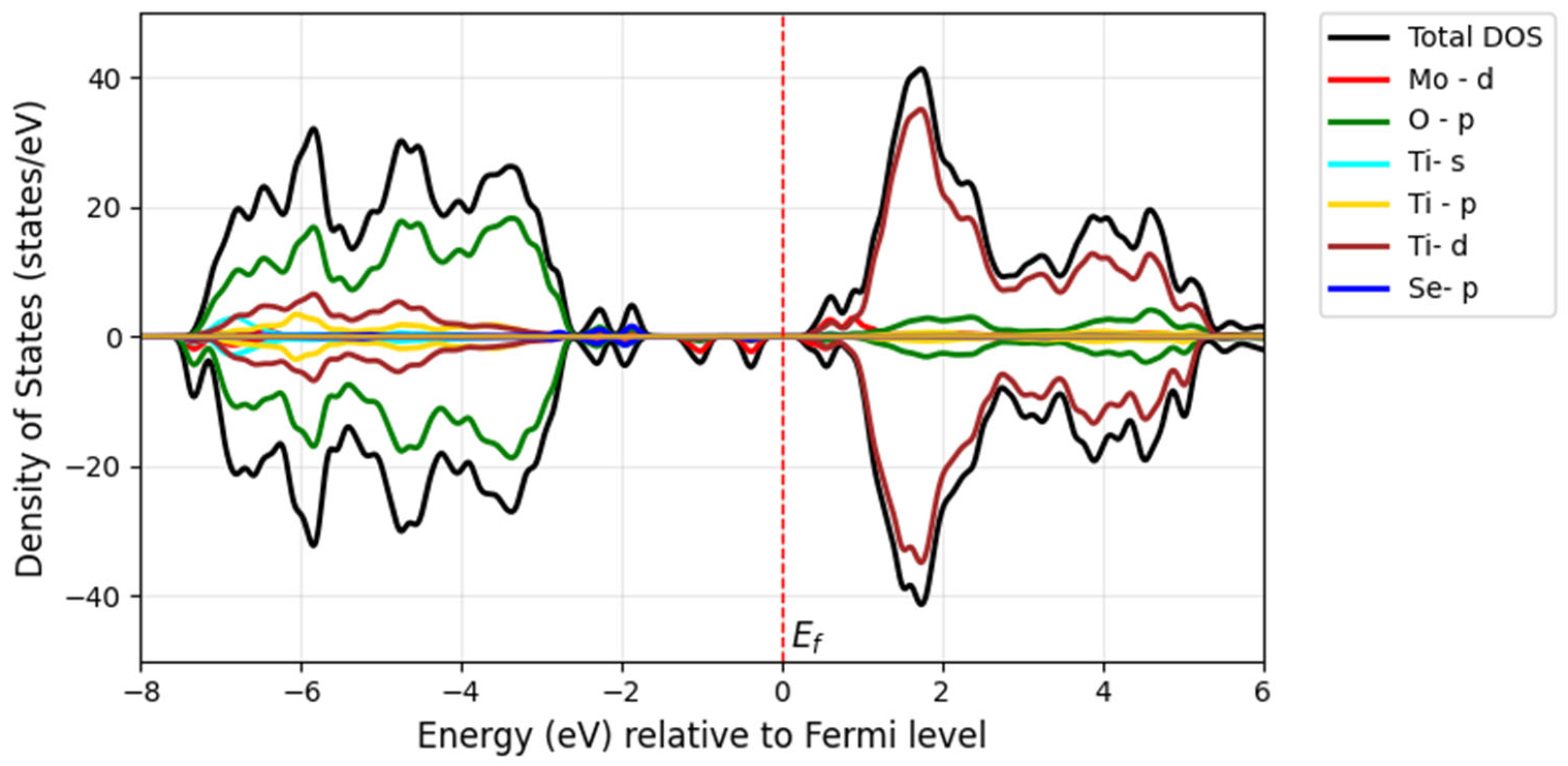
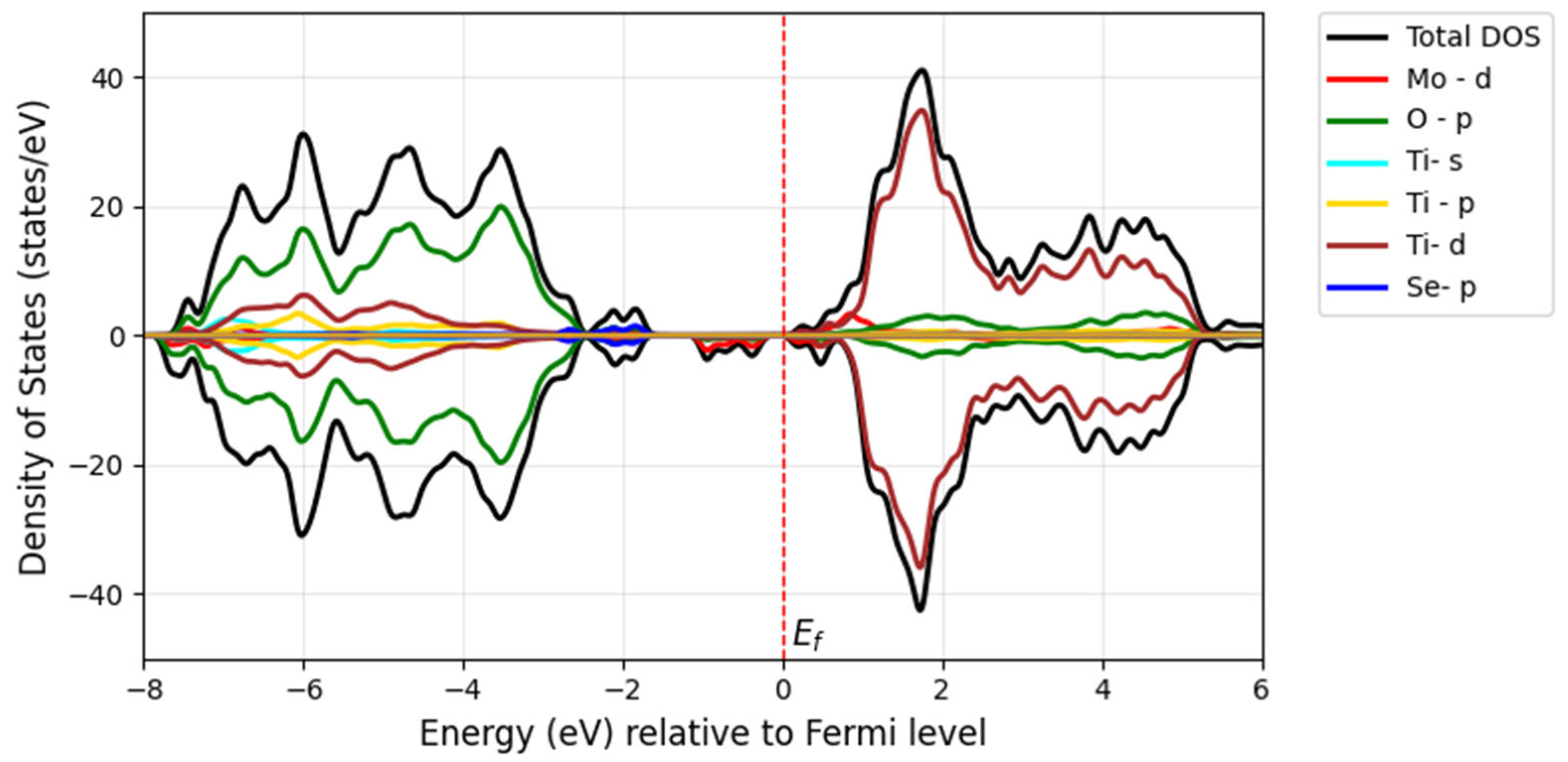
When Mo and Se are doped adjacently in TiO
2, their close proximity leads to strong interactions between the dopants and the TiO
2 lattice. The
d-orbitals of Mo and the
p-orbitals of Se strongly hybridize with the Ti-3
d and O-2
p orbitals of TiO
2. This hybridization creates new states in the band structures (
Figure 14), particularly near the Fermi level. The strong interaction introduces mid-gap states (defect states) within the bandgap of TiO
2, which can act as trapping centers for electrons or holes, thereby altering the material’s optical and electronic properties. The new states near the Fermi level increase the density of the free charge carriers, enhancing the material’s electrical conductivity. Model 1 (adjacent doping), shown in
Figure 14a, exhibits a bandgap of 2 eV with deeper mid-gap states due to strong dopant interactions. In contrast, Model 2 (far apart doping), which represents the electron distribution in the density of states (
Figure 14b), shows a bandgap of 2.2 eV with shallower mid-gap states due to weaker dopant interactions. These mid-gap states arise from the Mo-4
d and Se-4
p states in both models, as observed in the corresponding DOS figures of
Figure 15 and
Figure 16.
In Model 1, the band structure in
Figure 14a displays flatter bands near the Fermi level, suggesting localized states due to strong dopant interactions. Additional bands or split bands may appear due to hybridization between Mo, Se, and TiO
2 orbitals. According to Model 2, the DOS plot in
Figure 16 may exhibit broader peaks closer to the band edges, corresponding to shallow defect states resulting from weaker dopant interactions. The valence band and conduction band edges are closer together, indicating a smaller bandgap. The band structure in
Figure 14b shows more dispersed bands, indicating delocalized states caused by weaker dopant interactions. The bands may appear smoother, with fewer split bands or additional states.
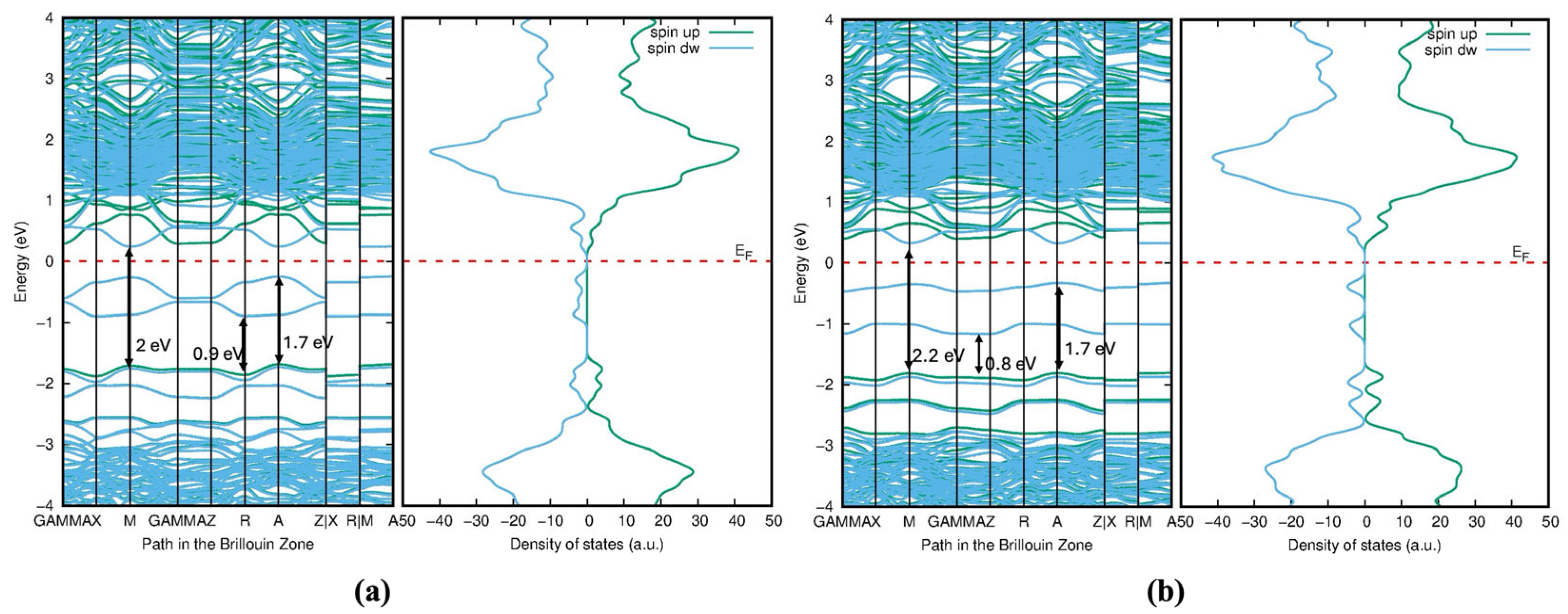
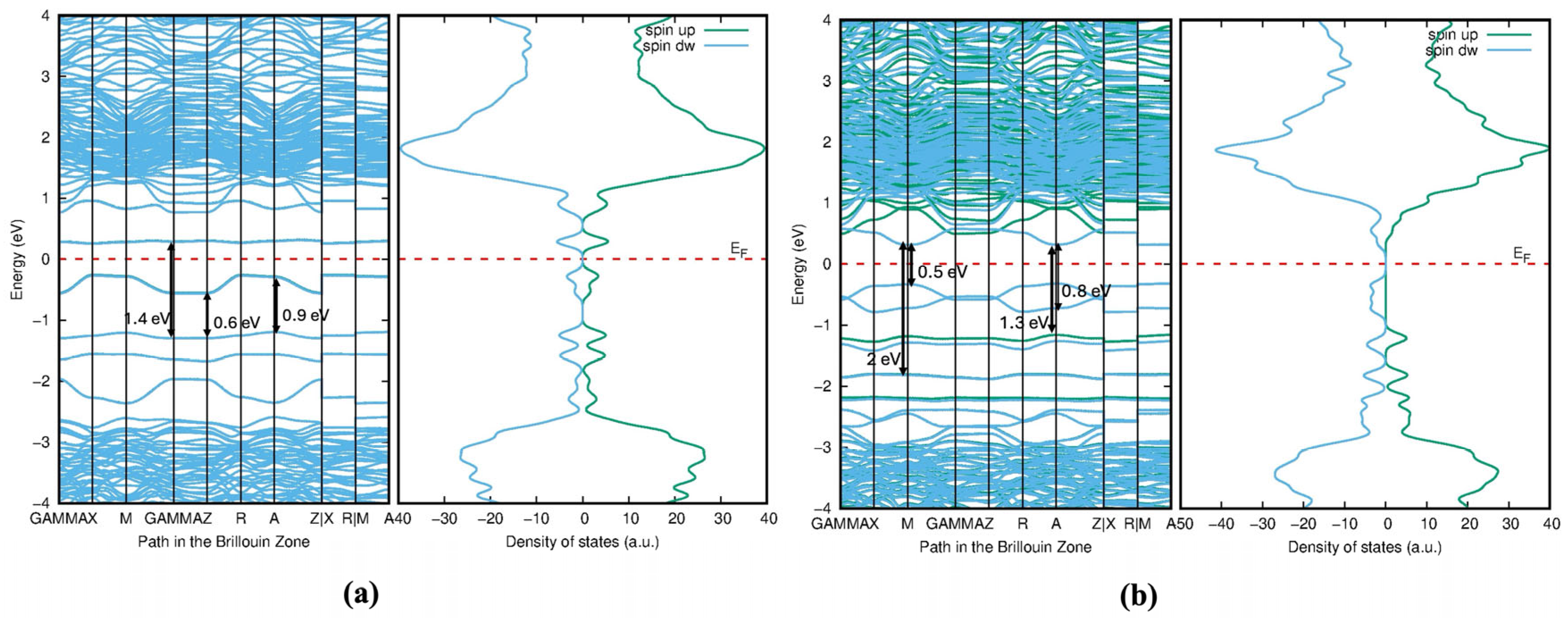
The significant reduction or near-closure of the bandgap in Mo- and Te-doped structures fundamentally alters the electronic properties of TiO2. Instead of behaving as a wide-bandgap semiconductor, the material becomes semi-metallic or highly conductive. This transformation may enhance its performance in applications requiring high electrical conductivity, such as electrocatalysis or sensing devices, but could compromise its effectiveness in photocatalytic processes, where a sizable bandgap is critical for efficient charge carrier separation.
The strong hybridization between Mo-4
d, Te-5
p, and Ti-3
d orbitals in Model 1,
Figure 17 leads to significant bandgap reduction and enhanced visible light absorption. However, the adjacent positioning of dopants may also introduce localized defect states (
Figure 18a) that could act as recombination centers for electron–hole pairs. The DOS plot is more symmetric and shows broader peaks, indicating a more uniform distribution of electronic states. The sharp peaks shown in
Figure 17 and
Figure 19 in the valence band are dominated by O-2
p and Te-5
p states. The introduction of Mo-4
d and Te-5
p states creates defect states within the bandgap of TiO
2, effectively reducing the bandgap. This is crucial for enhancing the material’s photocatalytic activity under visible light. The mid-gap states from Te-5
p orbitals are located just above the valence band, while the Mo-4
d states are near the conduction band. In Model 2,
Figure 18b and
Figure 19, the Mo-4
d orbitals contribute to the DOS, particularly near the conduction band. However, the peaks are broader and less sharp compared with Model 1, indicating weaker interactions due to the spatial separation of dopants. The Te-5
p and Mo-4
d orbitals introduce broader mid-gap states above the valence band maximum, as seen in
Figure 19. These states are more delocalized and lie closer to both the conduction and valence bands.
Overall, these findings highlight how Mo doping in combination with S, Se, and Te can create beneficial electronic states that improve the material’s ability to harness light energy for photocatalytic applications. The strong hybridization among the 5
p orbitals of Te, 3
p orbitals of S, and 4
p orbitals of Se, along with the 3
d orbitals of Ti and 4
d orbitals of Mo, produces intermediate peaks within the bandgap of the doped models. Zheng et al. reported that anionic doping with heavy chalcogen elements such as Te and Se significantly reduces the bandgap of TiO
2, extending its absorption into the visible light spectrum [
15]. Previous studies on mono-doping with S, Se, and Te align with our findings on bandgap reduction and localized electronic states in co-doped systems, as demonstrated by both DFT calculations and experimental results [
34,
35]. Due to the reduction in bandgap energy, the material can absorb visible light. However, partially occupied impurity states appear above the VBM and below the CBM, which can act as traps for excited electrons. This leads to faster electron–hole recombination, thereby limiting the efficiency of the compound in the visible light region. These intermediate states, except for those in the sulfur-doped models, are located near the VBM and function as shallow acceptor levels, primarily consisting of Se-4
p and Te-5
p orbitals. These states have the ability to capture photoexcited holes, which help to reduce the recombination rate of electron–hole pairs. Additionally, the presence of electron vacancies near the valence band can generate an anodic photocurrent, indicating a higher likelihood for electrons to be excited into the intermediate states. In these states, lower photon energy is sufficient to promote electrons into the conduction band. These results suggest that the degree of bandgap narrowing depends on the distance between the doped Mo atom and the chalcogen atom. Model 2, where the dopants are farther apart, exhibits a more significant bandgap reduction compared with Model 1, where the dopants are adjacent.
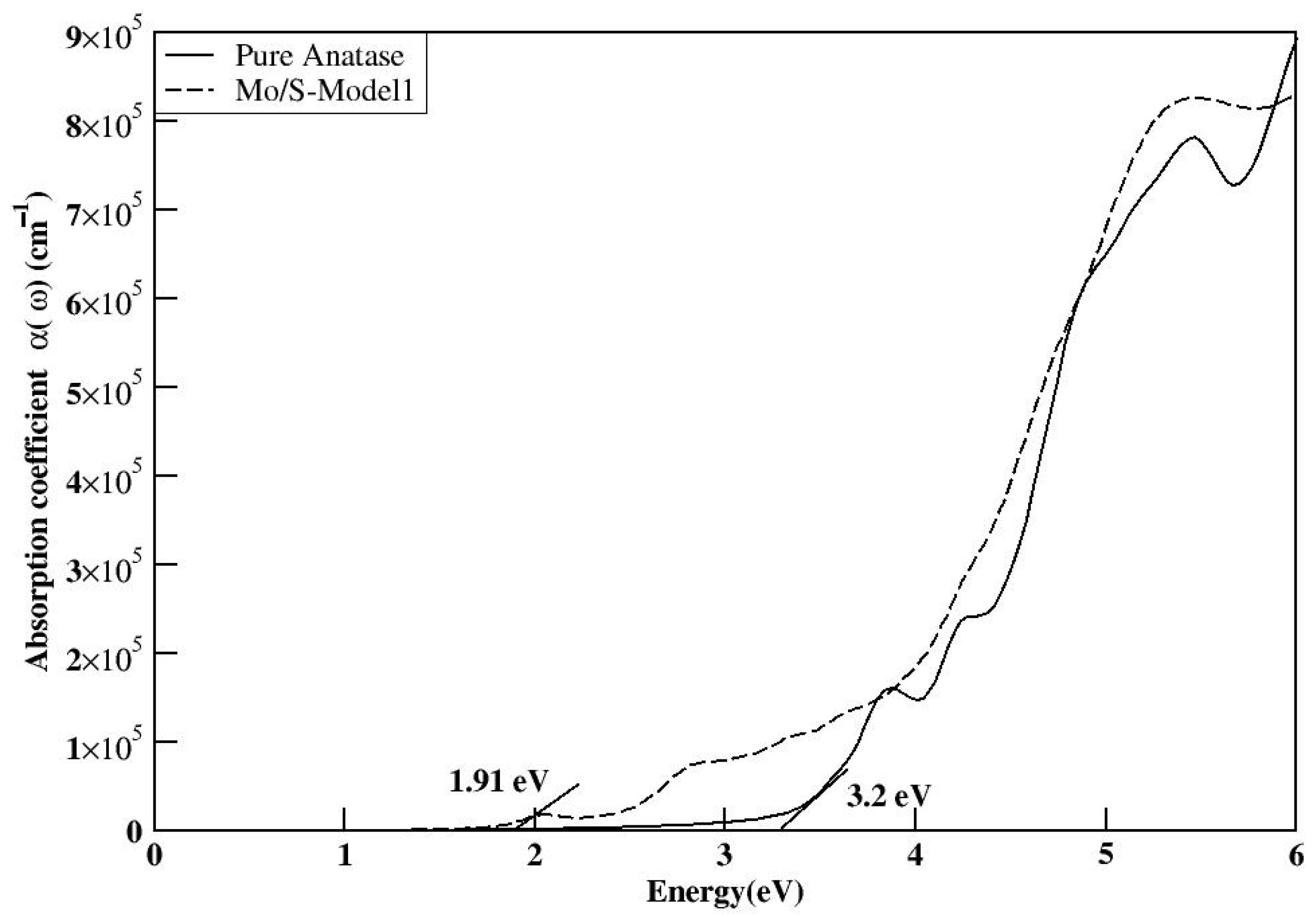
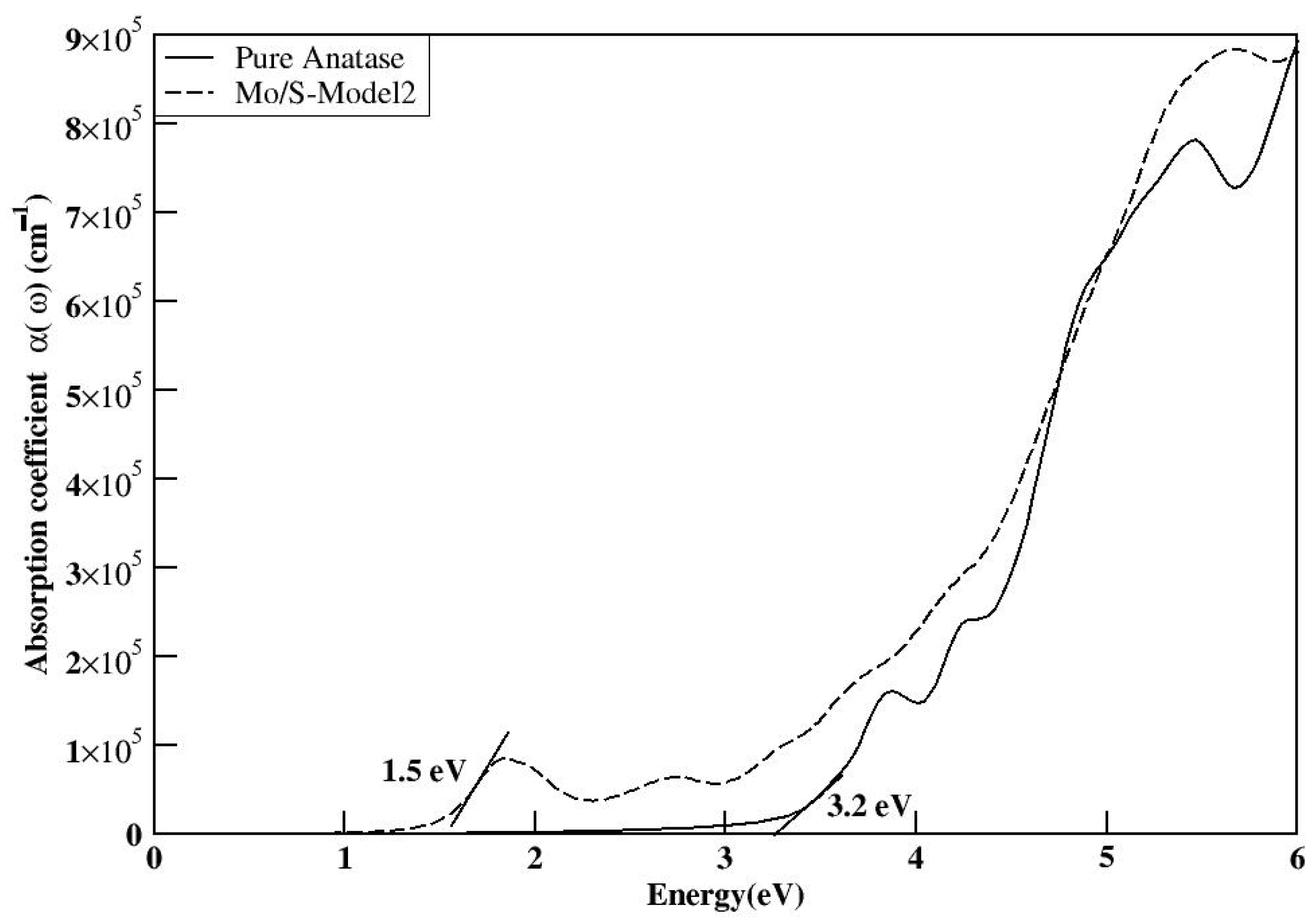
The electronic structures of the three co-doped models are nearly identical; however, the Mo and S co-doped structures exhibit fewer impurity states compared with the Se and Te co-doped structures. This reduction in impurity states may enhance the efficiency of the S-doped models by decreasing the recombination rate compared with the other doped models. The MoS-1 model exhibited a reduced bandgap of 2.52 eV and demonstrated optical absorption in
Figure 20 comparable to the other models. It was concluded that the adjacent positioning of the dopants in the MoS-1 model is the most optimal structure, compared with the other co-doped configurations.
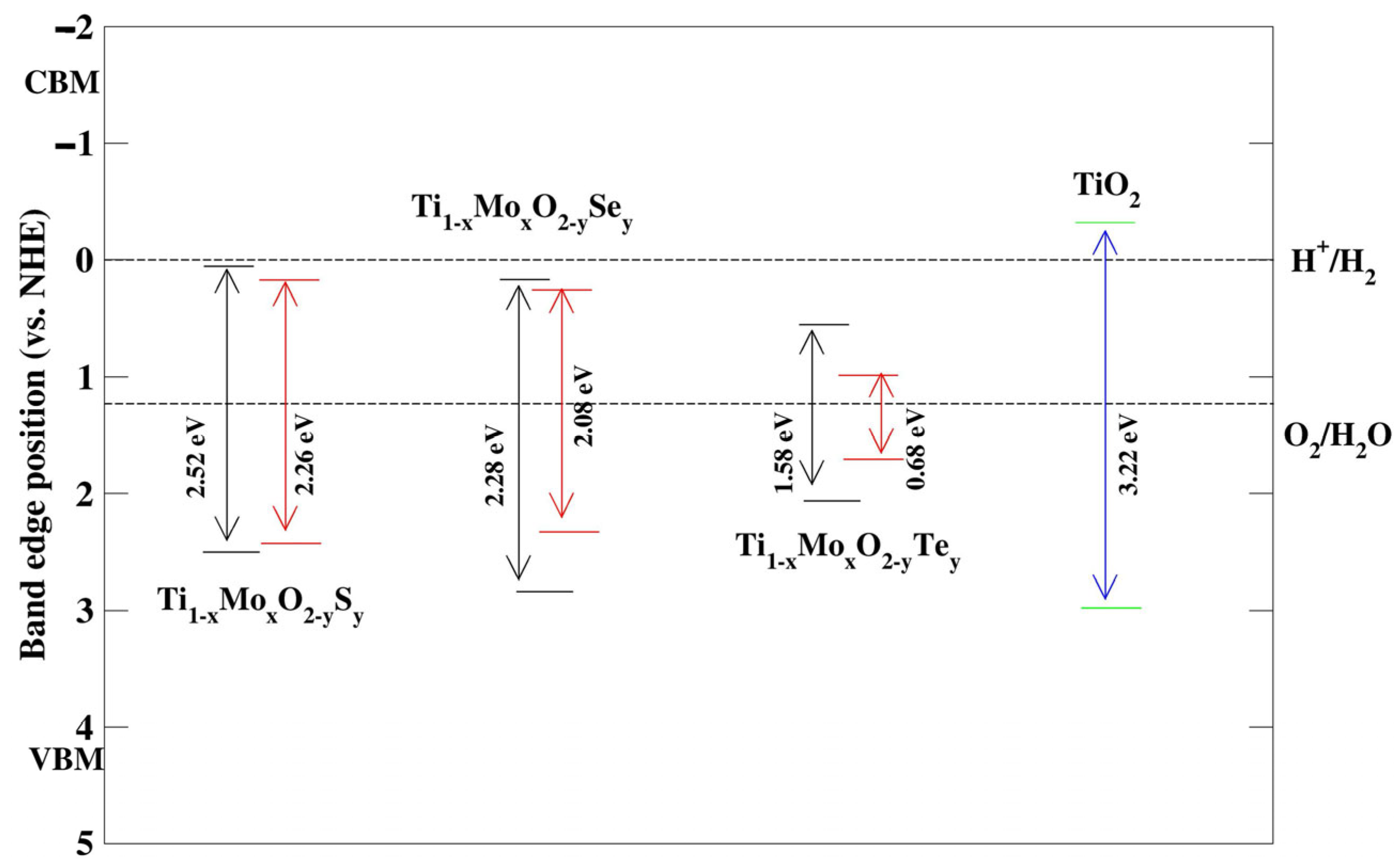
Photocatalytic Properties
The CBM and VBM potentials of the co-doped systems in both Model 1 and Model 2 can be determined as shown in
Figure 21 (
Table 2) relative to the water reduction potential of H
+/H
2 (0 eV against the normal hydrogen electrode, NHE) and the water oxidation potential of O
2/H
2O (1.23 eV). These calculations follow the formula established by Butler and Ginley [
36]. We determined the CBM and VBM potentials for both the pure and co-doped systems by applying the relevant Equations (5)–(9).
Here, E0 =−4.5 eV is the scale factor that bridges the absolute vacuum scale with the reference redox level of the normal hydrogen electrode (NHE), Eg signifies the bandgap energy, while X denotes the absolute electronegativity of the system, calculated using the following formulas:
For Mo-doped Te:
where (x = 0.0208, y = 0.0208) and
XTi,
XO,
XMo,
XS,
XSe, and
XTe are the absolute electronegativities of Ti, O, Mo, S, Se, and Te atoms, respectively, and their corresponding values are 3.45, 7.54, 3.9, 6.22, 5.89, and 5.49 eV [
37].
All the co-doped systems except the Mo and Te co-doped models show stronger oxidation power as much as pure TiO2 does compound under visible light irradiation. Mo and Te co-doped models have better oxidation abilities in the infrared region. However, they show a lower ability of oxidation power than the pure anatase. Our findings suggest that the Mo/S and Mo/Se co-doped anatase systems possess significant oxidation power, making them suitable candidates for applications in oxidative photocatalysis under visible light. However, their limited reduction ability indicates that they may be less effective for photocatalytic applications that require both oxidation and reduction capabilities such as overall water splitting. In this case, these co-doped systems are effective for oxidation-driven photocatalytic applications, such as the degradation of organic pollutants or water oxidation reactions, where oxidative power is critical.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}