




Graph-Theoretical Analysis of Biological Networks: A Survey

Abstract
:1. Introduction
- Topological Analysis: This analysis is based on the topological properties of the network, providing information to be used in further analysis, as described in the following sections.
- Clustering: This is the process of discovering dense regions of a biological network that may indicate important activity for the survival of the organism, or sometimes, disease states.
- Network Motifs: These are frequently repeating subgraph patterns in biological networks that may indicate some specific function performed by them.
- Network Alignment: The alignment of two networks shows the similarity between them, which may be used to deduce hereditary relationships. This affinity may help to discover conserved regions in organisms to aid with the understanding of the evolutionary process.
2. Biological Networks
2.1. Networks in the Cell
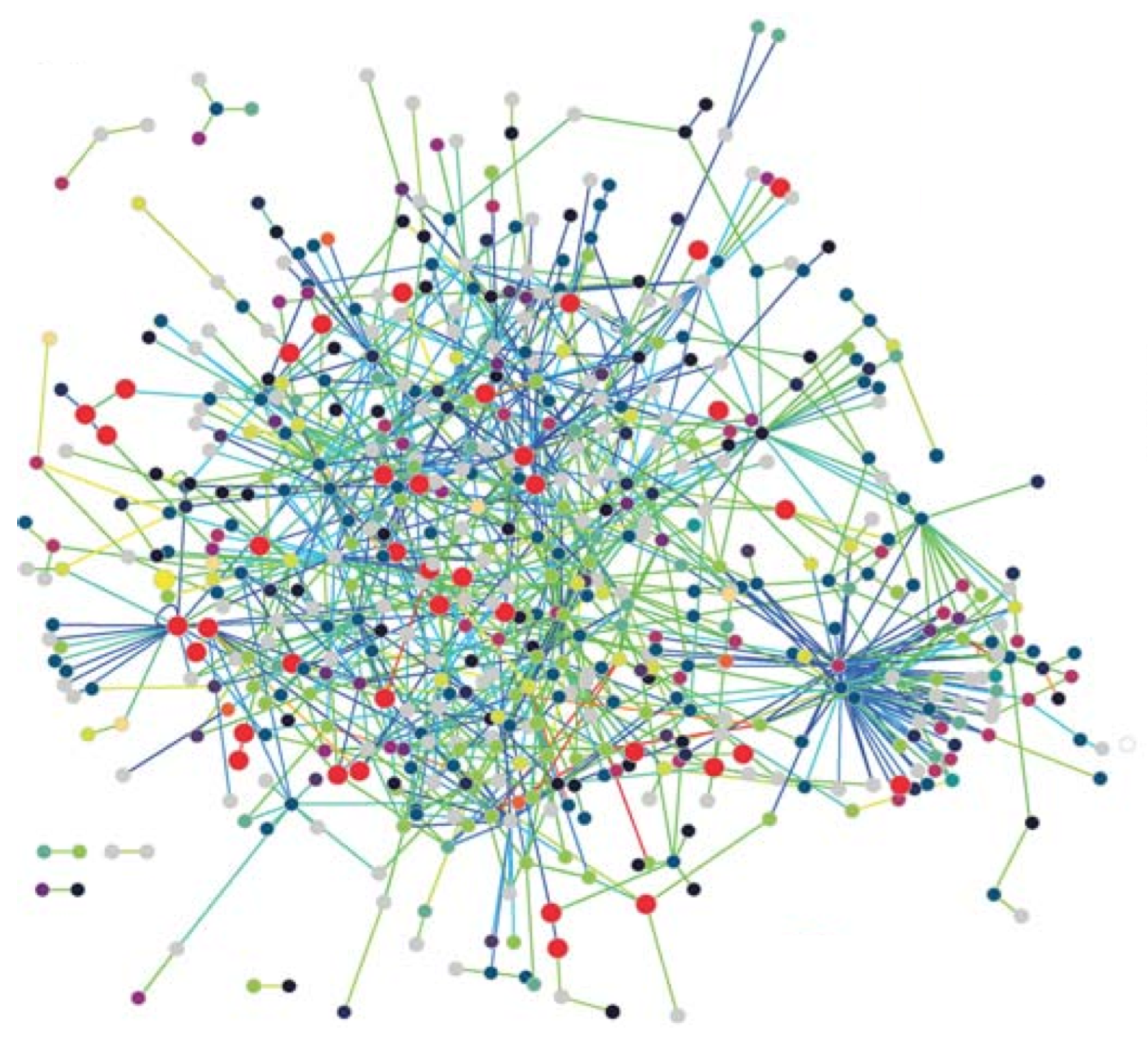
- Protein Networks: Proteins are the workhorses of the cell, performing the vital functions of organisms. A protein is basically a sequence of amino acids constructed by the code in a gene, which is part of the DNA. The 3-D structure of a protein plays an important role in its function, so that various drug treatment methods use this property to disable the functioning of a disease-causing virus such as the HIV. A protein interacts with various other proteins through biochemical reactions forming a protein–protein-interaction (PPI) network. Nodes with high degrees in a PPI network has fundamental functions in the cell [5]. The PPI network of T. pallidum is depicted in Figure 1, where proteins involved in DNA metabolism are shown as enlarged red circles.
- Gene Regulation Networks: The main function of a gene in DNA is to provide the code to be used through transcription and translation processes to produce a protein. This process is called gene expression, and the mechanism of specific gene expression is controlled and affected by proteins that are coded by other genes, denoted regularity interactions. For example, gene X regulates gene Y if a change in the expression of gene X results in a change in the expression of gene Y. A gene regulation network (GRN) is made of genes, proteins, and various other molecules, and it may be modeled using a directed graph, with nodes representing these entities and the edges showing their biochemical interactions leading to regulations, as shown in Figure 2. Typically, a GRN is a sparse graph with small-world and power-law properties, which means there are only a few nodes that have very high out-degrees that regulate the expression of other genes. Moreover, the distance between any two nodes in a GRN network is small compared to the size of the network as being consistent with small-world properties.Figure 1. The PPI network of T. pallidum, taken from [6].Figure 1. The PPI network of T. pallidum, taken from [6].
![Computation 11 00188 g001]()
- Metabolic Pathways: The main ingredients of the cell, such as sugars, amino acids, and lipids, are produced by the basic chemical system called metabolism that works on ingredients called metabolites. The biochemical reactions in the cell that result in metabolisms can be modeled by directed or undirected graphs, with nodes representing metabolites and edges showing biochemical reactions that transform one metabolite to another one [7,8,9]. An edge in such a graph may also represent an enzyme that catalyzes a biochemical reaction. An undirected edge in the graph model denotes a reversible reaction where a directed edge means an irreversible one. A metabolic pathway is a sequence of biochemical reactions to perform a specific metabolic function. An example of a metabolic function is glycolysis, in which a glucose molecule is divided into two sugars that generate adenosine triphosphates (ATPs) to produce energy. Graphs representing metabolic pathways have the small-world and scale-free properties. A study of metabolic pathways may provide insight into pathogens causing infections in search of cures for diseases [10].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2.2. Networks outside the Cell
- Brain Networks: We can analyze brain networks at the cell (neuron) level, or at a coarser functional level. A neuron in the brain fires when the sum of its input signal strengths exceeds a threshold. A neural network made of neurons performs various cognitive tasks such as problem solving, reasoning and, image processing. The artificial neural networks function similar to biological neural networks and have been used widely to implement various tasks in deep learning, which is a component of machine learning to be used for artificial intelligence tasks. At a coarser level, we can investigate the functions performed by the brain, using brain structural networks (BSNs) or brain functional networks (BFNs). A BSN basically reflects the structures of neural connections, whereas a BFN models the connnectedness of the functional regions of the brain. Studies of BFNs have shown that these networks are also small-world and scale-free networks, like most of the biological networks [11].
- Phylogenetic Networks: A phylogenetic tree shows evolutionary relationships among organisms, with leaves representing living organisms and the intermediate nodes, their common ancestors. A phylogenetic network is the general form of a phylogenetic tree where a node may have more than one parent.
- The Food Chain: Living organisms rely on food for survival. The food chain directed graph shows the relationships between the predators and the prey, where the direction of an edge is from the predator to the prey.
3. Large Graph Analysis
3.1. Degree Distribution
3.2. Density
3.3. Clustering Coefficient
3.4. Matching Index
3.5. Centrality
3.5.1. Closeness Centrality
3.5.2. Vertex Betweenness Centrality
3.5.3. Edge Betweenness Centrality
3.6. Topological Index
- The first Zagrep index
- The second Zagrep indexwhere is the degree of vertex v.
- The Wiener indexwhere is the distance as the number of edges between vertices u and v.
3.7. Network Perturbation Analysis
4. Large Network Models
- Random networks: These types of networks, proposed by Erdos and Renyi, assume that an edge between the vertices u and v is formed with the probability . The degree distribution in random networks is binomial, following a Poisson distribution. A random network has a short average path length and it has a clustering coefficient that is inversely proportional to the size of the network.
- Small-world networks: These types of networks are characterized by low average path lengths and short diameters. Biological networks such as PPI networks, GRNs, and metabolic pathways; and other complex networks such as social networks and the Internet exhibit this property. The diameter of a small-world network is proportional to , where n is the number of nodes in the network.
- Scale-free networks: Most biological networks have few high-degree nodes, with many low-degree ones. The PPI network of T. pallidum in Figure 1 exhibits small-world and scale-free network properties, as can be seen. These networks, along with various other complex networks, obey the power-law degree distribution shown by the following equation,where is known as the power-law exponent. These networks are called scale-free networks. The PPI networks of E. coli, D. melanogaster, C. elegans, and H. Pylori were shown to be scale-free. Barabasi and Albert provided a method to form a scale-free network with the following steps [18]:
- Growth: A new node is added to the network at each discrete time t.
- Preferential Attachment: A new node u is attached to any node v in the network with a probability proportional to the degree of v, which means that higher degree nodes tend to have more neighbors at each attachment.
- Hierarchical Networks: A study of biological networks shows that the clustering coefficients of nodes are inversely proportional to their degrees. This unexpected result means that lower degree nodes in these networks have higher clustering coefficients than the hubs. A hierarchical network model of a biological network captures all of the observed properties, such as small-world and scale-free, with an additional property that is exhibited by dense clusters of low-degree nodes, connected by high-degree hubs. That is, the neighbors of low-degree nodes in such networks are highly connected but the nodes around the high-degree nodes are sparsely connected.
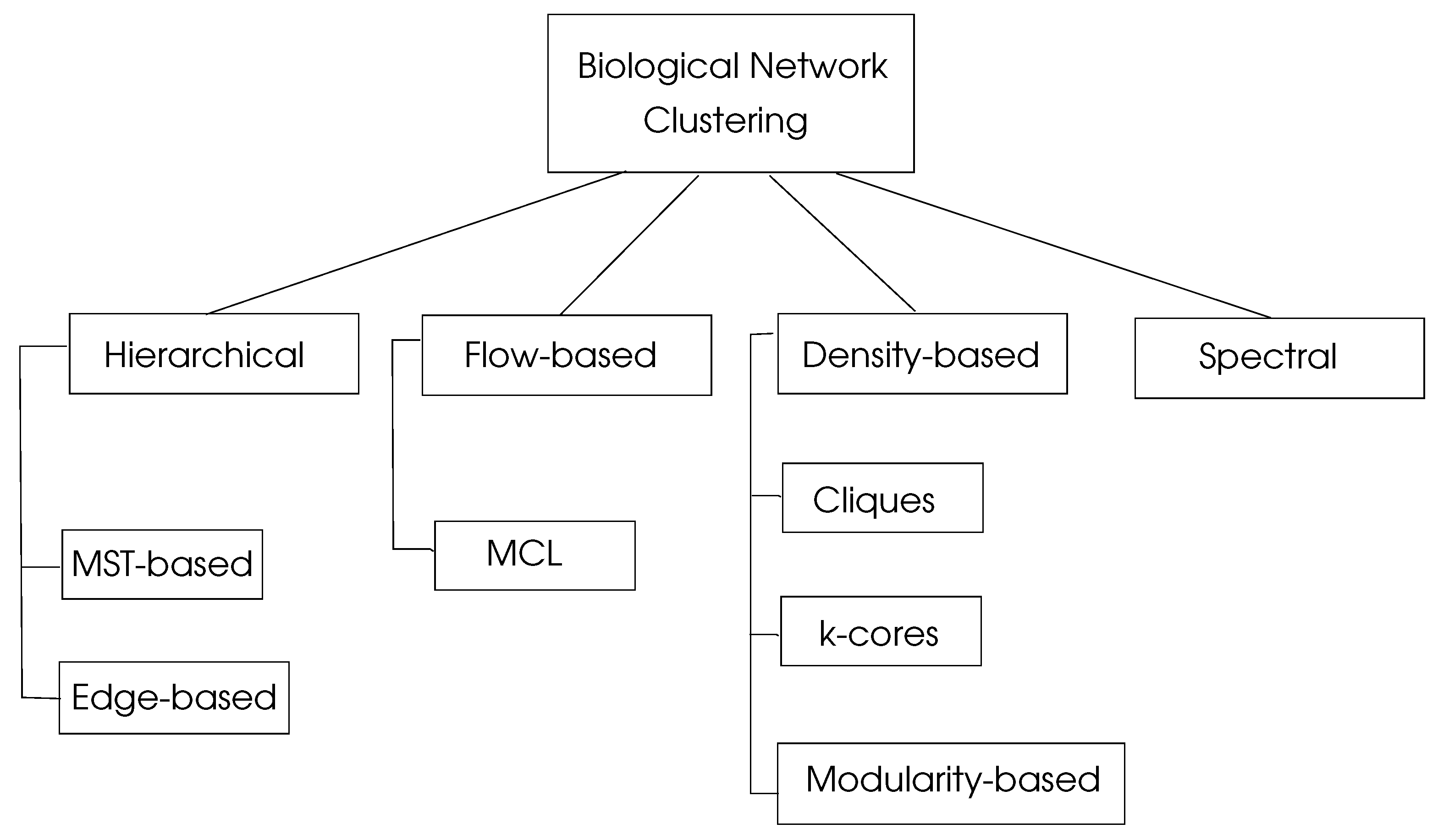
5. Cluster Discovery in Biological Networks
5.1. Hierarchical Clustering
- Single Link: The distance between two closest points, with one of them in and other in , is considered.
- Complete Link: The distance between the two points in two clusters that are farthest is used.
- Average Link: The average distance between every pair of points in and is considered.
5.2. Density-Based Clustering
5.3. Flow-Based Clustering
5.4. Spectral Clustering
5.5. Fuzzy Clustering
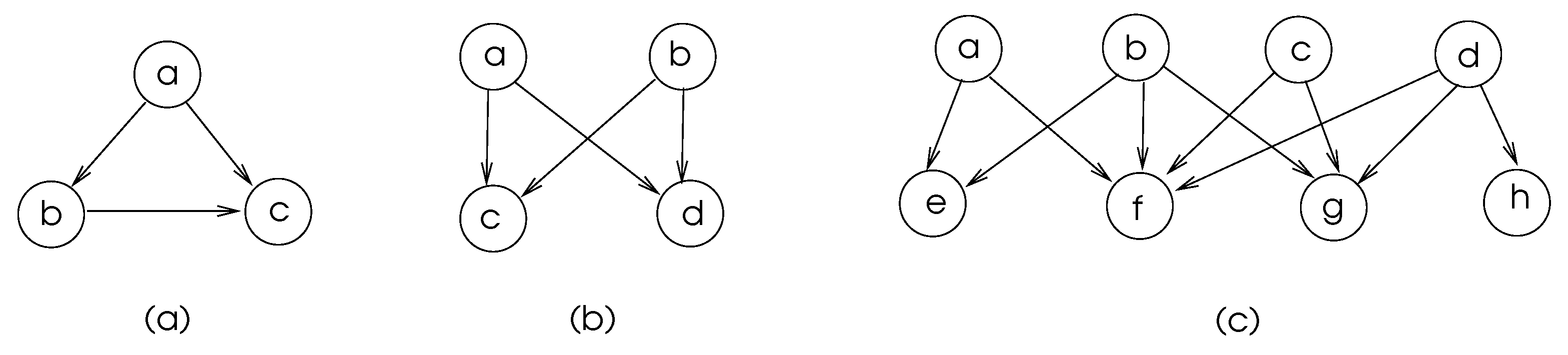
6. Network Motifs
6.1. Motif Discovery
- The detection of in G may be performed via exact counting, which involves the enumeration of all subgraphs of order k. This method evidently has a high time complexity; alternatively, sampling-based methods that work in a representative sample of the graph may provide approximate solutions.
- Isomorphic classes of the discovered motifs should be determined, since various motifs may be isomorphic to each other.
- Statistical significance of the discovered motifs in G should be determined. Commonly, a similar structured set of random graphs are generated, and motifs are searched in these graphs. If the motifs found in G are statistically higher in number than the ones found in the graphs of set , we can conclude that they do represent some biological function in the network represented by G.
6.2. Background
- P-value: This parameter is calculated by finding the number of elements of the randomly generated set that have a greater frequency of motif m than in the target graph G. A motif m is considered a significant motif if P-value of m, , given below, is less than 0.01.where is 1 if the occurrence of motif m in the random network is higher, and 0 if it is lower than that found in the target graph G.
- Z-score: The Z-score of a motif m, , in a graph G, is evaluated using the following formula:where is the number of discovered motifs m in G, and and are the mean and variance frequencies of m in a set of random networks. A motif m is significant if [43].
- Motif significance profile: The motif significance profile vector SP is structured with elements as Z-scores of motifs , and normalized to unity as below. Various graphs may then be compared for any common motifs contained in them.
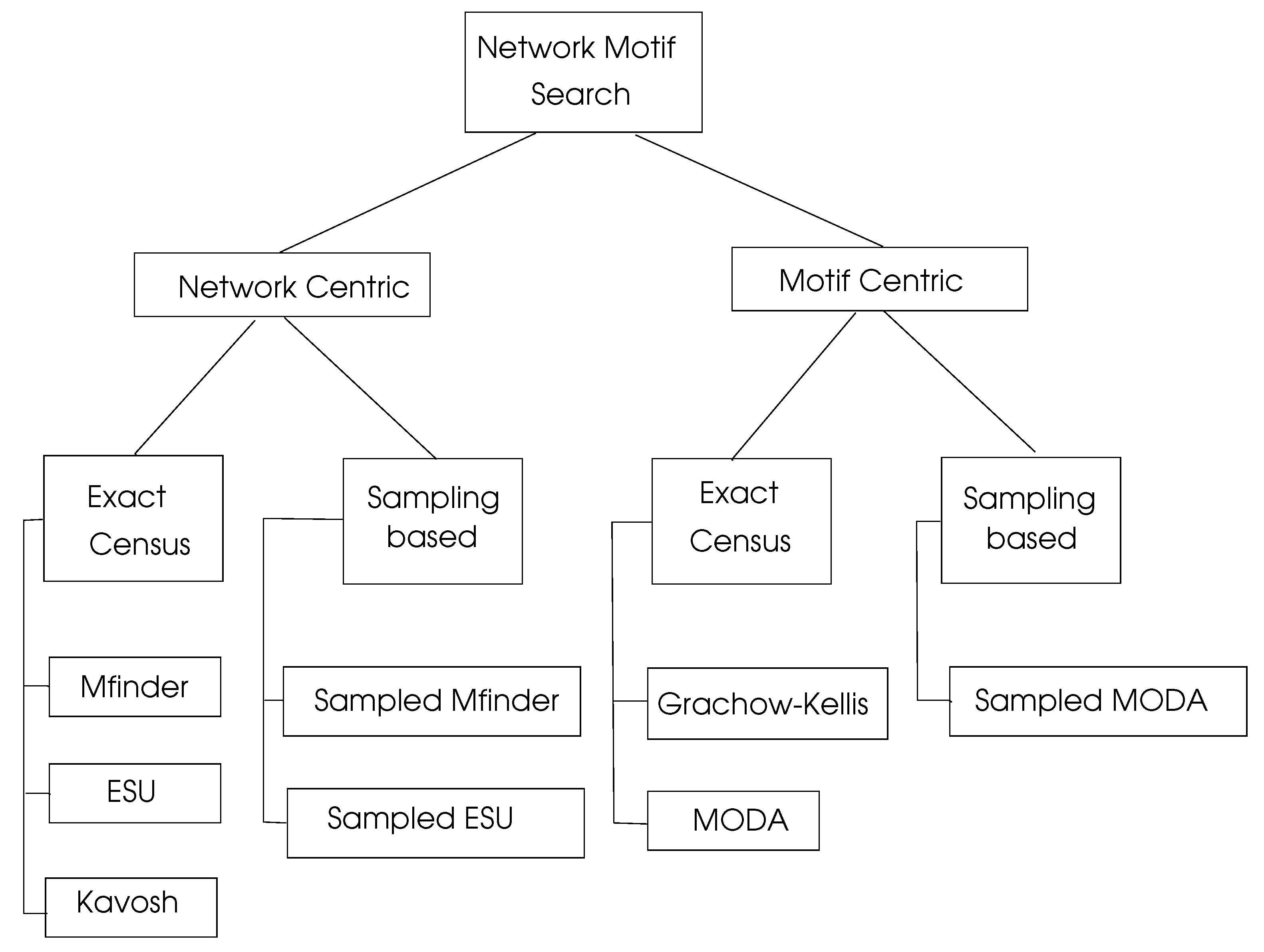
6.3. Review of Motif Searching Algorithms
6.3.1. Network Centric Search Algorithms
6.3.2. Motif Centric Search Algorithms
6.3.3. Parallel Motif Search Algorithms
7. Network Alignment
7.1. Background
- Form the similarity matrix R with entry showing the similarity score of the nodes and in input networks and , respectively.
- Implement a weighted matching algorithm to assess the similarity of the networks and .
7.2. Alignment Quality
7.3. Review of Network Alignment Algorithms
8. Discussion
Funding
Informed Consent Statement
Conflicts of Interest
References
- Vogelstein, B.; Lane, D.; Levine, A. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Carbonell, P.; Anne-Galle Planson, A.-G.; Davide Fichera, D.; Jean-Loup Faulon, J.-P. A retrosynthetic biology approach to metabolic pathway design for therapeutic production. BMC Syst. Biol. 2011, 5, 122. [Google Scholar] [CrossRef]
- Mason, O.; Verwoerd, M. Graph theory and networks in biology. IET Syst. Biol. 2007, 1, 89–119. [Google Scholar] [CrossRef]
- Erciyes, K. Distributed and Sequential Algorithms for Bioinformatics; Springer Computational Biology Series; Springer: Basel, Switzerland, 2013; Chapter 7 and Chapters 10–13. [Google Scholar]
- Jeong, H.; Mason, S.P.; Barabási, A.-L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2011, 411, 41–42. [Google Scholar] [CrossRef]
- Titz, B.; Rajagopala, S.V.; Goll, J.; Hauser, R.; McKevitt, M.T.; Palzkill, T.; Uetz, P. The binary protein interactome of Treponema pallidum, the syphilis spirochete. PLoS ONE 2008, 3, e2292. [Google Scholar] [CrossRef]
- He, Y.; Chen, Z.; Evans, A. Structural insights into aberrant topological patterns of large scale cortical networks in Alzheimers disease. J. Neurosci. 2008, 28, 4756–4766. [Google Scholar] [CrossRef]
- Schuster, S.; Fell, D.A.; Dandekar, T. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat. Biotechnol. 2000, 18, 326–332. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef]
- Junker, B. Analysis of Biological Networks; Wiley: Hoboken, NJ, USA, 2008; Chapter 9. [Google Scholar]
- Sporns, O. Networks of the Brain; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Ghorbani, M.; Khaki, A. A note on the fourth version of geometric-arithmetic index. Optoelectron. Adv. Mater. Rapid Commun. 2010, 4, 2212–2215. [Google Scholar]
- Gao, W.; Wu, H.; Siddiqui, M.K.; Baig, A.Q. Study of biological networks using graph theory. Saudi J. Biol. Sci. 2018, 25, 1212–1219. [Google Scholar] [CrossRef]
- Basavanagoud, B.; Barang, A.P. M-polynomial of some cactus chains and their topological indices. Open J. Discret. Appl. Math. 2019, 2, 59–67. [Google Scholar] [CrossRef]
- Dobrynin, A.A.; Estaji, E. Wiener index of hexago- nal chains under some transformations. Open J. Discret. Math. 2020, 3, 28–36. [Google Scholar] [CrossRef]
- Zhang, X.; Saleem, U.; Waheed, M.; Jamil, M.K.; Zeeshan, M. Comparative study of five topological invariants of supramolecular chain of different complexes of N-salicylidene-L-valine. AIMS Math. Biosci. Eng. 2023, 20, 11528–11544. [Google Scholar] [CrossRef]
- Santolini, M.; Barabási, A.L. Predicting perturbation patterns from the topology of biological networks. Proc. Natl. Acad. Sci. USA 2018, 115, E6375–E6383. [Google Scholar] [CrossRef]
- Albert, R.; Barabasi, A. The statistical mechanics of complex networks. Rev. Mod. Phys. 2002, 74, 47–97. [Google Scholar] [CrossRef]
- Newman, M.E.J. Fast algorithm for detecting community structure in networks. Phys. Rev. E 2004, 69, 066133. [Google Scholar] [CrossRef]
- Newman, M.E.J.; Girvan, M. Finding and evaluating community structure in networks. Phys. Rev. E 2004, 69, 026113. [Google Scholar] [CrossRef]
- Olman, V.; Mao, F.; Wu, H.; Xu, Y. Parallel clustering algorithm for large data sets with applications in bioinformatics. IEEE/ACM Trans. Comput. Biol. Bioinform. 2009, 6, 344–352. [Google Scholar] [CrossRef]
- Murtagh, F. Clustering in massive data sets. In Handbook of Massive Data Sets; Springer: Berlin/Heidelberg, Germany, 2002; pp. 501–543. [Google Scholar]
- Girvan, M.; Newman, M.E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef]
- Yang, Q.; Lonardi, S. A parallel edge-betweenness clustering tool for protein-protein interaction networks. Int. J. Data Min. Bioinform. (IJDMB) 2007, 1, 241–247. [Google Scholar] [CrossRef]
- Bron, C.; Kerbosch, J. Algorithm 457: Finding all cliques of an undirected graph. Commun. ACM 1973, 16, 575–577. [Google Scholar] [CrossRef]
- Schmidt, M.C.; Samatova, N.F.; Thomas, K.; Park, B.-H. A scalable, parallel algorithm for maximal clique enumeration. J. Parallel Distrib. Comput. 2009, 69, 417–428. [Google Scholar] [CrossRef]
- Mohseni-Zadeh, S.; Brezelec, P.; Risler, J.L. Cluster-C, an algorithm for the large-scale clustering of protein sequences based on the extraction of maximal cliques. Comput. Biol. Chem. 2004, 28, 211–218. [Google Scholar] [CrossRef]
- Jaber, K.; Rashid, N.A.; Abdullah, R. The parallel maximal cliques algorithm for protein sequence clustering. Am. J. Appl. Sci. 2009, 6, 1368–1372. [Google Scholar] [CrossRef]
- Batagelj, V.; Zaversnik, M. An O(m) algorithm for cores decomposition of networks. arXiv 2003, arXiv:0310049. [Google Scholar]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Montresor, A.; Pellegrini, F.D.; Miorandi, D. Distributed k-Core decomposition. IEEE Trans. Parallel Distrib. Syst. 2013, 24, 288–300. [Google Scholar] [CrossRef]
- Gehweiler, J.; Meyerhenke, H. A distributed diffusive heuristic for clustering a virtual P2P supercomputer. In Proceedings of the 7th High-Performance Grid Computing Workshop (HGCW10) in Conjunction with 24th International Parallel and Distributed Processing Symposium (IPDPS10), Atlanta, GA, USA, 19–23 April 2010. [Google Scholar]
- Riedy, J.; Bader, D.A.; Meyerhenke, H. Scalable multi-threaded community detection in social networks. In Proceedings of the IEEE 26th International Parallel and Distributed Processing Symposium Workshops and PhD Forum (IPDPSW), Shanghai, China, 21–25 May 2012; pp. 1619–1628. [Google Scholar]
- Dongen, S.V. Graph Clustering by Flow Simulation. Ph.D. Thesis, University of Utrecht, Utrecht, The Netherlands, 2000. [Google Scholar]
- Brohee, S.; van Helden, J. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinform. 2006, 7, 488. [Google Scholar] [CrossRef]
- Vlasblom, J.; Wodak, S.J. Markov clustering versus affinity propagation for the partitioning of protein interaction graphs. BMC Bioinform. 2009, 10, 99. [Google Scholar] [CrossRef]
- Bustamam, A.; Sehgal, M.S.; Hamilton, N.; Wong, S.; Ragan, M.A.; Burrage, K. An efficient parallel implementation of Markov clustering algorithm for large-scale protein-protein interaction networks that uses MPI. In Proceedings of the fifth IMT-GT International Conference Mathematics, Statistics, and Their Applications (ICMSA), Sumatra Barat, Indonesia, 9–11 June 2009; pp. 94–101. [Google Scholar]
- Bustamam, A.; Burrage, K.; Hamilton, N.A. Fast parallel Markov clustering in bioinformatics using massively parallel computing on GPU with CUDA and ELLPACK-R sparse format. IEEE/ACM Trans. Comp. Biol. Bioinform. 2011, 9, 679–691. [Google Scholar] [CrossRef]
- Fiedler, M. Laplacian of graphs and algebraic connectivity. Comb. Graph Theory 1989, 25, 57–70. [Google Scholar] [CrossRef]
- Chen, W.-Y.; Song, Y.; Bai, H.; Lin, C.-J.; Chang, E.Y. Parallel spectral clustering in distributed systems. IEEE Trans. Pattern. Anal. Mach. Intell. 2010, 33, 568–586. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Chan, K.C. Fuzzy clustering in a complex network based on content relevance and link structures. IEEE Trans. Fuzzy Syst. 2015, 24, 456–470. [Google Scholar] [CrossRef]
- Hu, L.; Yang, Y.; Tang, Z.; He, Y.; XLuo, X. FCAN-MOPSO: An Improved Fuzzy-based Graph Clustering Algorithm for Complex Networks with Multi-objective Particle Swarm Optimization. IEEE Trans. Fuzzy Syst. 2023. [Google Scholar] [CrossRef]
- Kashtan, N.; Itzkovitz, S.; Milo, R.; Alon, U. Mfinder Tool Guide; Technical Report; Department of Molecular Cell Biology and Computer Science and Applied Mathematics, Weizman Institute of Science: Rehovot, Israel, 2002. [Google Scholar]
- Kashtan, N.; Itzkovitz, S.; Milo, R.; Alon, U. Efficient sampling algorithm for estimating sub-graph concentrations and detecting network motifs. Bioinformatics 2004, 20, 1746–1758. [Google Scholar] [CrossRef]
- Wernicke, S. Efficient detection of network motifs. IEEE/ACM Trans. Comput. Biol. Bioinform. 2006, 3, 347–359. [Google Scholar] [CrossRef]
- Wernicke, S.; Rasche, F. FANMOD: A tool for fast network motif detection. Bioinformatics 2006, 22, 1152–1153. [Google Scholar] [CrossRef]
- Shen-Orr, S.S.; Milo, R.; Mangan, S.; Alon, U. Network motifs in the transcriptional regulation network of Escherichia Coli. Nat. Gen. 2002, 31, 64–68. [Google Scholar] [CrossRef]
- Milo, R.; Shen-Orr, S.; Itzkovitz, S.; Kashtan, N.; Chklovskii, D.; Alon, U. Network motifs: Simple building blocks of complex networks. Science 2004, 298, 824–827. [Google Scholar] [CrossRef]
- Williams, R.J.; Martinez, N.D. Simple rules yield complex food webs. Nature 2000, 404, 180–183. [Google Scholar] [CrossRef]
- Kashani, Z.R.; Ahrabian, H.; Elahi, E.; Nowzari-Dalini, A.; Ansari, E.S.; Asadi, S.; Mohammadi, S.; Schreiber, F.; Masoudi-Nejad, A. Kavosh: A new algorithm for finding network motifs. BMC Bioinform. 2009, 10, 318. [Google Scholar] [CrossRef] [PubMed]
- Grochow, J.; Kellis, M. Network motif discovery using subgraph enumeration and symmetry-breaking. In Proceedings of the 11th Annual International Conference Research in Computational Molecular Biology (RECOMB’07), Oakland, CA, USA, 21–25 April 2007; pp. 92–106. [Google Scholar]
- Han, J.-D.J.; Bertin, N.; Hao, T.; Goldberg, D.S.; Berriz, G.F.; Zhang, L.V.; Dupuy, D.; Walhout, A.J.M.; Cusick, M.E.; Roth, F.P.; et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 2004, 430, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.C.; Crawford, M.E.; Hirschman, J.E.; Kranz, J.E.; Olsen, P.; Robertson, L.S.; Skrzypek, M.S.; Braun, B.R.; Hopkins, K.L.; Kondu, P.; et al. Ypd(tm), pombepd(tm), and wormpd(tm): Model organism volumes of the bioknowledge(tm) library, an integrated resource for protein information. Nucleic Acids Res. 2001, 29, 75–79. [Google Scholar] [CrossRef]
- Omidi, S.; Schreiber, F.; Masoudi-Nejad, A. MODA: An efficient algorithm for network motif discovery in biological networks. Genes Genet. Syst. 2009, 84, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Touchman, J.W.; Zhang, W.; Suh, E.B.; Xue, G. A parallel algorithm for extracting transcription regulatory network motifs. In Proceedings of the IEEE International Symposium on Bioinformatics and Bioengineering, Minneapolis, MN, USA, 19–21 October 2005; IEEE Computer Society Press: Washington, DC, USA, 2005; pp. 193–200. [Google Scholar]
- Schatz, M.; Cooper-Balis, E.; Bazinet, A. Parallel Network Motif Finding; Technical Report; University of Maryland Insitute for Advanced Computer Studies: College Park, MD, USA, 2008. [Google Scholar]
- Ribeiro, P. Efficient and Scalable Algorithms for Network Motifs Discovery. Ph.D. Thesis, Doctoral Programme in Computer Science, Faculty of Science of the University of Porto, Porto, Portugal, 2009. [Google Scholar]
- Ribeiro, P.; Silva, F.; Lopes, L. A parallel algorithm for counting subgraphs in complex networks. In Proceedings of the 3rd International Conference on Biomedical Engineering Systems and Technologies, Valencia, Spain, 20–23 January 2010; pp. 380–393. [Google Scholar]
- Ribeiro, P.; Silva, F.; Lopes, L. Parallel discovery of network motifs. J. Parallel Distrib. Comput. 2012, 72, 144–154. [Google Scholar] [CrossRef]
- Ruzgar, E.; Erciyes, K.; Dalkilic, M.E. Parallelization of network motif discovery using star contraction. Parallel Comput. 2021, 101, 102734. [Google Scholar]
- Patra, S.; Mohapatra, A. Review of tools and algorithms for network motif discovery in biological networks. IET Syst. Biol. 2020, 14, 171–189. [Google Scholar] [CrossRef]
- Singh, R.; Xu, J.; Berger, B. Pairwise global alignment of protein interaction networks by matching neighbourhood topology. In Research in Computational Molecular Biology, Proceedings of the 11th Annunal International Conference, RECOMB 2007, Oakland, CA, USA, 21–25 April 2007; Springer: Berlin/Heidelberg, Germany, 2007; pp. 16–31. [Google Scholar]
- Patro, R.; Kingsford, C. Global network alignment using multiscale spectral signatures. Bioinformatics 2012, 28, 3105–3114. [Google Scholar] [CrossRef]
- Przulj, N. Graph theory analysis of protein-protein interactions. In A Chapter in Knowledge Discovery in Proteomics; Igor, J., Dennis, W., Eds.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Altschul, S.; Gish, W.; Miller, W.; Myers, E.; Lipman, D. Basic local alignment search tool. J. Mol. Biol 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kelley, B.P.; Sharan, R.; Karp, R.M.; Sittler, T.; Root, D.E.; Stockwell, B.R.; Ideker, T. Conserved pathways within bacteria and yeast as revealed by global protein network alignment. Proc. Natl. Acad. Sci. USA 2003, 100, 11394–11399. [Google Scholar] [CrossRef]
- Koyuturk, M.; Kim, Y.; Topkara, U.; Subramaniam, S.; Szpankowski, W.; Grama, A. Pairwise alignment of protein interaction networks. J. Comput. Biol. 2006, 13, 182–199. [Google Scholar] [CrossRef] [PubMed]
- El-Kebir, M.; Heringa, J.; Klau, G.W. Lagrangian relaxation applied to sparse global network alignment. In Proceedings of the 6th IAPR International Conference on Pattern Recognition in Bioinformatics (PRIB’11), Delft, The Netherlands, 2–4 November 2011; pp. 225–236. [Google Scholar]
- Klau, G.W. A new graph-based method for pairwise global network alignment. BMC Bioinform. 2009, 10, S59. [Google Scholar] [CrossRef] [PubMed]
- Kuchaiev, O.; Milenkovic, T.; Memisevic, V.; Hayes, W.; Przulj, N. Topological network alignment uncovers biological function and phylogeny. J. R. Soc. Interface 2010, 7, 1341–1354. [Google Scholar] [CrossRef] [PubMed]
- Aladag, A.E.; Erten, C. SPINAL: Scalable protein interaction network alignment. Bioinformatics 2013, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Preis, R. Linear time 2-approximation algorithm for maximum weighted matching in general graphs. In STACS99, Proceeedings of the 16th Annual Conference Theoretical Aspects of Computer Science, Trier, Germany, 4–6 March 1999; Lecture Notes in Computer, Science; Meinel, C., Tison, S., Eds.; Springer: Berlin/Heidelberg, Germany, 1999; pp. 259–269. [Google Scholar]
- Hoepman, J.H. Simple distributed weighted matchings. arXiv 2004, arXiv:cs/0410047v1. [Google Scholar]
- Manne, F.; Bisseling, R.H. A parallel approximation algorithm for the weighted maximum matching problem. In Proceedings of the Seventh International Conference on Parallel Processing and Applied Mathematics (PPAM 2007), Gdansk, Poland, 9–12 September 2007; Lecture Notes in Computer, Science. Wyrzykowski, R., Karczewski, K., Dongarra, J., Wasniewski, J., Eds.; pp. 708–717. [Google Scholar]
- Sathe, M.; Schenk, O.; Burkhart, H. An auction-based weighted matching implementation on massively parallel architectures. Parallel Comput. 2012, 38, 595–614. [Google Scholar] [CrossRef]
- Saribatir, M.B.; Erciyes, K. A Parallel Network Alignment Algorithm for Biological Networks. In Proceedings of the IEEE 3rd International Informatics and Software Engineering Conference (IISEC), Ankara, Turkey, 15–16 December 2022. [Google Scholar]
- Maskey, S.; Cho, Y.-R. Survey of biological network alignment: Cross-species analysis of conserved systems. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019; pp. 2090–2096. [Google Scholar]
- Erciyes, K. Algebraic Graph Algorithms, A Practical Approach Using Python; Springer Undergraduate Topics in Computer Science Series; Springer: Basel, Switzerland, 2021. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erciyes, K. Graph-Theoretical Analysis of Biological Networks: A Survey. Computation 2023, 11, 188. https://doi.org/10.3390/computation11100188
Erciyes K. Graph-Theoretical Analysis of Biological Networks: A Survey. Computation. 2023; 11(10):188. https://doi.org/10.3390/computation11100188
Chicago/Turabian StyleErciyes, Kayhan. 2023. "Graph-Theoretical Analysis of Biological Networks: A Survey" Computation 11, no. 10: 188. https://doi.org/10.3390/computation11100188
APA StyleErciyes, K. (2023). Graph-Theoretical Analysis of Biological Networks: A Survey. Computation, 11(10), 188. https://doi.org/10.3390/computation11100188