
Local Potential Functional Embedding Theory: A Self-Consistent Flavor of Density Functional Theory for Lattices without Density Functionals


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Theory
2.1. One-Dimensional Hubbard Lattice
2.2. Review of Ht-DMFET
2.2.1. Exact Non-Interacting Embedding
2.2.2. Non-Interacting Embedding Hamiltonian
2.2.3. Approximate Interacting Embedding
2.3. Exact Density-Functional Embedding
2.3.1. KS-DFT for Uniform Lattices
2.3.2. Density-Functional Interacting Cluster
2.4. Local Potential Functional Embedding Theory
2.5. Comparison with SDE
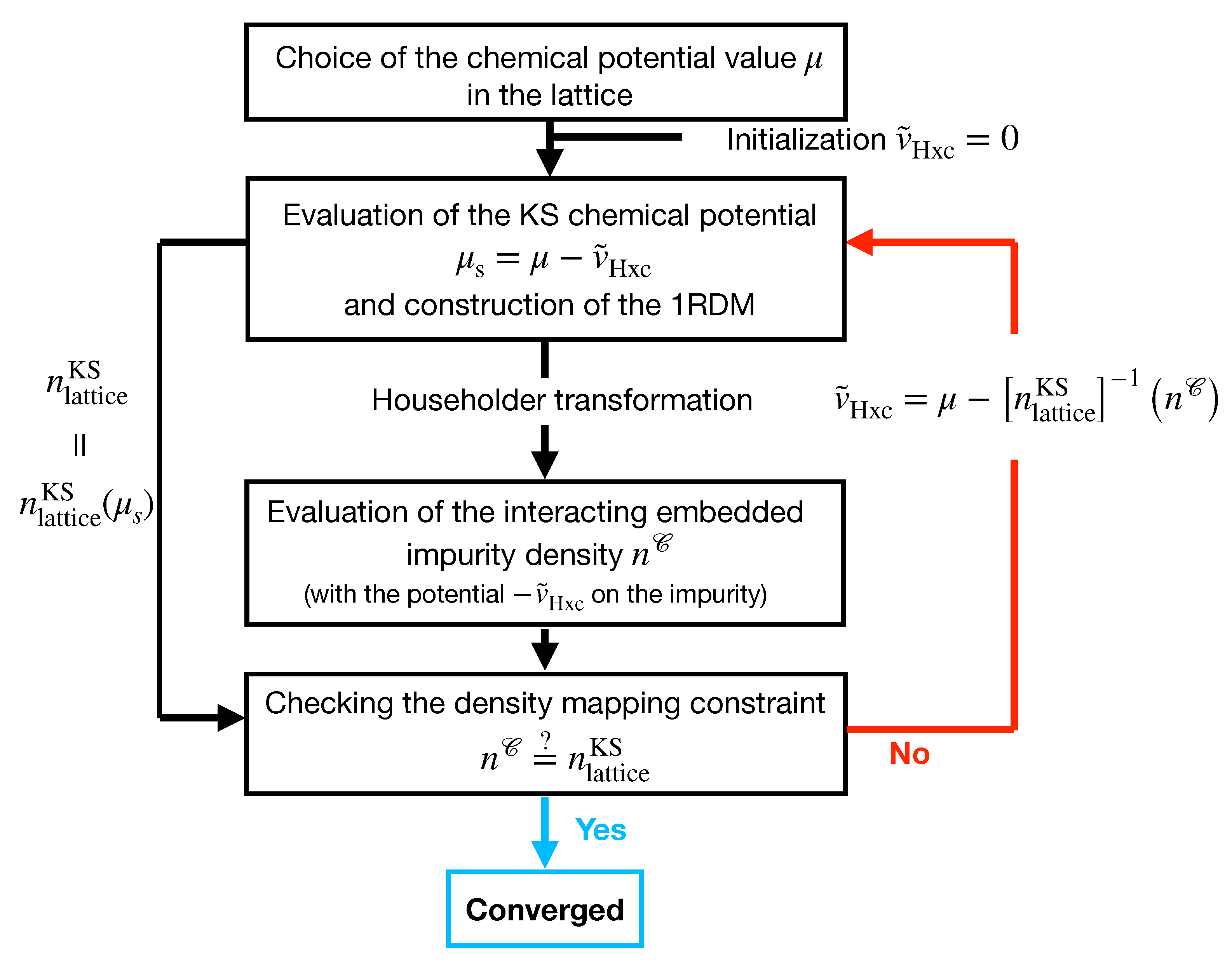
3. LPFET Algorithm
- We start by diagonalizing the one-electron Hamiltonian (i.e., the hopping in the present case) matrix (see Equation (7)). Thus, we obtain the “molecular” spin-orbitals and their corresponding energies. We fix the chemical potential of the interacting lattice to some value and (arbitrarily) initialize the Hxc potential to . Therefore, at the zeroth iteration, the KS chemical potential equals .
- We occupy all the molecular spin-orbitals with energies below and construct the corresponding density matrix (in the lattice representation). The latter provides the uniform KS density (denoted in Figure 2) and the embedding Householder cluster Hamiltonian (see Equation (46)) in which the impurity chemical potential is set to (see Equation (86)).
- We verify that the density in the KS lattice and the occupation of the interacting embedded impurity match (a convergence threshold has been set to 10). If this is the case, the calculation has converged and is interpreted as (an approximation to) the density in the true interacting lattice. If the two densities do not match, the Hxc potential is adjusted in the KS lattice such that the latter reproduces (see Equation (90)) or, equivalently, such that the KS lattice contains electrons. We then return to step 2.
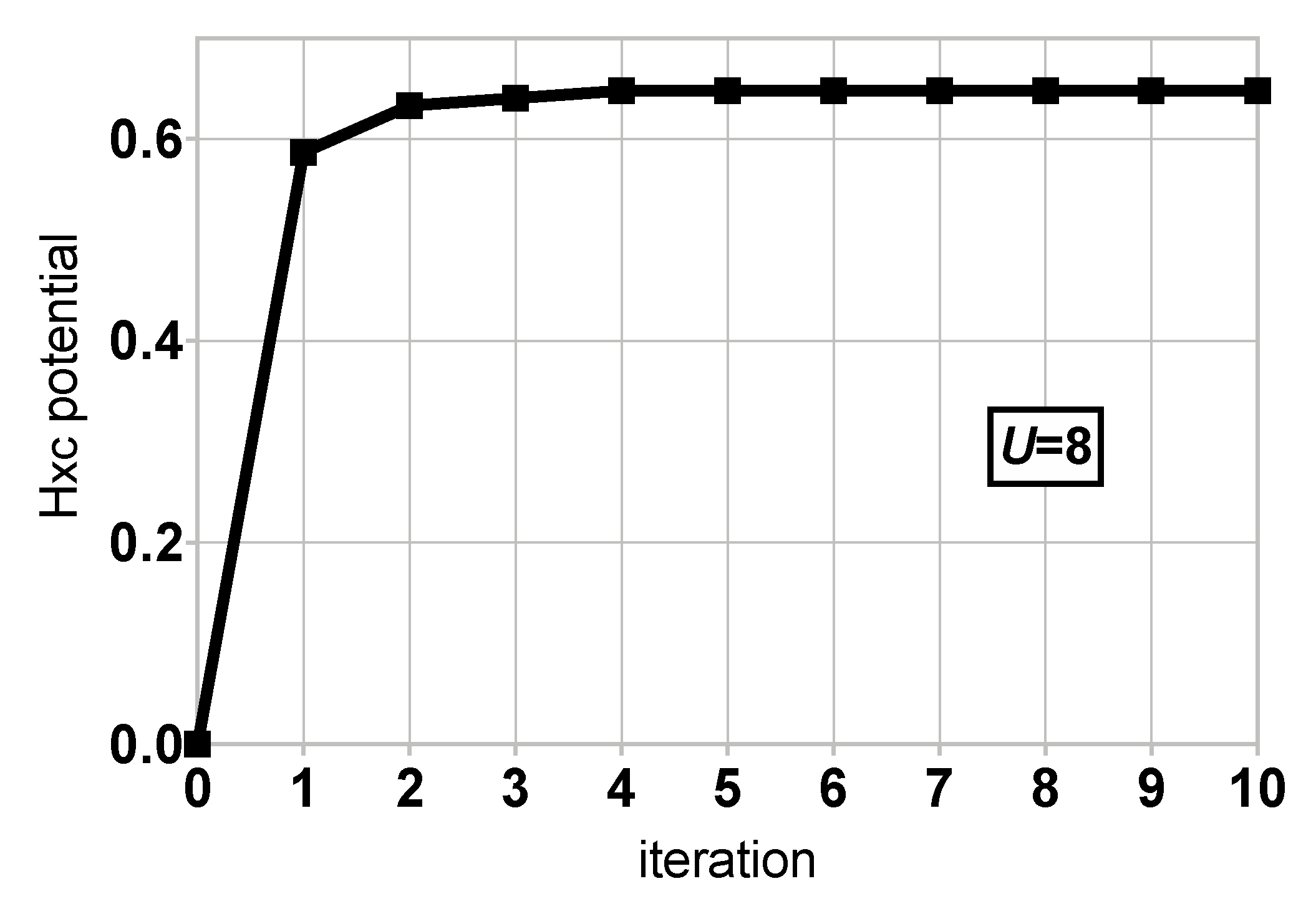
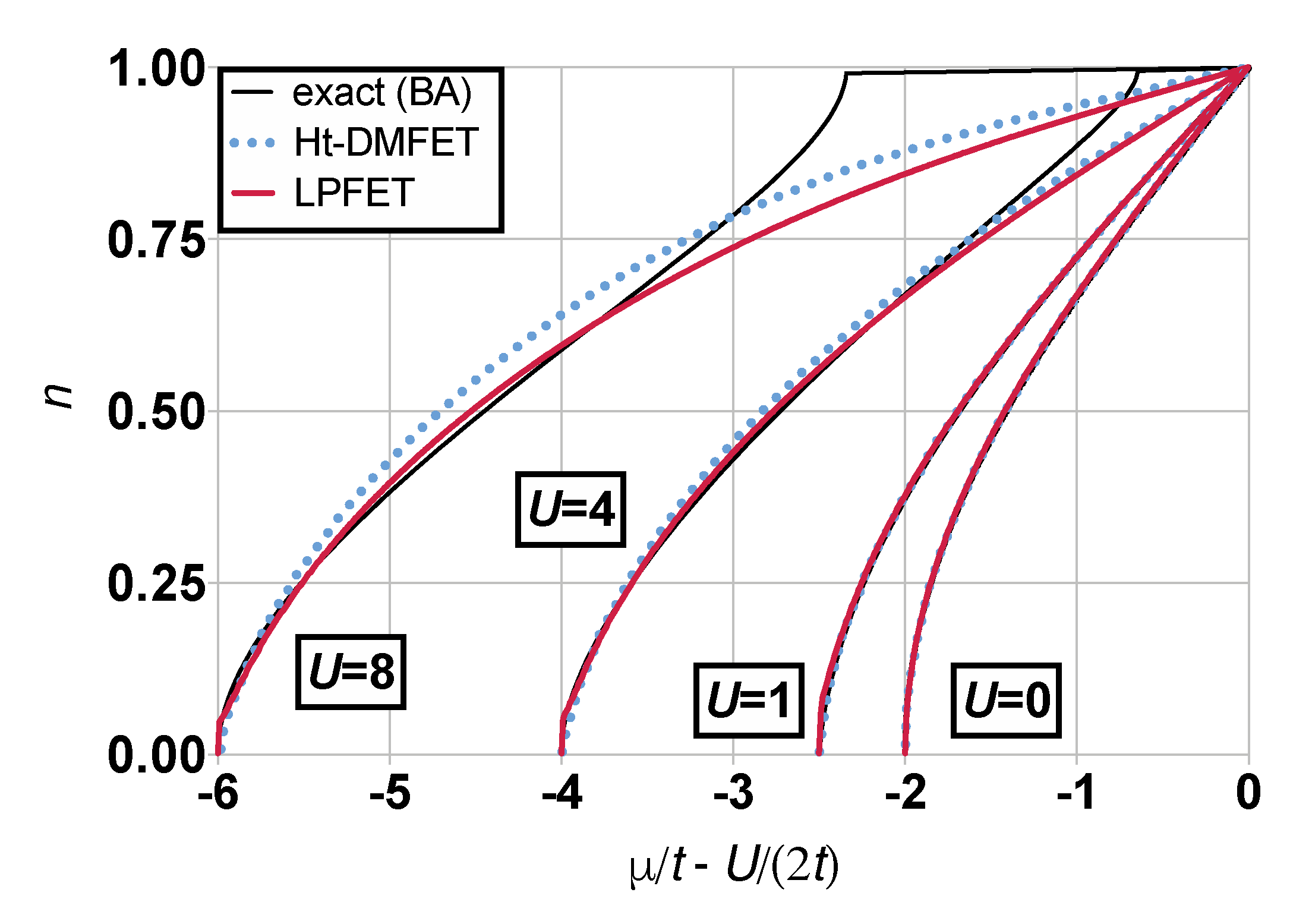
4. Results and Discussion
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Simplification of Density Matrix Elements in the Householder Representation
References
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Aryasetiawan, F.; Lichtenstein, A.I. First-principles calculations of the electronic structure and spectra of strongly correlated systems: The LDA +U method. J. Phys. Condens. Matter 1997, 9, 767–808. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Poteryaev, A.I.; Korotin, M.A.; Anokhin, A.O.; Kotliar, G. First-principles calculations of the electronic structure and spectra of strongly correlated systems: Dynamical mean-field theory. J. Phys. Condens. Matter 1997, 9, 7359–7367. [Google Scholar] [CrossRef]
- Lichtenstein, A.I.; Katsnelson, M.I. Ab initio calculations of quasiparticle band structure in correlated systems: LDA++ approach. Phys. Rev. B 1998, 57, 6884–6895. [Google Scholar] [CrossRef]
- Kotliar, G.; Savrasov, S.Y.; Haule, K.; Oudovenko, V.S.; Parcollet, O.; Marianetti, C.A. Electronic structure calculations with dynamical mean-field theory. Rev. Mod. Phys. 2006, 78, 865–951. [Google Scholar] [CrossRef]
- Haule, K. Exact Double Counting in Combining the Dynamical Mean Field Theory and the Density Functional Theory. Phys. Rev. Lett. 2015, 115, 196403. [Google Scholar] [CrossRef]
- Requist, R.; Gross, E.K.U. Model Hamiltonian for strongly correlated systems: Systematic, self-consistent, and unique construction. Phys. Rev. B 2019, 99, 125114. [Google Scholar] [CrossRef]
- Ghosh, S.; Verma, P.; Cramer, C.J.; Gagliardi, L.; Truhlar, D.G. Combining Wave Function Methods with Density Functional Theory for Excited States. Chem. Rev. 2018, 118, 7249–7292. [Google Scholar] [CrossRef]
- Savin, A. Recent Developments and Applications of Modern Density Functional Theory; Elsevier: Amsterdam, The Netherlands, 1996; p. 327. [Google Scholar]
- Toulouse, J.; Colonna, F.; Savin, A. Long-range–short-range separation of the electron-electron interaction in density-functional theory. Phys. Rev. A 2004, 70, 062505. [Google Scholar] [CrossRef]
- Sharkas, K.; Toulouse, J.; Savin, A. Double-hybrid density-functional theory made rigorous. J. Chem. Phys. 2011, 134, 064113. [Google Scholar] [CrossRef]
- Fromager, E. On the exact formulation of multi-configuration density-functional theory: Electron density versus orbitals occupation. Mol. Phys. 2015, 113, 419–434. [Google Scholar] [CrossRef]
- Senjean, B.; Nakatani, N.; Tsuchiizu, M.; Fromager, E. Site-occupation embedding theory using Bethe ansatz local density approximations. Phys. Rev. B 2018, 97, 235105. [Google Scholar] [CrossRef]
- Wasserman, A.; Pavanello, M. Quantum embedding electronic structure methods. Int. J. Quantum Chem. 2020, 120, e26495. [Google Scholar] [CrossRef]
- Knizia, G.; Chan, G.K.L. Density Matrix Embedding: A Simple Alternative to Dynamical Mean-Field Theory. Phys. Rev. Lett. 2012, 109, 186404. [Google Scholar] [CrossRef] [PubMed]
- Knizia, G.; Chan, G. Density matrix embedding: A strong-coupling quantum embedding theory. J. Chem. Theory Comput. 2013, 9, 1428–1432. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimochi, T.; Welborn, M.; Van Voorhis, T. Density matrix embedding in an antisymmetrized geminal power bath. J. Chem. Phys. 2015, 143, 024107. [Google Scholar] [CrossRef] [PubMed]
- Welborn, M.; Tsuchimochi, T.; Van Voorhis, T. Bootstrap embedding: An internally consistent fragment-based method. J. Chem. Phys. 2016, 145, 074102. [Google Scholar] [CrossRef]
- Sun, Q.; Chan, G.K.L. Quantum Embedding Theories. Accounts Chem. Res. 2016, 49, 2705–2712. [Google Scholar] [CrossRef]
- Wouters, S.; Jiménez-Hoyos, C.A.; Sun, Q.; Chan, G.K.L. A Practical Guide to Density Matrix Embedding Theory in Quantum Chemistry. J. Chem. Theory Comput. 2016, 12, 2706–2719. [Google Scholar] [CrossRef]
- Wu, X.; Cui, Z.H.; Tong, Y.; Lindsey, M.; Chan, G.K.L.; Lin, L. Projected density matrix embedding theory with applications to the two-dimensional Hubbard model. J. Chem. Phys. 2019, 151, 064108. [Google Scholar] [CrossRef]
- Cui, Z.H.; Zhu, T.; Chan, G.K.L. Efficient Implementation of Ab Initio Quantum Embedding in Periodic Systems: Density Matrix Embedding Theory. J. Chem. Theory Comput. 2020, 16, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Faulstich, F.M.; Kim, R.; Cui, Z.H.; Wen, Z.; Kin-Lic Chan, G.; Lin, L. Pure State v-Representability of Density Matrix Embedding Theory. J. Chem. Theory Comput. 2022, 18, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, L.; Maitra, N.T. Embedding via the Exact Factorization Approach. Phys. Rev. Lett. 2020, 124, 206401. [Google Scholar] [CrossRef]
- Requist, R.; Gross, E.K.U. Fock-Space Embedding Theory: Application to Strongly Correlated Topological Phases. Phys. Rev. Lett. 2021, 127, 116401. [Google Scholar] [CrossRef] [PubMed]
- Georges, A.; Kotliar, G. Hubbard model in infinite dimensions. Phys. Rev. B 1992, 45, 6479–6483. [Google Scholar] [CrossRef]
- Georges, A.; Kotliar, G.; Krauth, W.; Rozenberg, M.J. Dynamical mean-field theory of strongly correlated fermion systems and the limit of infinite dimensions. Rev. Mod. Phys. 1996, 68, 13–125. [Google Scholar] [CrossRef]
- Kotliar, G.; Vollhardt, D. Strongly Correlated Materials: Insights From Dynamical Mean-Field Theory. Phys. Today 2004, 57, 53–59. [Google Scholar] [CrossRef]
- Held, K. Electronic structure calculations using dynamical mean field theory. Adv. Phys. 2007, 56, 829–926. [Google Scholar] [CrossRef]
- Zgid, D.; Chan, G.K.L. Dynamical mean-field theory from a quantum chemical perspective. J. Chem. Phys. 2011, 134, 094115. [Google Scholar] [CrossRef]
- Sekaran, S.; Tsuchiizu, M.; Saubanère, M.; Fromager, E. Householder-transformed density matrix functional embedding theory. Phys. Rev. B 2021, 104, 035121. [Google Scholar] [CrossRef]
- Ayral, T.; Lee, T.H.; Kotliar, G. Dynamical mean-field theory, density-matrix embedding theory, and rotationally invariant slave bosons: A unified perspective. Phys. Rev. B 2017, 96, 235139. [Google Scholar] [CrossRef]
- Lee, T.H.; Ayral, T.; Yao, Y.X.; Lanata, N.; Kotliar, G. Rotationally invariant slave-boson and density matrix embedding theory: Unified framework and comparative study on the one-dimensional and two-dimensional Hubbard model. Phys. Rev. B 2019, 99, 115129. [Google Scholar] [CrossRef]
- Fertitta, E.; Booth, G.H. Rigorous wave function embedding with dynamical fluctuations. Phys. Rev. B 2018, 98, 235132. [Google Scholar] [CrossRef]
- Fertitta, E.; Booth, G.H. Energy-weighted density matrix embedding of open correlated chemical fragments. J. Chem. Phys. 2019, 151, 014115. [Google Scholar] [CrossRef] [PubMed]
- Sriluckshmy, P.V.; Nusspickel, M.; Fertitta, E.; Booth, G.H. Fully algebraic and self-consistent effective dynamics in a static quantum embedding. Phys. Rev. B 2021, 103, 085131. [Google Scholar] [CrossRef]
- Lee, T.H.; Lanatà, N.; Kim, M.; Kotliar, G. Efficient Slave-Boson Approach for Multiorbital Two-Particle Response Functions and Superconductivity. Phys. Rev. X 2021, 11, 041040. [Google Scholar] [CrossRef]
- Bulik, I.W.; Scuseria, G.E.; Dukelsky, J. Density matrix embedding from broken symmetry lattice mean fields. Phys. Rev. B 2014, 89, 035140. [Google Scholar] [CrossRef]
- Senjean, B. Projected site-occupation embedding theory. Phys. Rev. B 2019, 100, 035136. [Google Scholar] [CrossRef]
- Lima, N.A.; Silva, M.F.; Oliveira, L.N.; Capelle, K. Density Functionals Not Based on the Electron Gas: Local-Density Approximation for a Luttinger Liquid. Phys. Rev. Lett. 2003, 90, 146402. [Google Scholar] [CrossRef]
- Capelle, K.; Campo, V.L. Density functionals and model Hamiltonians: Pillars of many-particle physics. Phys. Rep. 2013, 528, 91–159. [Google Scholar] [CrossRef]
- Mordovina, U.; Reinhard, T.E.; Theophilou, I.; Appel, H.; Rubio, A. Self-Consistent Density-Functional Embedding: A Novel Approach for Density-Functional Approximations. J. Chem. Theory Comput. 2019, 15, 5209–5220. [Google Scholar] [CrossRef] [PubMed]
- Theophilou, I.; Reinhard, T.E.; Rubio, A.; Ruggenthaler, M. Approximations based on density-matrix embedding theory for density-functional theories. Electron. Struct. 2021, 3, 035001. [Google Scholar] [CrossRef]
- Rotella, F.; Zambettakis, I. Block Householder transformation for parallel QR factorization. Appl. Math. Lett. 1999, 12, 29–34. [Google Scholar] [CrossRef][Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Lieb, E.H.; Wu, F.Y. Absence of Mott Transition in an Exact Solution of the Short-Range, One-Band Model in One Dimension. Phys. Rev. Lett. 1968, 20, 1445–1448. [Google Scholar] [CrossRef]
- Vilela, L.N.P.; Capelle, K.; Oliveira, L.N.; Campo, V.L. Approximate expression for the ground-state energy of the two- and three-dimensional Hubbard model at arbitrary filling obtained from dimensional scaling. J. Phys. Condens. Matter 2019, 31, 455601. [Google Scholar] [CrossRef]
- Senjean, B.; Tsuchiizu, M.; Robert, V.; Fromager, E. Local density approximation in site-occupation embedding theory. Mol. Phys. 2017, 115, 48–62. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sekaran, S.; Saubanère, M.; Fromager, E. Local Potential Functional Embedding Theory: A Self-Consistent Flavor of Density Functional Theory for Lattices without Density Functionals. Computation 2022, 10, 45. https://doi.org/10.3390/computation10030045
Sekaran S, Saubanère M, Fromager E. Local Potential Functional Embedding Theory: A Self-Consistent Flavor of Density Functional Theory for Lattices without Density Functionals. Computation. 2022; 10(3):45. https://doi.org/10.3390/computation10030045
Chicago/Turabian StyleSekaran, Sajanthan, Matthieu Saubanère, and Emmanuel Fromager. 2022. "Local Potential Functional Embedding Theory: A Self-Consistent Flavor of Density Functional Theory for Lattices without Density Functionals" Computation 10, no. 3: 45. https://doi.org/10.3390/computation10030045
APA StyleSekaran, S., Saubanère, M., & Fromager, E. (2022). Local Potential Functional Embedding Theory: A Self-Consistent Flavor of Density Functional Theory for Lattices without Density Functionals. Computation, 10(3), 45. https://doi.org/10.3390/computation10030045