Abstract
Kapakahines A–G are natural products isolated from the marine sponge Carteriospongia sp., characterized by complex molecular architectures composed of fused rings and diverse functional groups. Preliminary studies have indicated that some of these peptides may exhibit cytotoxic and antitumor activities, which has prompted interest in further exploring their chemical and pharmacokinetic properties. Computational chemistry—particularly Conceptual Density Functional Theory (CDFT)-based Computational Peptidology (CP)—offers a valuable framework for investigating such compounds. In this study, the CDFT-CP approach is applied to analyze the structural and electronic properties of Kapakahines A–G. Alongside the calculation of global and local reactivity descriptors, predicted ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiles and pharmacokinetic parameters, including pKa and LogP, are evaluated. The integrated computational analysis provides insights into the stability, reactivity, and potential drug-like behavior of these marine-derived cyclopeptides and contributes to the theoretical groundwork for future studies aimed at optimizing their bioactivity and safety profiles.
1. Introduction
Kapakahines A–G are a group of cyclic peptides originally isolated from the marine sponge Carteriospongia sp. These compounds possess structurally complex frameworks featuring multiple fused rings and diverse functional groups. Their distinctive architecture has attracted interest for exploratory research into marine-derived bioactive compounds [1,2,3,4,5,6,7,8,9].
Preliminary studies have reported that some Kapakahines exhibit cytotoxic and antitumor properties, including potential mechanisms such as topoisomerase I inhibition and microtubule disruption. These observations suggest possible relevance for anticancer research, although no clinical development has yet been pursued, and their pharmacological potential remains under early-stage investigation. Some Kapakahines have also been reported to display antibacterial and antifungal activities, raising questions about their broader bioactivity profiles [9,10,11].
In this context, computational chemistry—particularly Conceptual Density Functional Theory (CDFT)—offers a valuable framework for characterizing the electronic structure, reactivity, and physicochemical behavior of peptide-based compounds such as the Kapakahines. The CDFT-based Computational Peptidology (CDFT-CP) approach provides insights into global and local reactivity patterns, which are useful for predicting molecular stability, interaction propensity, and drug-like features [12,13,14,15,16].
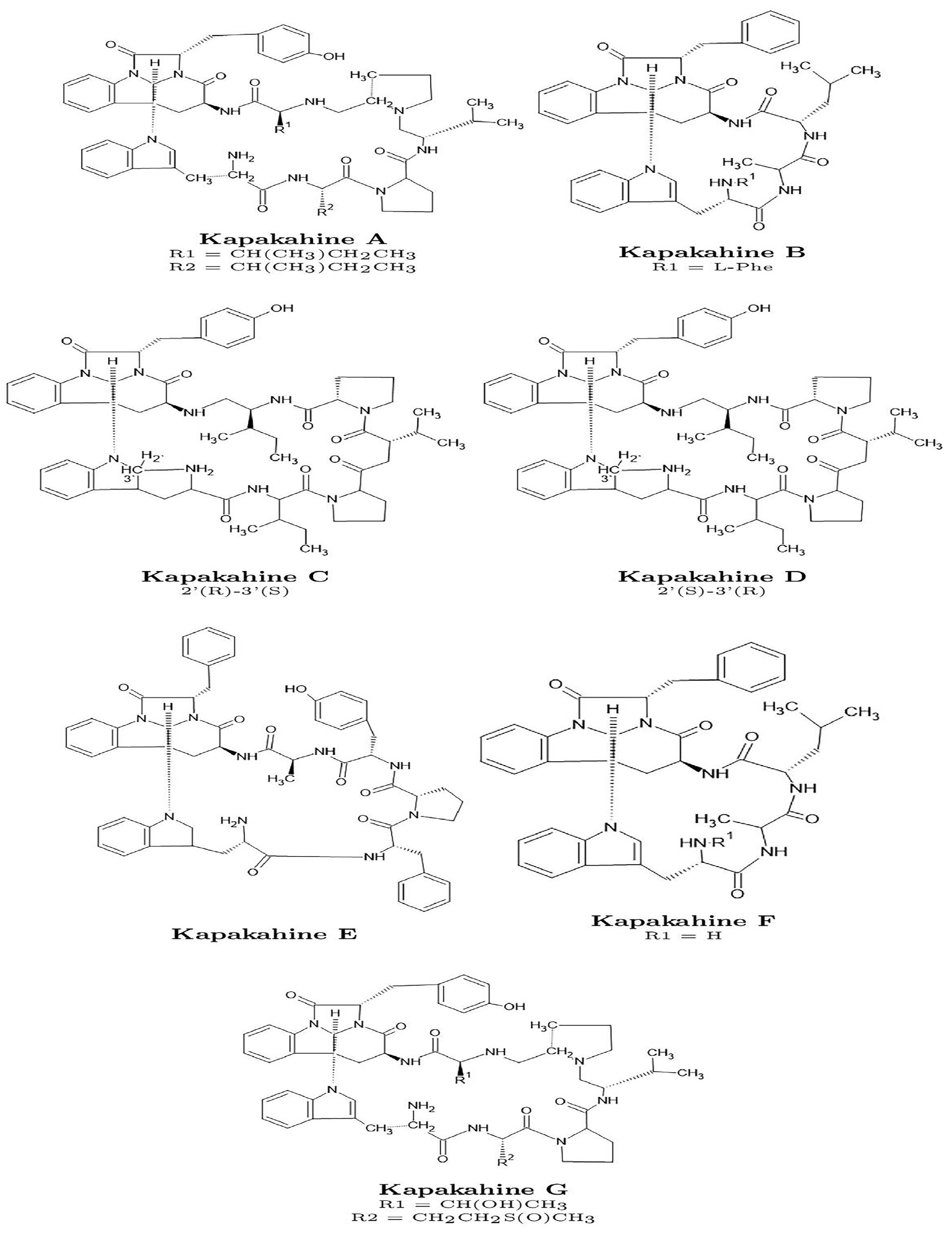
This study applies the CDFT-CP methodology [17] to analyze the Kapakahines A–G, whose structures are shown in Figure 1. In addition to conventional CDFT descriptors, less commonly reported properties such as hyperhardness, cubic electrophilicity, and the electrophilic descriptor are computed to deepen the understanding of molecular reactivity. The local hypersoftness and molecular electrostatic potential (MEP) maps are also visualized to explore nucleophilic and electrophilic domains. Furthermore, computational estimates of pKa and LogP, derived from CDFT parameters, are presented. Finally, predicted ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiles are compiled and discussed to evaluate key pharmacokinetic and toxicological aspects [18,19,20,21]. Overall, this integrative study aims to provide a comprehensive theoretical perspective on the chemical behavior and potential pharmaceutical relevance of this class of marine cyclopeptides.
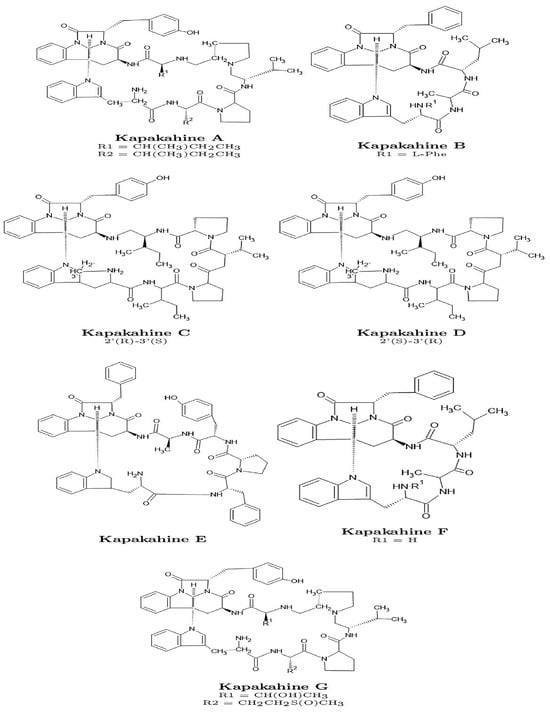
Figure 1.
Graphical sketches of the molecular structures of the Kapakahines A–G.
2. Materials and Methods
2.1. Conceptual DFT Studies
In this research, the Kohn–Sham (KS) framework [22,23,24,25], combined with the Conceptual Density Functional Theory (CDFT) methodology [12,13,14,15,16], was applied to evaluate the molecular energies, electronic densities, and orbital characteristics of the studied ligands. This analysis included the determination of the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO). The molecular conformers were generated using MarvinView 17.15 from ChemAxon [http://www.chemaxon.com] (accessed 25 March 2025) by conducting Molecular Mechanics calculations based on the MMFF94 force field [26,27,28,29,30].
Subsequently, geometry optimization and vibrational frequency calculations were performed using the Density Functional Tight Binding (DFTBA) methodology [31]. Further refinement of the molecular geometry, frequency analysis, and evaluation of electronic properties and chemical reactivity descriptors were conducted. The MN12SX/Def2SVP/H2O computational model was employed for these calculations, incorporating the MN12SX screened-exchange density functional [32], and the Def2SVP basis set [33,34]. The molecular charge was assumed to be neutral, while both the radical anion and cation were examined in the doublet spin state. These computations were executed using the Gaussian 16 Rev. C01 software package [29] in conjunction with the SMD solvation model [35]. To confirm that the optimized structure represented a true energy minimum, the absence of imaginary frequencies was verified. Notably, this computational model has been validated in previous studies for its adherence to the “Koopmans in DFT” (KID) approach [17].
2.2. Computational ADMET
Understanding how a molecule behaves within the body is essential for the development of new therapeutic drugs. The Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties serve as key indicators in this process. Instead of relying solely on experimental methods, computational models are frequently employed to assess these parameters [18,19,20,21]. For this analysis, Chemicalize, a tool developed by ChemAxon (http://www.chemaxon.com) (accessed 25 March 2025), was utilized. Furthermore, additional insights into pharmacokinetic parameters and ADMET characteristics were gathered using pkCSM, a predictive software that analyzes these properties based on SMILES (https://biosig.lab.uq.edu.au/pkcsm/) (accessed 25 March 2025), together with its associated AI-based derived software Deep-PK (https://biosig.lab.uq.edu.au/deeppk/) (accessed 25 March 2025).
3. Results and Discussion
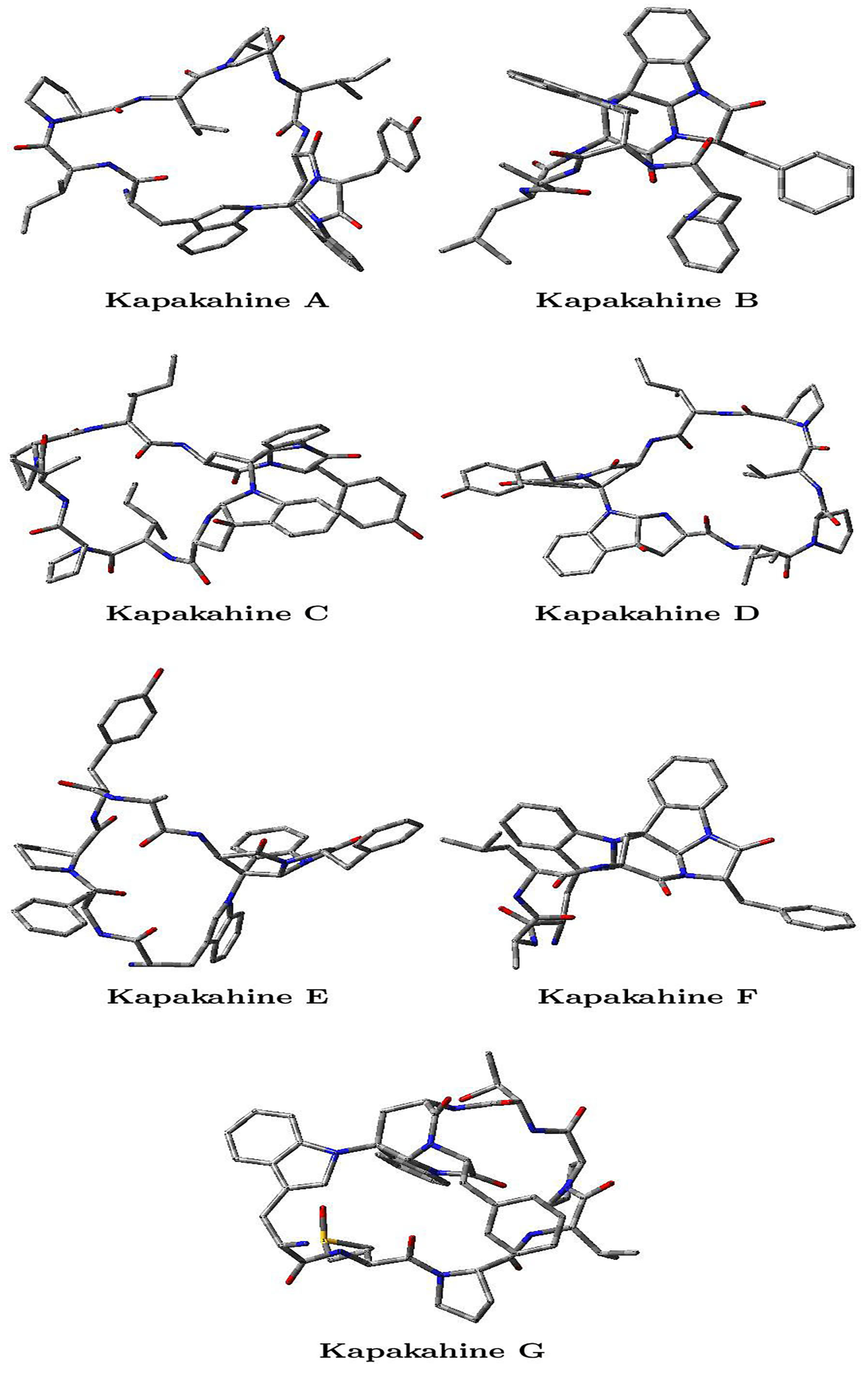
As indicated in Section 2, the molecular structures of the optimized conformers of the Kapakahine A–G peptides were obtained by using the Density Functional Tight-Binding Approximation (DFTBA) model available in the Gaussian 16 software to achieve our research goals. Then, the reoptimization of the selected conformers was conducted by resorting to the MN12SX density functional with the Def2SVP basis set and the SMD solvent model, with water as the solvent. To ensure accuracy, the authors used frequency calculation analysis to verify that all the structures matched the lowest energy conformations. Once verified, the electronic properties of the same model chemistry were calculated. This methodology allowed for a detailed analysis of the electronic properties of the model chemistry, which can be useful in understanding the behavior and function of the studied system. The findings of this study may provide insights into future research in the field of chemistry and may have practical applications in material science, drug design, and other areas. The optimized molecular structures of the studied cyclopeptides are displayed in Figure 2.
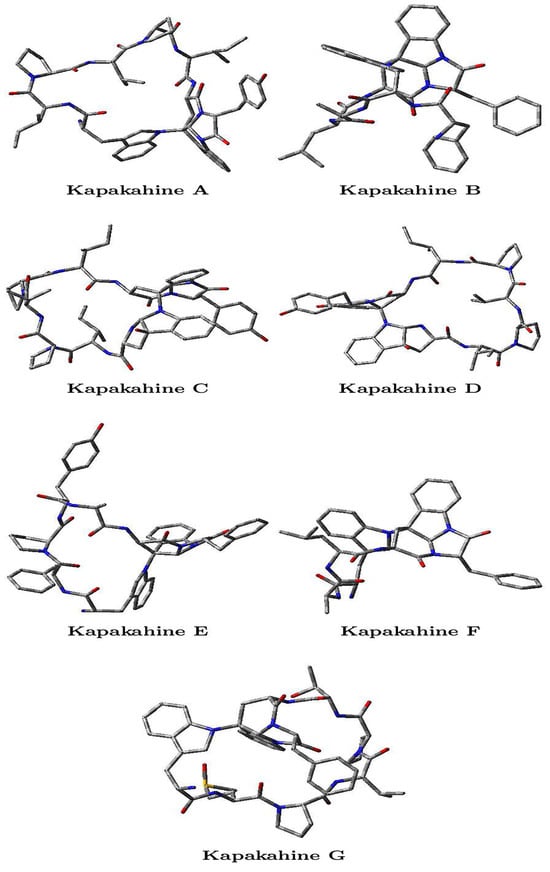
Figure 2.
Optimized molecular structures of the Kapakahines A–G, where the oxygens O are marked in red, the nitrogens N are marked in blue, the carbons C are gray, and the hydrogens H are not shown.
The Cartesian coordinates of the optimized Kapakahines A–G are provided in Tables S1–S7 of the Supplementary Materials.
3.1. Chemical Reactivity Properties
This section of the study focuses on evaluating the results to verify compliance with the KID (Koopmans in DFT) protocol. In previous analyses, various descriptors related to the outcomes of HOMO and LUMO calculations were examined in relation to those obtained through vertical ionization energy (I) and electron affinity (A) using the SCF procedure, where SCF stands for the Self-Consistent Field method. A correlation exists between these three fundamental descriptors and Koopmans’ theorem by linking to -I, to -A, and their interactions, which define the HOMO-LUMO gap: , , and . It is important to highlight that the descriptor is based on an approximation that holds true only when the radical anion’s HOMO (SOMO) closely resembles the LUMO of the neutral system. To enhance the accuracy of this approximation, our research group has introduced an additional descriptor, SL, to aid in its validation. In order to condense all this information, a final descriptor has been defined as , which must be equal to zero for the exact density functional and represents a measure of the quality of the density functional used [17]. The results of this analysis are shown in Table 1.

Table 1.
Orbital energies (HOMO, LUMO, and SOMO), HOMO-LUMO energy gap, and KID descriptors (all in eV) used to evaluate the Koopmans-like behavior of the MN12SX density functional in the context of the Kapakahine A–G marine cyclopeptides.
As can be seen from the results presented in Table 1, the agreement with the Koopmans-like behavior of the MN12SX/Def2SVP/H2O model chemistry is excellent. Thus, we can be confident that the CDFT descriptors calculated in the next step will be of high quality. It is well known that the value of the HOMO-LUMO gap is dependent on the choice of the exchange-correlation density functional. Thus, the results from Table 1 reinforce the election of the MN12SX density functional because its observed Koopmans-complaining behavior warrants the quality and accuracy of the HOMO and LUMO orbitals, and consequently of the HOMO-LUMO gap in all the studied cases.
The working formulae for the global CDFT descriptors are as follows [12,13,14,15,16,36,37,38]:
- Electronegativity
- Global Hardness
- Global Electrophilicity
- Global Hyperhardness
- Global Softness
- Nucleophilicity
- Electroaccepting Power
- Electrodonating Power
- Net Electrophilicity
While the global electrophilicity has been derived by considering a second-order Taylor expansion of the system energy, there has been recently presented two new descriptors of electrophilicity based on a third-order Taylor expansion of the energy of the molecular system. Thus, they are dependent on the N-2 state as well as on the global hyperhardness . The first one is the cubic electrophilicity [39] that has been defined as follows:
and in practice is evaluated with the aid of the Multiwfn 3.8 software [40,41,42] as
where = , , and .
The second one is the electrophilic descriptor (ED) [43,44], which is also related to the N-2 state and the global hyperhardness and may be evaluated through the following working equation [40,41,42]:
where . However, it must be noted that , , , and in the previous equation must be calculated in a different way as before [40,41,42]. Thus, = a, = −a, = 2 (b − a c), and = −3 c (b − a c). Following this, c = , b = , and a = . Finally, A′ = E(N + 1) − E(N), that is, the electron affinity, but differs with standard definition by the sign.
In this study, the energies of the optimized molecular structures of every peptide have been calculated in the neutral state, as well as for the N + 1, N − 1, N + 2, and N − 2 states, so as to estimate the values of the first and second ionization energies (I1 and I2) and electron affinities (A1 and A2) whose values are displayed in Table 2:
Table 2.
First and second ionization energies (I1 and I2) and electron affinities (A1 and A2) for the Kapakahine A–G marine cyclopeptides (all in eV) calculated through energy differences.
The results presented in Table 2 were fed into Equations (1)–(12) in order to obtain the values of the CDFT descriptors for the Kapakahine A–G marine cyclopeptides and the results are shown in Table 3:
Table 3.
Key global reactivity descriptors for the Kapakahine A–G marine cyclopeptides including electronegativity (), hardness (), electrophilicity () (all measured in eV), softness (S) (expressed in eV−1), nucleophilicity (N), electrodonating power (), electroaccepting power (), net electrophilicity(), cubic electrophilicity (), and the electrophilic descriptor (ED) (also in eV).
From Table 3, it can be seen that the electronegativity [12,13,45] attains a maximum value for Kapakahine C, followed by the B and G variants, while the minimum value is observed for Kapakahine F. Additionally, Kapakahine C displays the largest value of chemical hardness , making it the most stable molecule. Similarly, for electronegativity , Kapakahines B and F follow, although the values for the other peptides also indicate high stability. Conversely, global softness S is determined as the inverse of , allowing for a direct comparison of the various molecules analyzed in this study, with values approximately within the range of 0.214–0.224 eV−1.
Global electrophilicity is a descriptor used to assess the reactivity of a molecular system toward electron-donating species. In CDFT, it is defined as a function of the chemical potential and global hardness of the system [12,13]. Upon inspecting Table 3, it can be concluded that the maximum value of this descriptor is associated with Kapakahine G, while the minima correspond to the E and F variants.
According to the scale proposed by Domingo et al. [16] for the nucleophilicity N, which is calculated through Equation (6) and the results from Table 3, it can be concluded that Kapakahines A, D, E, and F can be considered strong nucleophiles, while the remaining peptides will be moderate nucleophiles.
Within CDFT, the concepts of electrodonating and electroaccepting powers ( and , respectively) [37] play a crucial role in understanding molecular properties and reactivity. By comparing the results for the Kapakahines A–G in Table 3, it can be concluded that as is usual for these kinds of molecules, the values of the electrodonating power will be larger than the electroaccepting . The concept of net electrophilicity was introduced by Chattaraj [38] as a way to compare the relative magnitudes of and . From Table 3, it can be seen that the results for this descriptor will display the largest value for Kapakahine G, with the results for Kapakahines B and C being very similar, with Kapakahine D presenting the smallest value of all the considered peptides.
Finally, the results for the cubic electrophilicity presented in Table 3 show the maximum value for Kapakahine G, with nearly identical values for Kapakahines B and C, and smaller values for Kapakahines A, D, E, and F. On the other hand, Kapakahine C has the largest value for the electrophilic descriptor ED, while Kapakahine D represents the smallest value. Further studies will be needed to evaluate the ability of these two new CDFT descriptors in explaining the chemical reactivity of the studied compounds.
3.2. The Molecular Electrostatic Potential
The molecular electrostatic potential (MEP) is a crucial concept in computational chemistry, providing valuable insights into the charge distribution and reactivity of a molecule. It represents the electrostatic potential at a point in space, generated by the distribution of electron density within a molecule. The MEP is often visualized in 3D, showing regions of positive, negative, and neutral potential, which correspond to areas of electrostatic attraction or repulsion around the molecule [46,47,48,49,50]. In computational studies, the MEP is typically calculated using methods like density functional theory (DFT) or Hartree–Fock theory, where the electron density distribution is used to evaluate the electrostatic potential at various points in the surrounding space. This potential can be interpreted to predict various molecular properties such as dipole moments, reactive sites, and interactions with other molecules or solvents.
The MEP plays an important role in understanding molecular reactivity, particularly in the context of nucleophilic and electrophilic reactions. Regions of negative electrostatic potential correspond to electron-rich areas, which tend to attract electrophiles, while regions of positive potential correspond to electron-poor areas, which are more likely to interact with nucleophiles. This makes the MEP an invaluable tool for predicting reaction mechanisms, molecular recognition, and the behavior of molecules in complex environments. Furthermore, the MEP is used in a variety of applications, such as the design of new drugs, materials, and catalysts, where understanding how a molecule will interact with other entities is critical. By visualizing the electrostatic potential, researchers can gain a deeper understanding of the factors governing molecular interactions and reactivity, facilitating more efficient and targeted designs in computational chemistry.
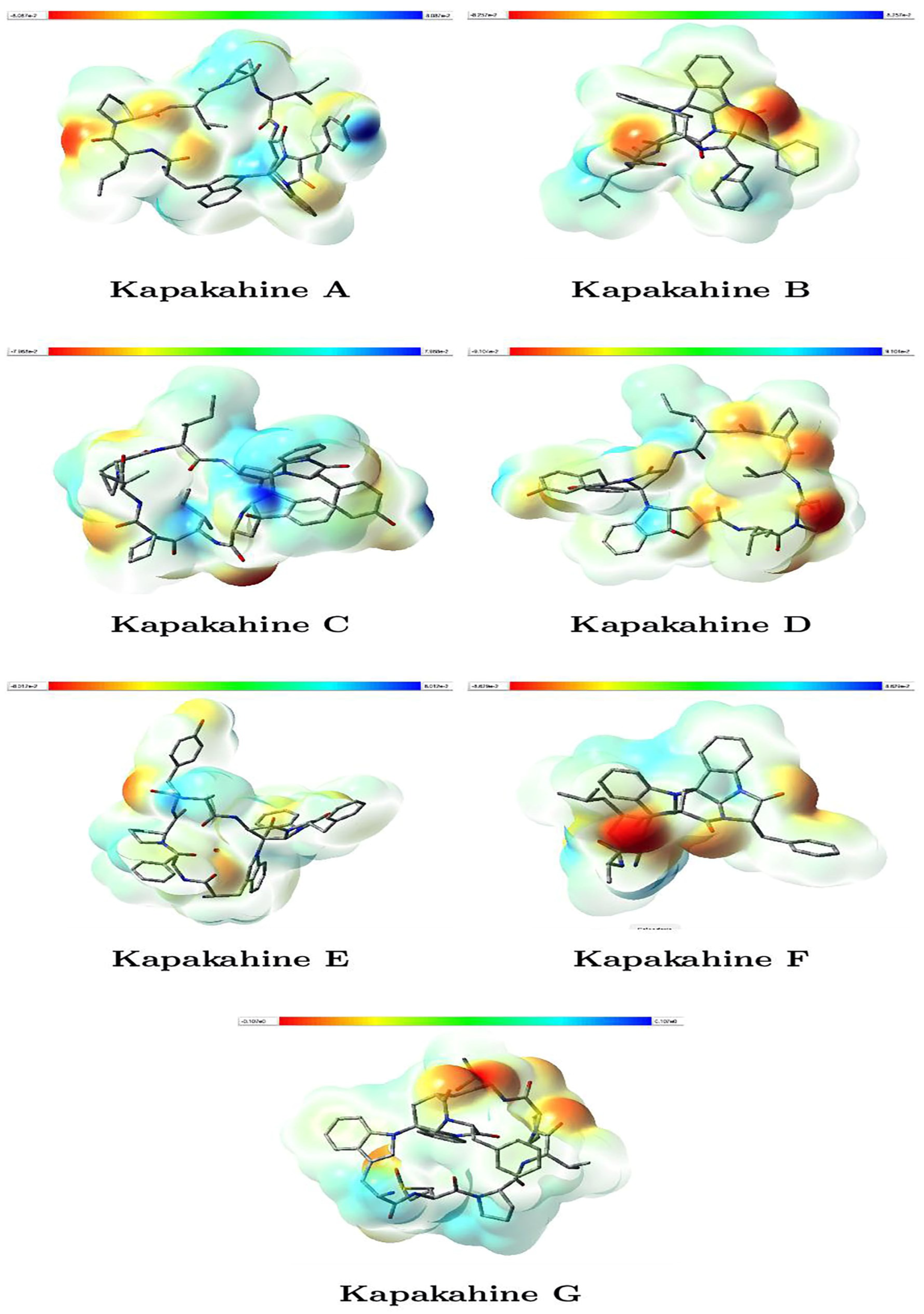
Figure 3 illustrates the molecular electrostatic potential (MEP) maps for Kapakahines A–G. The reddish regions indicate areas of negative electrostatic potential (electron-rich, likely nucleophilic sites), while bluish zones represent positive potential (electron-deficient, likely electrophilic sites). The isovalue was set at 0.020000, enabling consistent comparisons, while the range shown in the color bars is expressed in atomic units (a.u.).
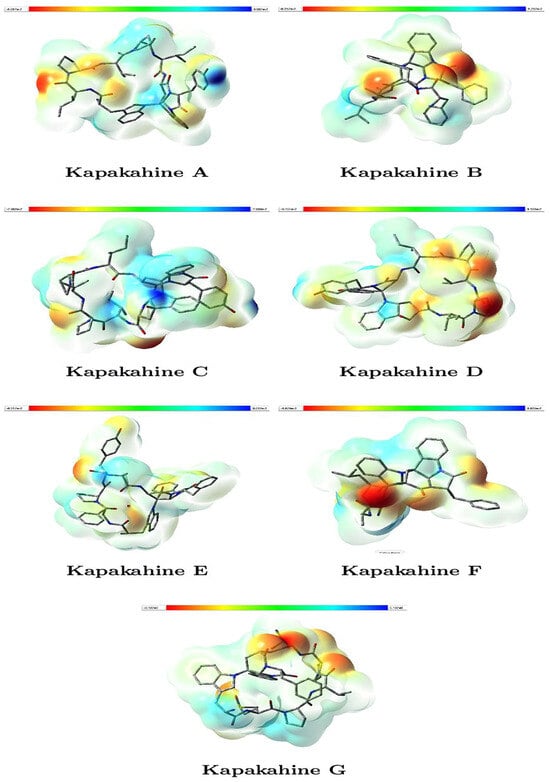
Figure 3.
Graphical representations of the MEP of the Kapakahines A–G. (isovalues = 0.020000; atomic units (a.u.)).
- Kapakahines display prominent red regions localized on carbonyl oxygens and amide groups, suggesting that these zones are strong nucleophilic centers likely to interact with electrophilic species or metal ions.
- The electrophilic (blue) areas are generally smaller, often surrounding hydrogen atoms or less electron-rich zones, consistent with typical peptide behavior.
- Kapakahine G, with the highest electrophilicity index ( = 1.423 eV) and net electrophilicity ( = 6.258 eV) (Table 3), shows extended blue regions, supporting its pronounced electron-acceptor character and potential interaction with nucleophilic residues in biological systems.
- Kapakahine D, with the lowest electrophilic descriptor (ED = 2.538 eV), presents a more balanced MEP with limited blue zones, suggesting lower reactivity, which matches its milder predicted ADMET profile.
3.3. The Local Hyper-Softness
Global descriptors provide an overall assessment of a molecule’s chemical reactivity [12,13,14,15,16]. However, local reactivity descriptors have been introduced to examine variations in reactivity within different regions of a molecule. One of the most significant local reactivity descriptors is the Dual Descriptor (DD), which is mathematically expressed as DD = . This descriptor is closely linked to Fukui functions, another set of essential local reactivity indicators [51,52].
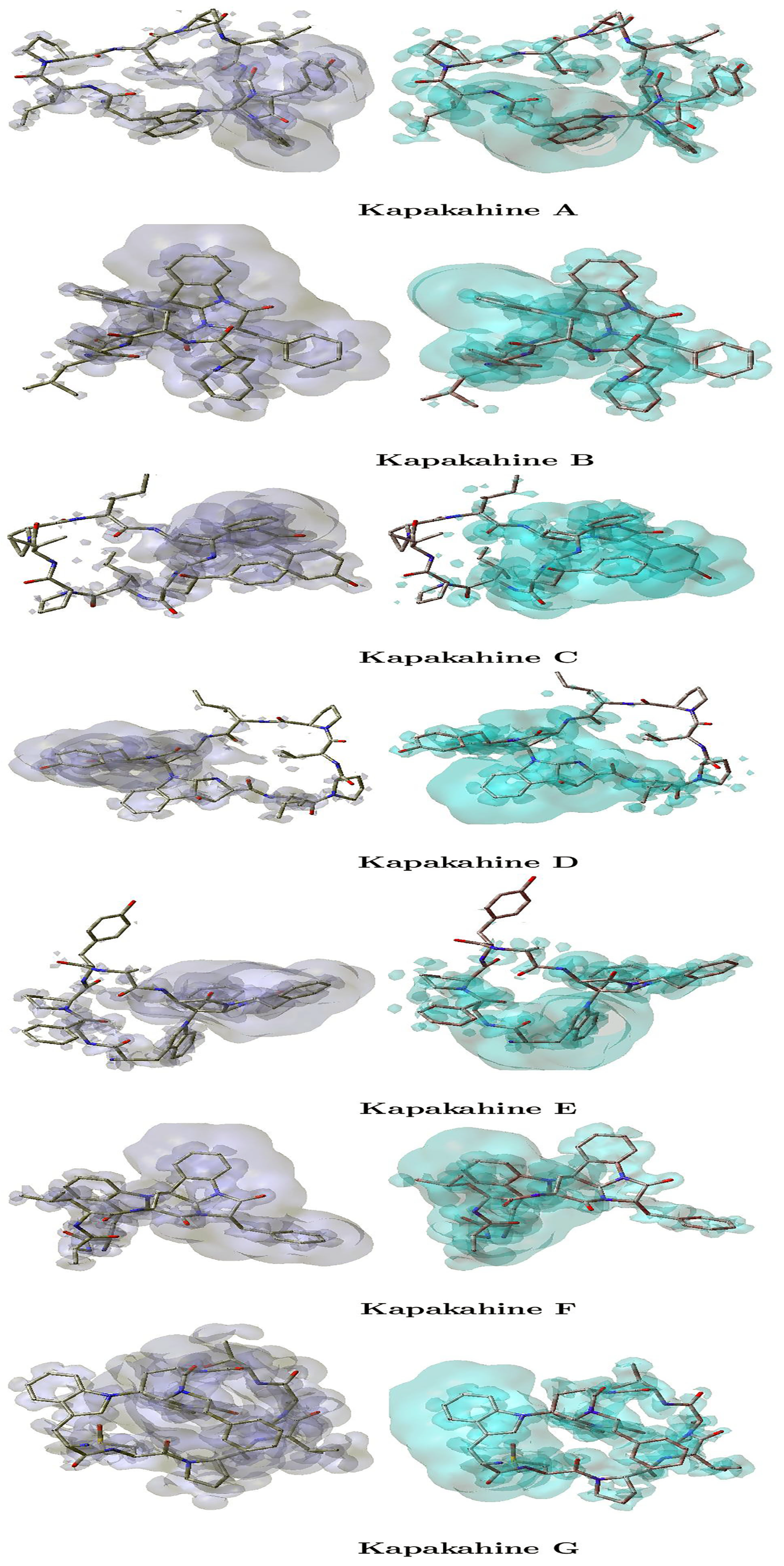
Within the framework of Density Functional Theory (DFT), the dual descriptor serves as a crucial tool for analyzing molecular reactivity. However, it has been shown that the local hyper-softness LHS [53,54] is a better descriptor of the chemical reactivity than the DD when the comparison of molecular systems of different size is being carried out [36]. According to the analysis performed by Martínez-Araya [36], the working formula for the local hyper-softness is LHS ≈ S2 × DD, where S is the global softness; it can be estimated from our calculations with the aid of the Multiwfn 3.8 software [40,41,42].
The local hyper-softness (LHS) graphical representations for the Kapakahines A–G are displayed in Figure 4, where the left panels are regions where LHS > 0 indicating electrophilic reactivity, i.e., areas susceptible to nucleophilic attack, and the right panels are zones with LHS < 0 reflecting nucleophilic reactivity, i.e., favorable for electrophilic attack.
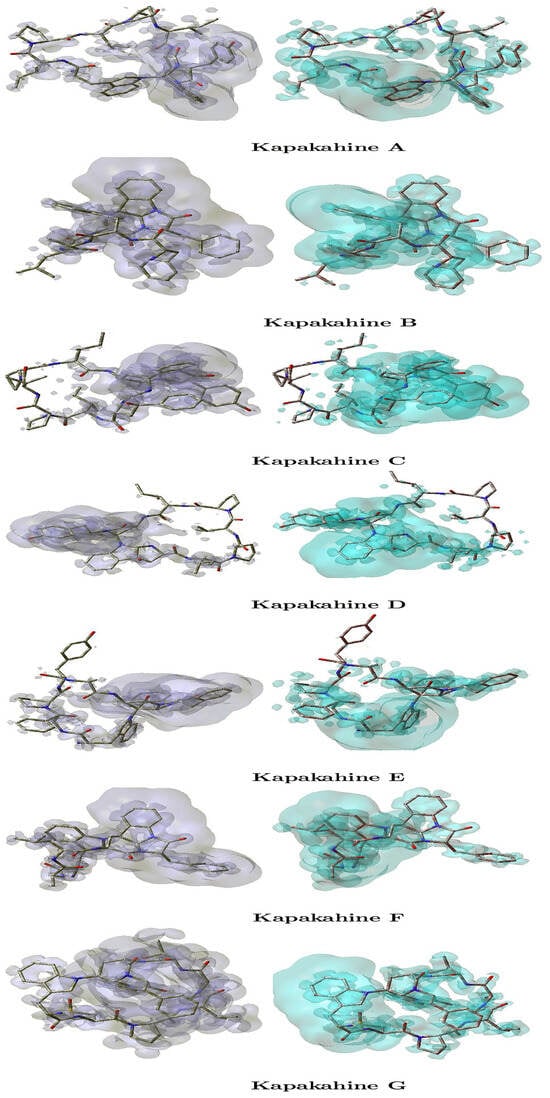
Figure 4.
Graphical representations of the local hyper-softness of the Kapakahines A–G. Left: LHS > 0. Right: LHS < 0.
An analysis of Figure 4 reveals the following:
- In all peptides, LHS > 0 regions (electrophilic centers) are mainly found near amide hydrogen atoms and electron-deficient carbons.
- LHS < 0 zones (nucleophilic centers) overlap with the carbonyl and ether oxygens—corroborating the MEP findings.
- Kapakahine C, which has the highest electronegativity ( = 3.695 eV) and electrophilic descriptor (ED = 7.466 eV), exhibits the most intense and spatially distinct LHS regions. This suggests highly polarized local reactivity, making it a strong candidate for site-selective interactions.
- Kapakahine F, with moderate nucleophilicity N and low , shows more diffuse LHS regions, implying lower spatial reactivity density.
- The intensity and localization of LHS maps also appear to correlate with chemical hardness (). For example, Kapakahine C ( = 5.033 eV) has sharply confined reactive zones, consistent with high chemical rigidity.
3.4. Computational Pharmacokinetic Properties and ADMET
The process of discovering and developing new drugs requires significant time and financial investment. Computational ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) refers to the use of computer-based models and simulations to predict the pharmacokinetic and toxicological properties of potential drug candidates. These in silico approaches help streamline drug discovery by assessing a compound’s behavior in the human body early in development, reducing the need for extensive laboratory testing. By leveraging machine learning, molecular modeling, and database-driven predictions, computational ADMET enhances efficiency, lowers costs, and minimizes the risk of late-stage failures in drug development [18,19,20,21].
pKa, the acid dissociation constant, plays a crucial role in ADMET predictions, as it influences a drug’s ionization state at physiological pH. This, in turn, affects solubility, membrane permeability, and protein binding, all of which impact a compound’s absorption and distribution. Additionally, pKa can influence metabolic stability and excretion pathways, as ionized drugs often exhibit different interactions with enzymes and transporters. Accurate pKa predictions are essential in computational ADMET modeling to optimize drug design and improve pharmacokinetic properties. We recently conducted a study on the computational prediction of the pKa values of small peptides using conceptual DFT descriptors. Our findings indicate that the equation pKa = 16.3088 − 0.8268 can serve as an effective starting point for predicting the pKa of larger peptides [55]. Building on the methodology from our previous work, the global hardness of each peptide in Table 3 was examined and we applied the same equation to calculate the pKa values of these molecules. The results are presented in Table 4.
Table 4.
pKa and LogP results based on CDFT for the Kapakahine A–G marine cyclopeptides.
LogP, the partition coefficient between octanol and water, is a key parameter in ADMET studies, as it reflects a compound’s lipophilicity. Lipophilicity influences drug absorption, membrane permeability, and distribution in the body. Highly lipophilic drugs may have better membrane penetration but can also exhibit poor solubility and increased metabolic instability, leading to potential toxicity. Conversely, overly hydrophilic compounds may struggle with cell permeability and bioavailability. Optimizing LogP is essential in drug design to balance solubility, permeability, and metabolic stability for improved pharmacokinetic properties. An electronic analogue of LogP based on CDFT descriptors has been recently presented [56]. According to the proposed methodology, LogP = log QP, where QP is defined through the following working equation:
where A is the molecule being studied (in this case, any of the Kapakahines A–G), while = 3.9907 eV, = 8.7640 eV, = −3.4103 eV, and = 8.1104 eV, whose values were obtained through calculations performed with the same model chemistry as for the cyclopeptides considered in this work.
By feeding the results from Table 4 for each peptide into Equation (13), the corresponding LogP values were obtained, which are also shown in Table 4.
Finally, as part of the supplementary information, the key ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties [21] calculated for the studied cyclopeptides are summarized in Table 5. These predictions were generated using the Chemicalize platform (http://www.chemaxon.com) (accessed 25 March 2025) as well as the pkCSM tool (https://biosig.lab.uq.edu.au/pkcsm/) (accessed 25 March 2025) and its AI-enhanced extension, Deep-PK (https://biosig.lab.uq.edu.au/deeppk/) (accessed 25 March 2025) The analyses were based on the SMILES (Simplified Molecular Input Line Entry System) representations of the compounds. These ADMET descriptors offer crucial insights into the pharmacokinetic behavior and toxicity profiles of the molecules, contributing to the assessment of their drug-likeness. It is important to highlight that some parameters yield categorical outcomes such as “Yes” or “No”, while others provide quantitative values with specific units, which will be clearly identified in the subsequent discussion.
Table 5.
Computed ADMET parameters for the Kapakahine A–G marine cyclopeptides.
The computed ADMET parameters presented in Table 5 offer essential insights into the pharmacokinetic behavior and toxicological risks associated with the Kapakahine A–G cyclopeptides:
- Absorption: All Kapakahines are substrates and dual inhibitors (I and II) of P-glycoprotein, suggesting they may interact with efflux transporters and influence oral bioavailability and multidrug resistance. This aligns with their moderate to high LogP values (Table 4), indicating sufficient lipophilicity for membrane permeability.
- Distribution:
- −
- BBB permeability values range from −0.626 (F) to −1.106 (G), indicating poor blood–brain barrier penetration, typical of larger peptides.
- −
- CNS permeability values (all < −2) also support their limited access to central nervous system tissues. This correlates with relatively high electronegativity () and electrophilicity () in Table 3, implying a tendency toward hydrophilicity and reduced passive diffusion through lipid membranes.
- Metabolism: All peptides are substrates of CYP3A4, a key metabolic enzyme, while none are substrates of CYP2D6. Notably, Kapakahines A, C, D, and G inhibit CYP3A4, which may pose risks for drug–drug interactions. Their strong nucleophilicity (N) and variable global hardness () suggest potential reactivity with metabolic enzymes.
- Excretion: Total clearance ranges from slightly negative to slightly positive values (F: +0.079), suggesting relatively slow elimination. No Kapakahine is a substrate for OCT2, a renal transporter, implying hepatic over renal clearance. This is consistent with their moderate LogP (Table 4) and relatively large size.
- Toxicity: All peptides are non-AMES toxic and non-inhibitors of hERG I, indicating a favorable genotoxicity and cardiac safety profile. However, all are hERG II inhibitors and predicted to be hepatotoxic, raising concerns for cardiotoxicity and liver injury. These risks may be linked to their net electrophilicity (), especially for Kapakahines G and B, which display the highest values in Table 3, suggesting strong electron-accepting capabilities that could impact interactions with biological macromolecules.
- Kapakahine G: Highest electrophilicity ( = 1.423 eV) and net electrophilicity ( = 6.258 eV); matches its broader ADMET liability (lowest BBB and CNS permeability, high hepatotoxicity and clearance). These values suggest high reactivity with biological targets, enhancing potency but possibly increasing toxicity.
- Kapakahine C: Highest hardness ( = 5.033 eV) and electronegativity ( = 3.695 eV); exhibits lowest total clearance and poor CNS penetration, supporting its chemical stability but reduced excretion. Also, the highest ED value (7.466 eV) may be linked to its specific metabolic behavior.
- Kapakahine D: Lowest net electrophilicity ( = 5.785 eV) and electrophilic descriptor (ED = 2.538 eV); presents lower predicted toxicity risk and moderate metabolic profile. These values could suggest a safer ADMET profile among the set.
The ADMET profiles [21] of Kapakahines A–G complement and reinforce the insights provided by their global reactivity descriptors. Compounds like Kapakahine G, with high electrophilic indices, show increased toxicity and metabolic complexity, while more chemically stable peptides like Kapakahine D display relatively benign pharmacokinetic profiles. These results underscore the relevance of Conceptual DFT parameters in anticipating pharmacokinetic and toxicological behavior during early-stage drug discovery.
4. Conclusions
In this study, we examined the structural, electronic, and pharmacokinetic properties of the marine-derived Kapakahine A–G peptides using Conceptual Density Functional Theory (CDFT)-based Computational Peptidology (CP). This theoretical framework, combined with ADMET analysis, has enabled the estimation of key descriptors relevant to chemical reactivity, molecular stability, and drug-like behavior.
The CDFT-based approach allowed for the evaluation of both global and local reactivity indicators, including less conventional descriptors such as hyperhardness and the electrophilic descriptor. Additionally, molecular electrostatic potential (MEP) and local hypersoftness (LHS) maps provided spatial insight into nucleophilic and electrophilic sites across the peptide structures. Estimates of pKa and LogP based on CDFT descriptors, alongside computational ADMET predictions, offered further context for assessing the pharmacokinetic and toxicological properties of the peptides.
While the Kapakahines show interesting chemical features and predicted interactions with metabolic and transport proteins, they also present certain ADMET-related limitations—such as hepatotoxicity and hERG II inhibition—that warrant further investigation. These results contribute to a foundational understanding of the molecular behavior of Kapakahines and illustrate the value of CDFT in guiding early-stage drug discovery efforts. However, experimental validation and additional optimization are necessary to assess their viability as therapeutic candidates.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/computation13050111/s1. Tables S1–S7: Cartesian coordinates of the optimized Kapakahines A–G.
Author Contributions
Conceptualization, D.G.-M., N.F.-H., and J.F.; methodology, D.G.-M.; software, D.G.-M.; validation, D.G.-M., N.F.-H., and J.F.; formal analysis, D.G.-M.; investigation, D.G.-M., N.F.-H., and J.F.; resources, D.G.-M.; data curation, D.G.-M.; writing—original draft preparation, D.G.-M.; writing—review and editing, D.G.-M.; visualization, D.G.-M. and N.F.-H.; supervision, D.G.-M.; project administration, D.G.-M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
N.F.-H. and D.G.-M. are researchers affiliated with Centro de Investigación en Materiales Avanzados (CIMAV), and Sistema Nacional de Investigadoras e Investigadores (SNII) of the Secretaría de Ciencia, Humanidades, Tecnología e Innovación (SECIHTI), Mexico, while J.F. is a researcher associated with Universitat de les Illes Balears (UIB), Palma de Mallorca, Spain.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| DFT | Density Functional Theory |
| CDFT | Conceptual Density Functional Theory |
| CDFT-CP | Conceptual Density Functional Theory-based Computational Peptidology |
| KID | Koopmans in DFT |
| ADMET | Absorption, Distribution, Metabolism, Excretion and Toxicity |
References
- Agrawal, S.; Adholeya, A.; Deshmukh, S.K. The Pharmacological Potential of Non-ribosomal Peptides from Marine Sponge and Tunicates. Front. Pharmacol. 2016, 7, 333. [Google Scholar] [CrossRef] [PubMed]
- Gogineni, V.; Hamann, M.T. Marine Natural Product Peptides with Therapeutic Potential: Chemistry, Biosynthesis, and Pharmacology. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 81–196. [Google Scholar] [CrossRef] [PubMed]
- Kamihira, R.; Nakao, Y. Small-Scale Preparation of Fluorescently Labeled Chemical Probes from Marine Cyclic Peptides, Kapakahines A and F. Mar. Drugs 2021, 19, 76. [Google Scholar] [CrossRef]
- Nakao, Y.; Yeung, B.K.S.; Yoshida, W.Y.; Scheuer, P.J.; Kelly-Borges, M. Kapakahine B, a Cyclic Hexapeptide with an α-Carboline Ring System from the Marine Sponge Cribrochalina olemda. J. Am. Chem. Soc. 1995, 117, 8271–8272. [Google Scholar] [CrossRef]
- Nakao, Y.; Kuo, J.; Yoshida, W.Y.; Kelly, M.; Scheuer, P.J. More Kapakahines from the Marine Sponge Cribrochalina olemda. Org. Lett. 2003, 5, 1387–1390. [Google Scholar] [CrossRef]
- Negi, B.; Kumar, D.; Rawat, D.S. Marine Peptides as Anticancer Agents: A Remedy to Mankind by Nature. Curr. Protein Pept. Sci. 2017, 18, 885–904. [Google Scholar] [CrossRef]
- Newhouse, T.; Lewis, C.A.; Baran, P.S. Enantiospecific Total Syntheses of Kapakahines B and F. J. Am. Chem. Soc. 2009, 131, 6360–6361. [Google Scholar] [CrossRef]
- Espejo, V.R.; Rainier, J.D. Total Synthesis of Kapakahine E and F. Org. Lett. 2010, 12, 2154–2157. [Google Scholar] [CrossRef]
- Yeung, B.K.S.; Nakao, Y.; Kinnel, R.B.; Carney, J.R.; Yoshida, W.Y.; Scheuer, P.J.; Kelly-Borges, M. The Kapakahines, Cyclic Peptides from the Marine Sponge Cribrochalina olemda. J. Org. Chem. 1996, 61, 7168–7173. [Google Scholar] [CrossRef]
- Phyo, Y.Z.; Ribeiro, J.; Fernandes, C.; Kijjoa, A.; Pinto, M.M.M. Marine Natural Peptides: Determination of Absolute Configuration Using Liquid Chromatography Methods and Evaluation of Bioactivities. Molecules 2018, 23, 306. [Google Scholar] [CrossRef]
- Varijakzhan, D.; Loh, J.Y.; Yap, W.S.; Yusoff, K.; Seboussi, R.; Lim, S.H.E.; Lai, K.S.; Chong, C.M. Bioactive Compounds from Marine Sponges: Fundamentals and Applications. Mar. Drugs 2021, 19, 246. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; Chamorro, E.; Chattaraj, P.K.; Proft, F.D.; Gázquez, J.L.; Liu, S.; Morell, C.; Toro-Labbé, A.; Vela, A.; Ayers, P. Conceptual Density Functional Theory: Status, Prospects, Issues. Theor. Chem. Accounts 2020, 139. [Google Scholar] [CrossRef]
- Geerlings, P. From Density Functional Theory to Conceptual Density Functional Theory and Biosystems. Pharmaceuticals 2022, 15, 1112. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Chattaraj, P.K. Conceptual Density Functional Theory Based Electronic Structure Principles. Chem. Sci. 2021, 12, 6264–6279. [Google Scholar] [CrossRef]
- Chakraborty, A.; Pan, S.; Chattaraj, P.K. Biological Activity and Toxicity: A Conceptual DFT Approach. In Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2012; pp. 143–179. [Google Scholar]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Virtual Screening of Marine Natural Compounds by Means of Chemoinformatics and CDFT-Based Computational Peptidology. Mar. Drugs 2020, 18, 478. [Google Scholar] [CrossRef]
- van de Waterbeemd, H.; Gifford, E. ADMET in silico Modelling: Towards Prediction Paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Wang, Y.; Xing, J.; Xu, Y.; Zhou, N.; Peng, J.; Xiong, Z.; Liu, X.; Luo, X.; Luo, C.; Chen, K.; et al. In silicoADME/T Modelling for Rational Drug Design. Q. Rev. Biophys. 2015, 48, 488–515. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Tsaioun, K.; Kates, S.A. (Eds.) ADMET for Medicinal Chemists; Wiley-Blackwell: Hoboken, NJ, USA, 2011. [Google Scholar]
- Lewars, E. Computational Chemistry—Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003. [Google Scholar]
- Young, D. Computational Chemistry—A Practical Guide for Applying Techniques to Real-World Problems; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Cramer, C.J. Essentials of Computational Chemistry—Theories and Models, 2nd ed.; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. II. MMFF94 van der Waals and Electrostatic Parameters for Intermolecular Interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s Option for Energy Minimization Studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Peverati, R.; Truhlar, D.G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Phys. Chem. Chem. Phys. 2012, 14, 16187. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Why are the Local Hyper-Softness and the Local Softness more Appropriate Local Reactivity Descriptors than the Dual Descriptor and the Fukui Function, Respectively? J. Math. Chem. 2023, 62, 461–475. [Google Scholar] [CrossRef]
- Gázquez, J.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Chattaraj, P.; Chakraborty, A.; Giri, S. Net Electrophilicity. J. Phys. Chem. A 2009, 113, 10068–10074. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.; Tognetti, V.; Joubert, L. Electrophilicity Indices and Halogen Bonds: Some New Alternatives to the Molecular Electrostatic Potential. J. Phys. Chem. A 2020, 124, 2090–2101. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T. A Comprehensive Electron Wavefunction Analysis Toolbox for Chemists, Multiwfn. J. Chem. Phys. 2024, 161. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Realization of Conceptual Density Functional Theory and Information-Theoretic Approach in Multiwfn Program. In Conceptual Density Functional Theory: Towards a New Chemical Reactivity Theory; John Wiley & Sons: Hoboken, NJ, USA, 2022; Volume 2, pp. 631–647. [Google Scholar] [CrossRef]
- Figueredo, S.; Páez, M.; Torres, F. The Electrophilic Descriptor. Comput. Theor. Chem. 2019, 1157, 34–39. [Google Scholar] [CrossRef]
- Figueredo, S.F.; Quintero, M.A. Electrophilic Descriptor from Third-order Taylor Expansion: The Role of Hyperhardness. Int. J. Quantum Chem. 2024, 124. [Google Scholar] [CrossRef]
- Glossman-Mitnik, D. (Ed.) Density Functional Theory—Recent Advances, New Perspectives and Applications; IntechOpen: London, UK, 2022. [Google Scholar] [CrossRef]
- Srebrenik, S.; Weinstein, H.; Pauncz, R. Analytical Calculation of Atomic and Molecular Electrostatic Potentials from the Poisson Equation. Chem. Phys. Lett. 1973, 20, 419–423. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The Fundamental Nature and Role of the Electrostatic Potential in Atoms and Molecules. In Theoretical Chemistry Accounts: Theory, Computation, and Modeling (Theoretica Chimica Acta); Springer: Berlin/Heidelberg, Germany, 2002; Volume 108, pp. 134–142. [Google Scholar] [CrossRef]
- Politzer, P. Atomic and Molecular Energies as Functionals of the Electrostatic Potential. Theor. Chem. Accounts 2004, 111, 395–399. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. The Electrostatic Potential: An Overview. WIREs Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Electrostatic Potentials at the Nuclei of Atoms and Molecules. Theor. Chem. Accounts 2021, 140, 7. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Grand, A.; Toro-Labbé, A. Theoretical Support for Using the Δf(r) Descriptor. Chem. Phys. Lett. 2006, 425, 342–346. [Google Scholar] [CrossRef]
- Cárdenas, C.; Rabi, N.; Ayers, P.W.; Morell, C.; Jaramillo, P.; Fuentealba, P. Chemical Reactivity Descriptors for Ambiphilic Reagents: Dual Descriptor, Local Hypersoftness, and Electrostatic Potential. J. Phys. Chem. A 2009, 113, 8660–8667. [Google Scholar] [CrossRef]
- Sandoval-Yañez, C.; Mascayano, C.; Martínez-Araya, J.I. A Theoretical Assessment of Antioxidant Capacity of Flavonoids by Means of Local Hyper–Softness. Arab. J. Chem. 2018, 11, 554–563. [Google Scholar] [CrossRef]
- Frau, J.; Hernández-Haro, N.; Glossman-Mitnik, D. Computational Prediction of the pKas of Small Peptides through Conceptual DFT Descriptors. Chem. Phys. Lett. 2017, 671, 138–141. [Google Scholar] [CrossRef]
- Halabi Diaz, A.; Duque-Noreña, M.; Chamorro, E. Unveiling an Electronic LogP Analogue within the Conceptual Density Functional Theory Framework. Chem. Phys. 2024, 584, 112346. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).