
A Parallel Computing Approach to Gene Expression and Phenotype Correlation for Identifying Retinitis Pigmentosa Modifiers in Drosophila



Abstract
1. Introduction
2. Materials and Methods
2.1. Input Datasets
2.2. Correlation Analysis
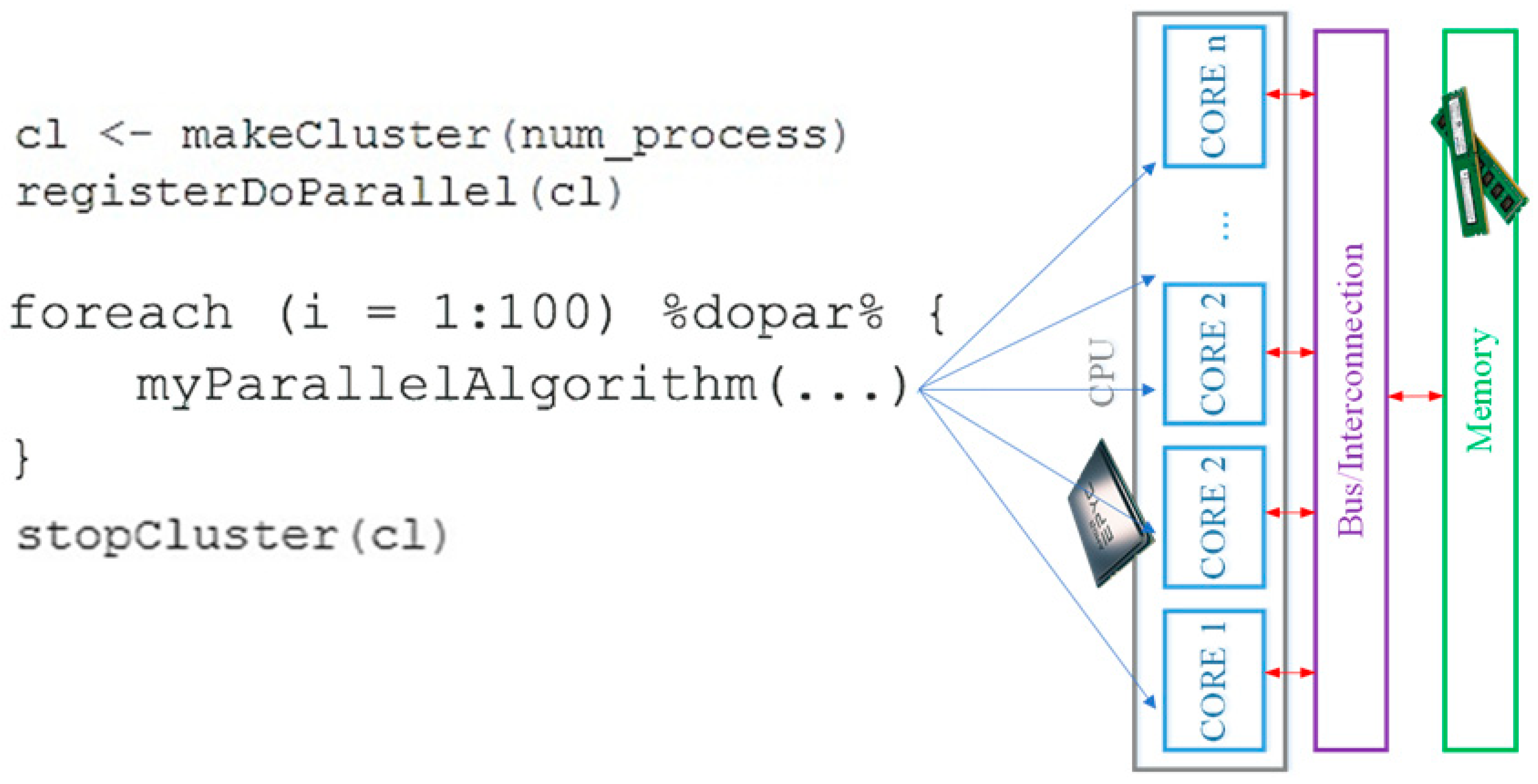
2.3. Parallel Computation in R
2.4. The Computational Approach
| Algorithm 1. Main Algorithm |
| Input: Aes—Average eye sizes Expr—Expression-level matrix low_quantile—Bottom quantile of eye sizes high_quantile—Top quantile of eye sizes C—Correlation threshold value num_process—Number of parallel processes Output: List of candidate genes |
| Begin Filterout strains in Expr with only one replicate. Filterout strains in Expr with no matching values in Aes. selSizes ⟵ eye sizes in Aes less than low_quantile or greater than high_quantile values extreme_strains ⟵ strains in Aes corresponding to selSizes rep_combs ⟵ Algorithm2 (extreme_strains) best_rep_comb ⟵ Algorithm3 (selSizes, Expr, rep_combs, C, num_ process) candidate_genes ⟵ Algorithm4 (SelSizes, Expr, rep_combs, best_rep_comb, C) Print candidate_genes, their correlation coefficients, and p-values. End |
| Algorithm 2. Generate replicate combinations |
| Input: selStrain—Selected extreme strains. Output: replicate_comb—Replicate combinations matrix. |
| Begin N ⟵ Length(selStrain) for i ⟵ 1 to 2N do binary ⟵ DecimalToBinary(i) for every binary digit d at position j in binary do if d is 0 then replicate_comb[i] ⟵ first replicate of strain j else replicate_comb[i] ⟵ second replicate of strain j return replicate_comb End |
| Algorithm 3. Find the best replicate combination |
| Input: selSizes—Average eye sizes of selected strains Expr—Expression level matrix replicate_comb—Replicate combinations matrix C—Correlation threshold value num_process—Number of parallel processes Output: bestCombination—Best replicate combination |
| Begin Create a parallel team with num_process processes. Let scoreVec and combVec be empty vectors. foreach replicate combination c[i] in replicate_comb do for every gene j do Score ⟵ 0 selExprs[j] ⟵ expression levels of j in the selected combination c[i] temp[j] ⟵ correlation value between selExprs[j] and selSizes if temp[j] < -C OR temp[j] > C then Score ⟵ Score + 1 end end Append Score to ScoreVec Append c[i] to combVec end Terminate the parallel session. bestCombination ⟵ Replicate combination in combVec associated with Max(ScoreVec) return bestCombination End |
| Algorithm 4. Find candidate genes |
| Input: selSizes—Average eye sizes of selected strains Expr—Expression level matrix best_rep_comb—Best replicate combination C—Correlation threshold value Output: sorted_genes—List of candidate genes with their correlation coefficients and p-values |
| Begin Let m be the number of genes in Expr Let Results be a list of length m foreach gene j do Results[j] ⟵ correlation coefficient and p-values of selSize and gene expression levels of j for best_rep_comb in Expr end Filterout genes in Results’ with p-values less than 0.05 Filterout genes in Results’ with correlation coefficients not in the range [C, 1] nor [−1, -C] sorted_genes ⟵ Sort Results’ in descending order based on the absolute values of correlation s coefficients return sorted_genes End |
3. Results
3.1. Experimental Setup
3.1.1. Datasets
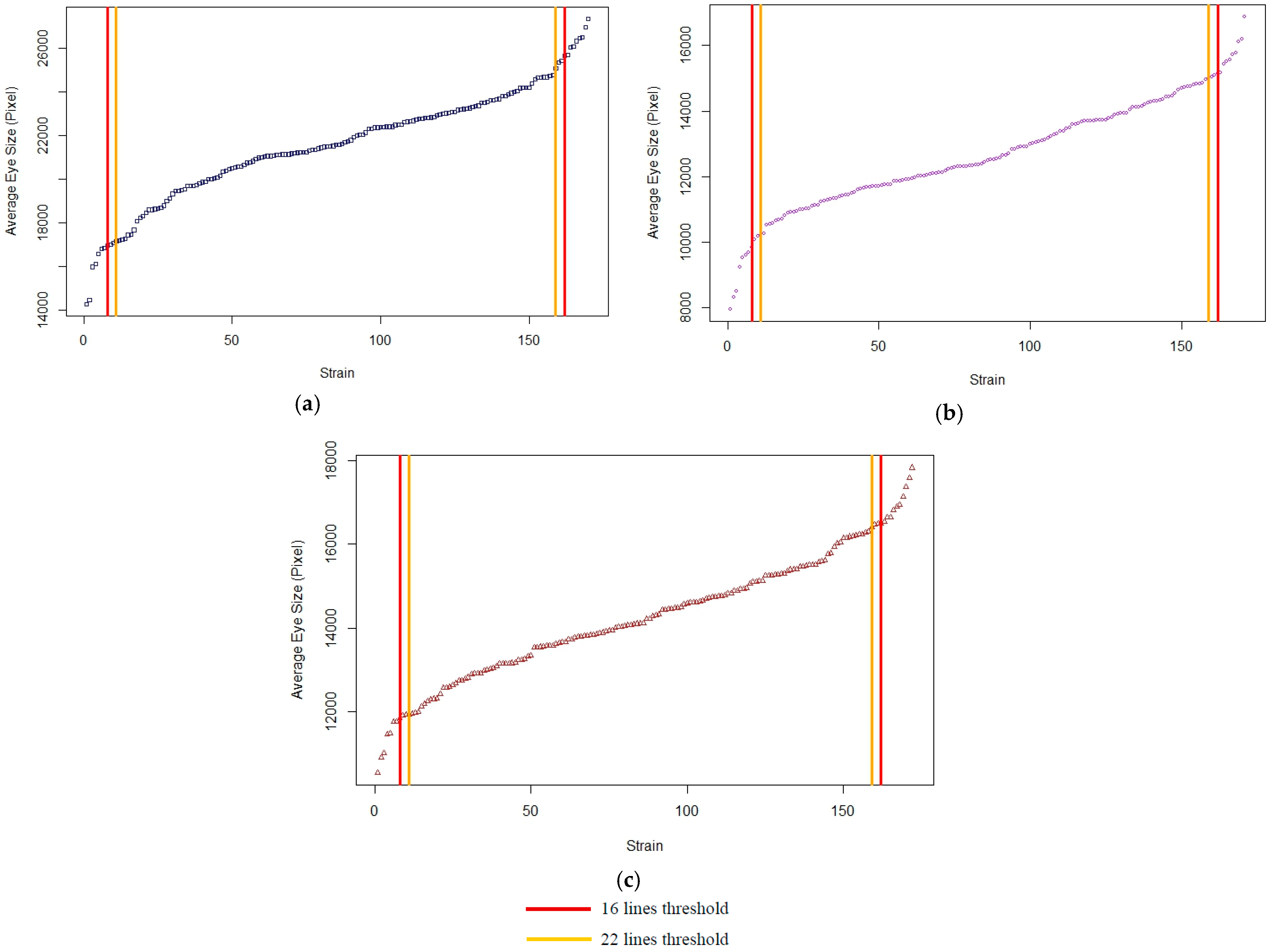
3.1.2. Quantile Thresholds
3.1.3. Hardware & Software Specifications
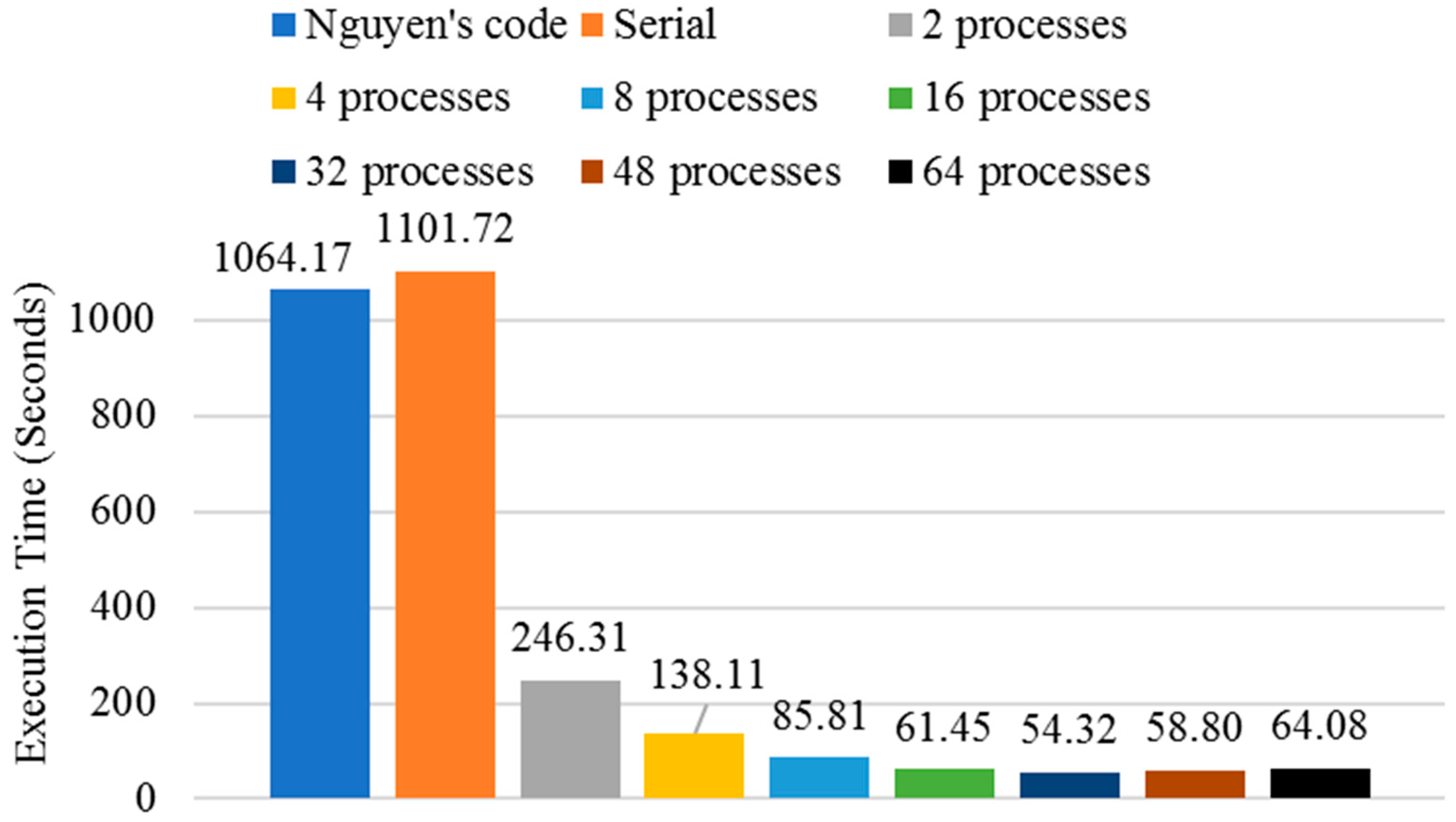
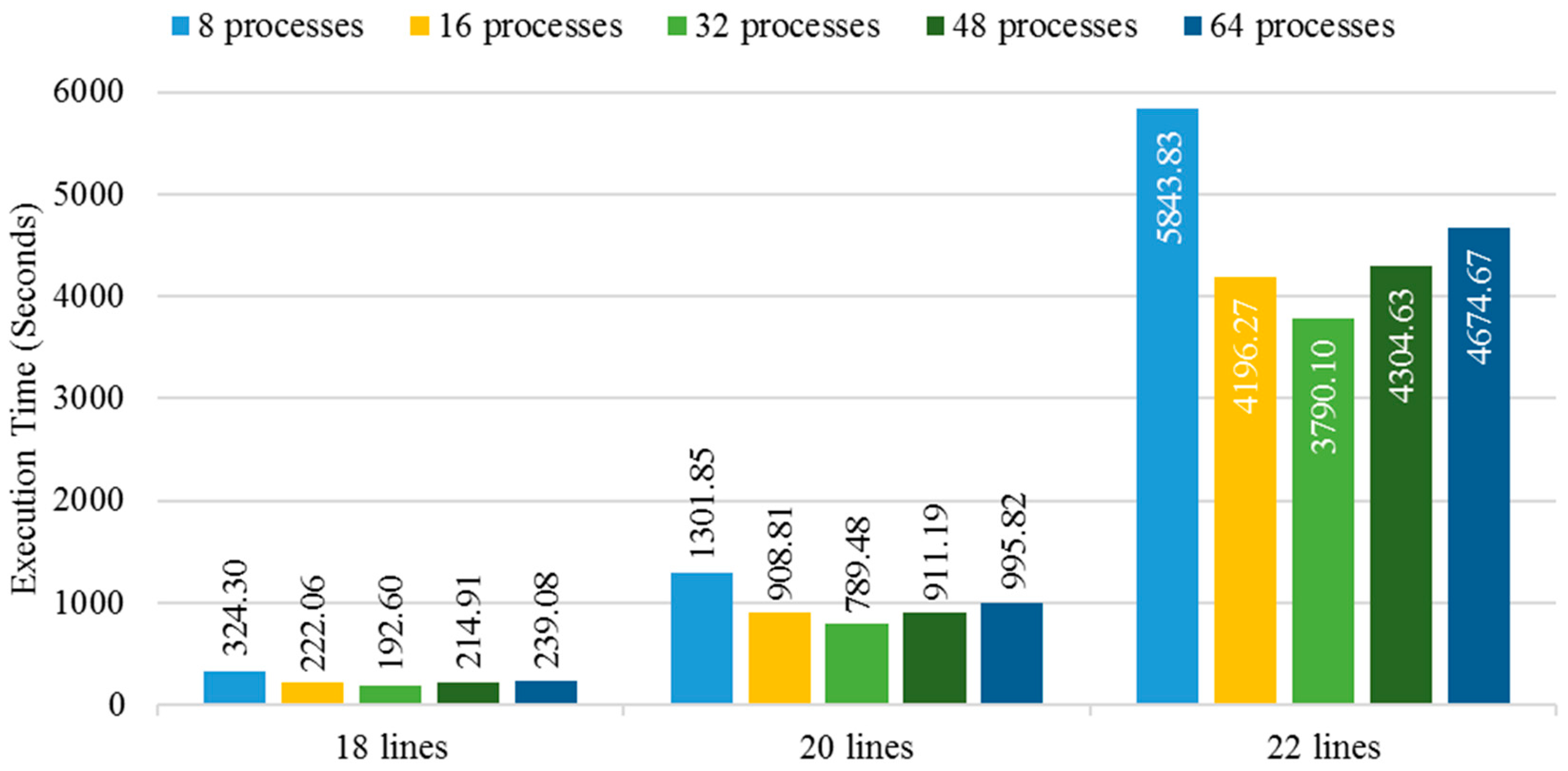
3.2. Execution Time Analysis
3.3. Suspected Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Massouras, A.; Inoue, Y.; Peiffer, J.; Ràmia, M.; Tarone, A.M.; Turlapati, L.; Zichner, T.; Zhu, D.; Lyman, R.F.; et al. Natural variation in genome architecture among 205 Drosophila melanogaster Genetic Reference Panel lines. Genome Res. 2014, 24, 1193–1208. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F.C.; Richards, S.; Stone, E.A.; Barbadilla, A.; Ayroles, J.F.; Zhu, D.; Casillas, S.; Han, Y.; Magwire, M.M.; Cridland, J.M.; et al. The Drosophila melanogaster Genetic Reference Panel. Nature 2012, 482, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Kelsey, K.J.P.; Wolfner, M.F.; Clark, A.G. Candidate genetic modifiers of retinitis pigmentosa identified by exploiting natural variation in Drosophila. Hum. Mol. Genet. 2016, 25, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Karademir, D.; Todorova, V.; Ebner, L.J.A.; Samardzija, M.; Grimm, C. Single-cell RNA sequencing of the retina in a model of retinitis pigmentosa reveals early responses to degeneration in rods and cones. BMC Biol. 2022, 20, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Du, W.; Xu, N.; Tao, T.; Tang, X.; Huang, L. RNA-Seq Analysis for Exploring the Pathogenesis of Retinitis Pigmentosa in P23H Knock-In Mice. Ophthalmic Res. 2021, 64, 798–810. [Google Scholar] [CrossRef]
- Robinson, M.D.; Smyth, G.K. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 2007, 23, 2881–2887. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Hardcastle, T.J.; Kelly, K.A. baySeq: Empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinform. 2010, 11, 422. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tibshirani, R. Finding consistent patterns: A nonparametric approach for identifying differential expression in RNA-Seq data. Stat. Methods Med. Res. 2013, 22, 519–536. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Delorenzi, M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinform. 2013, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Pardee, A.B. Analysing differential gene expression in cancer. Nat. Rev. Cancer 2003, 3, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Esteban, R.; Jiang, X. Differential gene expression in disease: A comparison between high-throughput studies and the literature. BMC Med. Genom. 2017, 10, 59. [Google Scholar] [CrossRef]
- Amstutz, J.; Khalifa, A.; Palu, R.; Jahan, K. Cluster-Based Analysis of Retinitis Pigmentosa Modifiers Using Drosophila Eye Size and Gene Expression Data. Genes 2022, 13, 386. [Google Scholar] [CrossRef]
- Nguyen, T.; Khalifa, A.; Palu, R. Identifying Genes Related to Retinitis Pigmentosa in Drosophila melanogaster Using Eye Size and Gene Expression Data. BioMedInformatics 2022, 2, 625–636. [Google Scholar] [CrossRef]
- Huang, W.; Carbone, M.A.; Magwire, M.M.; Peiffer, J.A.; Lyman, R.F.; Stone, E.A.; Anholt, R.R.H.; Mackay, T.F.C. Genetic basis of transcriptome diversity in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2015, 112, E6010–E6019. [Google Scholar] [CrossRef]
- Palu, R.A.S.; Ong, E.; Stevens, K.; Chung, S.; Owings, K.G.; Goodman, A.G.; Chow, C.Y. Natural Genetic Variation Screen in Drosophila Identifies Wnt Signaling, Mitochondrial Metabolism, and Redox Homeostasis Genes as Modifiers of Apoptosis. G3 Genes Genomes Genet. 2019, 9, 3995–4005. [Google Scholar] [CrossRef]
- Posnien, N.; Hopfen, C.; Hilbrant, M.; Ramos-Womack, M.; Murat, S.; Schönauer, A.; Herbert, S.L.; Nunes, M.D.S.; Arif, S.; Breuker, C.J.; et al. Evolution of eye morphology and rhodopsin expression in the Drosophila melanogaster species subgroup. PLoS ONE 2012, 7, e37346. [Google Scholar] [CrossRef]
- Greenland, S.; Senn, S.J.; Rothman, K.J.; Carlin, J.B.; Poole, C.; Goodman, S.N.; Altman, D.G. Statistical tests, P values, confidence intervals, and power: A guide to misinterpretations. Eur. J. Epidemiol. 2016, 31, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Weston, S.; Calaway, R. Getting Started with doParallel and Foreach. 2022. Available online: https://CRAN.R-project.org/package=doParallel (accessed on 6 January 2023).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Expression Level | |||
|---|---|---|---|---|
| RAL021:1 | RAL021:2 | RAL026:1 | RAL026:2 | |
| FBgn0000014 | 4.244723137 | 4.216353088 | 4.028685457 | 3.965513774 |
| FBgn0000015 | 3.234859699 | 3.199773952 | 3.266073855 | 3.514853684 |
| FBgn0000017 | 8.066864662 | 7.962031505 | 8.016965853 | 8.081375654 |
| FBgn0000018 | 5.317033088 | 5.268665083 | 5.583749674 | 4.949218486 |
| FBgn0000022 | 3.000683083 | 3.000127343 | 4.033542617 | 3.364429304 |
| FBgn0000024 | 6.120670813 | 6.023183171 | 6.363472661 | 6.83930746 |
| FBgn0000028 | 4.101309578 | 4.050933404 | 4.581349626 | 4.276622648 |
| FBgn0000032 | 7.460913282 | 7.68689799 | 7.782455553 | 7.635495636 |
| FBgn0000036 | 3.988090417 | 3.789139103 | 3.979189512 | 3.95396714 |
| FBgn0000037 | 4.475747359 | 4.323271618 | 4.457239171 | 4.378994365 |
| … | … | … | … | … |
| XLOC_006439 | 2.414951288 | 2.612959863 | 3.717652528 | 2.090561202 |
| Strain ID | Average Eye Size (Pixels × 103) |
|---|---|
| RAL021 | 19,976.8 |
| RAL026 | 21,473.2 |
| RAL038 | 19,981.5 |
| RAL040 | 16,992.9 |
| RAL042 | 21,481.4 |
| … | … |
| RAL913 | 19,488.5 |
| Dataset | Filtered Strains | Mean | Median | Minimum | Maximum | Quartile | |
|---|---|---|---|---|---|---|---|
| 1st | 3rd | ||||||
| Rh1G69D | 170 | 21,540.20 | 21,561.65 | 14,254.60 | 27,349.11 | 19,995.88 | 23,199.63 |
| rpr | 171 | 12,666.09 | 12,486.20 | 79,57.20 | 16,883.50 | 11,620.60 | 13,906.10 |
| p53 | 172 | 14,244.73 | 14,166.00 | 10,542.20 | 17834.60 | 13,160.12 | 15,278.90 |
| Dataset | Number of Selected Lines | Quantile (%) | |
|---|---|---|---|
| Bottom | Top | ||
| Rh1G69D | 16 | 20.90 | 87.20 |
| 18 | 21.00 | 87.00 | |
| 20 | 21.50 | 85.30 | |
| 22 | 22.10 | 84.60 | |
| rpr | 16 | 21.10 | 83.80 |
| 18 | 23.80 | 80.80 | |
| 20 | 25.10 | 80.40 | |
| 22 | 25.30 | 79.90 | |
| p53 | 16 | 17.80 | 83.68 |
| 18 | 18.90 | 83.60 | |
| 20 | 19.10 | 82.20 | |
| 22 | 19.20 | 81.68 | |
| Nguyen’s | Ours | ||||
|---|---|---|---|---|---|
| Serial | 2 Processes | 4 Processes | 8 Processes | ||
| Algorithm 1 | 3.763 | 21.210 | 25.710 | 23.130 | 24.810 |
| Algorithm 2 | 862.640 | 901.010 | 322.110 | 215.600 | 166.960 |
| Algorithm 3 | 71.342 | 76.362 | 23.960 | 17.868 | 17.562 |
| No. of Selected Lines | Gene ID | Correlation Coefficient | p-Value |
|---|---|---|---|
| 16 | FBgn0027378 | −0.863661390 | 1.624926332 × 10−5 |
| FBgn0263005 | −0.856324086 | 2.297889577 × 10−5 | |
| XLOC_006268 | −0.849992594 | 3.053251949 × 10−5 | |
| FBgn0032847 | −0.840543849 | 4.560179069 × 10−5 | |
| FBgn0033782 | 0.840068819 | 4.649931586 × 10−5 | |
| FBgn0036761 | −0.834562369 | 5.802852891 x 10−5 | |
| FBgn0037531 | −0.828535797 | 7.329098346 × 10−5 | |
| FBgn0086679 | −0.826073369 | 8.042371477 × 10−5 | |
| FBgn0031233 | −0.814773350 | 1.210237900 × 10−4 | |
| FBgn0038639 | −0.810804945 | 1.388117979 × 10−4 | |
| 18 | FBgn0085376 | 0.859283205 | 4.923670750 × 10−6 |
| FBgn0033782 | 0.854529969 | 6.322511262 × 10−6 | |
| FBgn0037531 | −0.850683118 | 7.692063225 × 10−6 | |
| FBgn0003345 | 0.849498533 | 8.161927414 × 10−6 | |
| FBgn0036299 | −0.824116242 | 2.609375328 × 10−5 | |
| FBgn0086679 | −0.820352399 | 3.052250605 × 10−5 | |
| FBgn0032847 | −0.809207602 | 4.758311930 × 10−5 | |
| FBgn0037016 | 0.804435051 | 5.705515305 × 10−5 | |
| FBgn0263005 | −0.799150447 | 6.936945025 × 10−5 | |
| FBgn0263659 | 0.798357906 | 7.139778718 × 10−5 | |
| 20 | FBgn0032847 | −0.835162086 | 4.614128240 × 10−6 |
| FBgn0086679 | −0.810201591 | 1.489680678 × 10−5 | |
| XLOC_004892 | 0.808596485 | 1.596926917 × 10−5 | |
| FBgn0037531 | −0.798402859 | 2.447648614 × 10−5 | |
| FBgn0037770 | −0.795138465 | 2.792299682 × 10−5 | |
| FBgn0033087 | −0.782129177 | 4.616635661 × 10−5 | |
| FBgn0038039 | −0.777530252 | 5.470964221 × 10−5 | |
| FBgn0261703 | 0.777148268 | 5.547684002 × 10−5 | |
| FBgn0263602 | −0.776940029 | 5.589897080 × 10−5 | |
| FBgn0023513 | −0.77665287 | 5.648562369 × 10−5 | |
| 22 | FBgn0032847 | −0.819701175 | 3.041416193 × 10−6 |
| FBgn0003345 | 0.806371734 | 5.849733373 × 10−6 | |
| FBgn0039125 | −0.798479606 | 8.420737122 × 10−6 | |
| FBgn0027378 | −0.798116202 | 8.559914937 × 10−6 | |
| FBgn0037770 | −0.775234661 | 2.259984672 × 10−5 | |
| FBgn0036299 | −0.773718232 | 2.400671440 × 10−5 | |
| FBgn0038039 | −0.766653185 | 3.162045710 × 10−5 | |
| FBgn0030817 | −0.759197474 | 4.186542502 × 10−5 | |
| FBgn0033087 | −0.758610329 | 4.278306102 × 10−5 | |
| FBgn0263602 | −0.757552951 | 4.447992560 × 10−5 |
| No. of Selected Lines | Gene ID | Correlation Coefficient | p-Value |
|---|---|---|---|
| 16 | FBgn0053017 | 0.878155178 | 7.701469269 × 10−6 |
| FBgn0032225 | 0.864824355 | 1.535297878 × 10−5 | |
| FBgn0033244 | −0.863862383 | 1.609129485 × 10−5 | |
| FBgn0015513 | −0.863578519 | 1.631477307 × 10−5 | |
| FBgn0027601 | −0.863380084 | 1.647253890 × 10−5 | |
| FBgn0004620 | 0.859876472 | 1.947654735 × 10−5 | |
| XLOC_003703 | 0.857112086 | 2.215964129 × 10−5 | |
| FBgn0052451 | −0.854030715 | 2.550919501 × 10−5 | |
| FBgn0030394 | 0.853756622 | 2.582664724 × 10−5 | |
| FBgn0015024 | −0.853296712 | 2.636675031 × 10−5 | |
| 18 | FBgn0030394 | 0.863043231 | 4.013975720 × 10−6 |
| FBgn0052451 | −0.861824973 | 4.291403231 × 10−6 | |
| FBgn0053017 | 0.857064474 | 5.539380529 × 10−6 | |
| FBgn0015513 | −0.854016815 | 6.492125538 × 10−6 | |
| XLOC_001754 | 0.85284074 | 6.895646685 × 10−6 | |
| FBgn0027601 | −0.851538255 | 7.367464694 × 10−6 | |
| FBgn0032225 | 0.848190573 | 8.709105665 × 10−6 | |
| FBgn0040508 | 0.846060487 | 9.667524618 × 10−6 | |
| FBgn0051523 | −0.839435144 | 1.324811250 × 10−5 | |
| XLOC_003703 | 0.838478248 | 1.384887686 × 10−5 | |
| 20 | FBgn0052451 | −0.865408608 | 8.358927794 × 10−7 |
| FBgn0027601 | −0.856270053 | 1.457998848 × 10−6 | |
| XLOC_003703 | 0.846002475 | 2.608233863 × 10−6 | |
| FBgn0037223 | 0.842028538 | 3.230636630 × 10−6 | |
| FBgn0053017 | 0.841496599 | 3.323055691 × 10−6 | |
| FBgn0033244 | −0.834443888 | 4.784948638 × 10−6 | |
| FBgn0051523 | −0.834340071 | 4.810090828 × 10−6 | |
| XLOC_004120 | 0.832821352 | 5.191263979 × 10−6 | |
| XLOC_006378 | 0.832584454 | 5.253030551 × 10−6 | |
| FBgn0039491 | 0.828453174 | 6.437918905 × 10−6 | |
| 22 | FBgn0027601 | −0.848794066 | 5.948318658 × 10−7 |
| FBgn0053017 | 0.828428119 | 1.924641707 × 10−6 | |
| FBgn0004620 | 0.819129408 | 3.131318980 × 10−6 | |
| FBgn0015513 | −0.816721873 | 3.536085022 × 10−6 | |
| FBgn0036874 | 0.815971236 | 3.671371942 × 10−6 | |
| FBgn0039491 | 0.815006786 | 3.851872618 × 10−6 | |
| FBgn0033244 | −0.812433525 | 4.372265832 × 10−6 | |
| FBgn0036017 | 0.80726159 | 5.608563439 × 10−6 | |
| FBgn0085692 | 0.806424546 | 5.835169750 × 10−6 | |
| XLOC_003128 | 0.804285096 | 6.451350215 × 10−6 |
| No. of Selected Lines | Gene ID | Correlation Coefficient | p-Value |
|---|---|---|---|
| 16 | FBgn0263110 | 0.914150185 | 7.326821639 × 10−7 |
| FBgn0030089 | 0.912212484 | 8.520806952 × 10−7 | |
| FBgn0051804 | −0.893134917 | 3.203841392 × 10−6 | |
| XLOC_002940 | −0.880664278 | 6.703825055 × 10−6 | |
| FBgn0262148 | −0.84019034 | 4.626832053 × 10−5 | |
| FBgn0004373 | 0.824802374 | 8.432604659 × 10−5 | |
| FBgn0029952 | −0.824030001 | 8.677364856 × 10−5 | |
| XLOC_006034 | −0.816964908 | 1.120425958 × 10−4 | |
| FBgn0034624 | −0.813349043 | 1.271757728 × 10−4 | |
| FBgn0026369 | 0.809052147 | 1.473328296 × 10−4 | |
| 18 | FBgn0030089 | 0.90872731 | 1.812918682 × 10−7 |
| FBgn0051804 | −0.884756312 | 1.083361127 × 10−6 | |
| XLOC_002940 | −0.857875241 | 5.307141864 × 10−6 | |
| FBgn0004373 | 0.823245148 | 2.706658753 × 10−5 | |
| FBgn0263598 | 0.820901298 | 2.983927843 × 10−5 | |
| FBgn0085478 | 0.813059623 | 4.094743159 × 10−5 | |
| FBgn0005632 | 0.811169207 | 4.409799798 × 10−5 | |
| XLOC_001981 | −0.810258107 | 4.568867256 × 10−5 | |
| FBgn0029952 | −0.808005811 | 4.983200012 × 10−5 | |
| FBgn0015024 | 0.800553196 | 6.589932229 × 10−5 | |
| 20 | XLOC_002940 | −0.838696327 | 3.848574574 × 10−6 |
| FBgn0004373 | 0.830822887 | 5.732735463 × 10−6 | |
| FBgn0050039 | −0.814340058 | 1.241477643 × 10−5 | |
| FBgn0259146 | −0.810730528 | 1.455735618 × 10−5 | |
| FBgn0005649 | 0.805892929 | 1.792731166 × 10−5 | |
| FBgn0037770 | 0.799502932 | 2.340117744 × 10−5 | |
| FBgn0027338 | 0.799283244 | 2.361258995 × 10−5 | |
| FBgn0037327 | 0.792106903 | 3.149179899 × 10−5 | |
| XLOC_003332 | −0.790428741 | 3.363187698 × 10−5 | |
| FBgn0085478 | 0.789154503 | 3.533971171 × 10−5 | |
| 22 | XLOC_002940 | −0.857556268 | 3.401980564 × 10−7 |
| FBgn0030089 | 0.84421251 | 7.858326004 × 10−7 | |
| FBgn0085478 | 0.802630148 | 6.966511250 × 10−6 | |
| FBgn0029976 | 0.799946333 | 7.878992278 × 10−6 | |
| FBgn0005632 | 0.799262002 | 8.127819248 × 10−6 | |
| FBgn0004373 | 0.789236473 | 1.265143068 × 10−5 | |
| FBgn0029952 | −0.788090936 | 1.328762771 × 10−5 | |
| FBgn0263598 | 0.786198336 | 1.440028907 × 10−5 | |
| FBgn0021760 | 0.78617631 | 1.441370482 × 10−5 | |
| XLOC_004713 | −0.784049252 | 1.576201243 × 10−5 |
| Gene ID | Shared Datasets/Lines | Gene Symbol | Gene Name | Human Ortho. | Link to RP |
|---|---|---|---|---|---|
| FBgn0032847 | Rh1G69D: 16, 18, 20, 22 | CG10756 | TBP-associated factor 13 | TAF13; SUPT3H | Unknown |
| FBgn0037531 | Rh1G69D: 16, 18, 20 | CG10445 | N/A | TTF2; HLTF | Unknown |
| FBgn0086679 | Rh1G69D: 16, 18, 20 | CG9770 | pink | HPS5; TECPR2 | Eye expression and primary function. |
| FBgn0027601 | rpr: 16, 18, 20, 22 | CG9009 | pudgy | ACSF2/ACSF3 | Fatty acid metabolism influences mitochondrial function and cell death. |
| FBgn0053017 | rpr: 16, 18, 20, 22 | CG33017 | N/A | GPATCH8 | Unknown |
| FBgn0052451 | rpr: 16, 18, 20 | CG32451 | secretory pathway calcium atpase | ATP2C1/ATP2C2 | Calcium influx can be a trigger for apoptosis. Loss in humans is associated with various diseases, including some atrophy/degeneration. |
| XLOC_003703 | rpr: 16, 18, 20 | N/A | N/A | N/A | Unknown |
| FBgn0015513 | rpr: 16, 18, 22 | CG10379 | myoblast city | DOCK1/DOCK2/ DOCK5 | Associated in (DOCK2) with immunodeficiency 40 (OMIM 616433). More distant orthologue (DOCK3) associated with neurodevelopmental disorder with autophagy and degenerative axons. |
| FBgn0033244 | rpr: 16, 20, 22 | CG8726 | N/A | PXK | Loss in humans associated with susceptibility to lupus. |
| FBgn0004373 | p53: 16, 18, 20, 22 | CG7004 | four-wheel drive | PI4KB | Connection to deafness and to insulin signaling in human/rodents. |
| XLOC_002940 | p53: 16, 18, 20, 22 | N/A | N/A | N/A | Unknown |
| FBgn0030089 | p53: 16, 18, 22 | CG9113 | adaptor protein complex 1, gamma subunit | AP1G1/AP1G2 | Associated with USRISR, a neurodevelopmental disorder (AP1G1) (OMIM 619548). |
| FBgn0029952 | p53: 16, 18, 22 | CG12689 | N/A | N/A | Unknown |
| FBgn0085478 | p53: 18, 20, 22 | CG34449 | zinc finger DHHC-type containing 8 | ZDHHC5/ ZDHHC8 | Linked to learning and memory (neuronal function) in mouse models. |
| FBgn0037770 | Rh1G69D: 20, 22 p53: 20 | CG5358 | arginine methyltransferase 4 | CARM1; METTL27/7B/7A; PRMT9/3/7/6/8/2/1; NDUFAF5; ALKBH8; BUD23; ATPSCKMT; GSTCD; TRMT9B; ANTKMT | Unknown |
| FBgn0015024 | rpr: 16 p53: 18 | CG2028 | casein kinase Iα | Hsap\CSNK1A1, Hsap\CSNK1A1L | a biomarker for Alzheimer’s Disease |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metah, C.; Khalifa, A.; Palu, R. A Parallel Computing Approach to Gene Expression and Phenotype Correlation for Identifying Retinitis Pigmentosa Modifiers in Drosophila. Computation 2023, 11, 118. https://doi.org/10.3390/computation11060118
Metah C, Khalifa A, Palu R. A Parallel Computing Approach to Gene Expression and Phenotype Correlation for Identifying Retinitis Pigmentosa Modifiers in Drosophila. Computation. 2023; 11(6):118. https://doi.org/10.3390/computation11060118
Chicago/Turabian StyleMetah, Chawin, Amal Khalifa, and Rebecca Palu. 2023. "A Parallel Computing Approach to Gene Expression and Phenotype Correlation for Identifying Retinitis Pigmentosa Modifiers in Drosophila" Computation 11, no. 6: 118. https://doi.org/10.3390/computation11060118
APA StyleMetah, C., Khalifa, A., & Palu, R. (2023). A Parallel Computing Approach to Gene Expression and Phenotype Correlation for Identifying Retinitis Pigmentosa Modifiers in Drosophila. Computation, 11(6), 118. https://doi.org/10.3390/computation11060118