
Metaverse Applications in Bioinformatics: A Machine Learning Framework for the Discrimination of Anti-Cancer Peptides

 ,
,  ,
,  , and
, and 
Abstract
1. Introduction
- A computational framework is proposed that extracts significant information from complex biological sequences, improving therapy for critical diseases. Leveraging the metaverse’s immersive features, it enables precise identification of potential cancer treatments, revolutionizing disease management.
- Existing models face overfitting problems caused by class imbalances. To this end, we contribute by employing the synthetic minority oversampling technique (SMOTE) for preprocessing, reducing error scores, and fostering equitable feature learning. Additionally, we enhance overall model performance through a majority-voting ensemble decision-making technique in the final output, collectively advancing the robustness of our approach.
- We refined the existing pseudo amino acid composition (PseAAC) method for sequence classification by systematically incorporating additional physicochemical properties, addressing limitations arising from heterogeneous peptide patterns. This enhancement aims to generate context-rich features, improving the robustness and informativeness of obtained features and ultimately enhancing the methodology’s effectiveness in capturing the complexity of peptide sequences.
- Comprehensive results from tests are obtained through analyses conducted on two sets of benchmark datasets, proving that the recommended trustworthy framework achieves new SOTA accuracy. Furthermore, ablation research is conducted to measure the effectiveness of each feature descriptor technique separately and evaluate the complementary strength produced from the diverse combinations of information.
2. Literature Review
- The imbalanced nature of datasets in anticancer peptide classification poses a significant hurdle for many existing machine learning methods. Biased models can emerge, favoring classes with a higher number of instances, thereby compromising the model’s ability to accurately identify and classify less-represented classes. This imbalance issue is particularly critical in the context of anticancer peptides, where a thorough understanding of diverse instances is crucial for effective classification.
- Prevailing methods often lean towards simplicity, employing single-feature extractions and classifiers. While this simplicity aids in model interpretability and computational efficiency, it may fall short of capturing the intricate and nuanced patterns inherent in anticancer peptides. The complex nature of these peptides demands more sophisticated approaches that can discern subtle variations and relationships within the data, enhancing the model’s discriminatory power.
- Current strategies aimed at enhancing classification accuracy often resort to fusion techniques. While these techniques offer potential improvements, they may inadvertently introduce homogeneity in the utilized information, leading to limiting the model’s ability to discern diverse and subtle characteristics crucial for accurate anticancer peptide classification. Striking a balance between fusion for improved accuracy and preserving the diversity of information remains a key challenge in developing robust models.
- Some machine learning-based methods in anticancer peptide classification may exhibit a tendency to overlook the expansive landscape of feature extraction models and selection techniques. A more comprehensive exploration of this landscape is imperative to ensure that potentially more effective approaches are not neglected. The diversity among anticancer peptides demands a thorough examination of various feature extraction methods and selection techniques to uncover the most suitable combination for accurate classification.
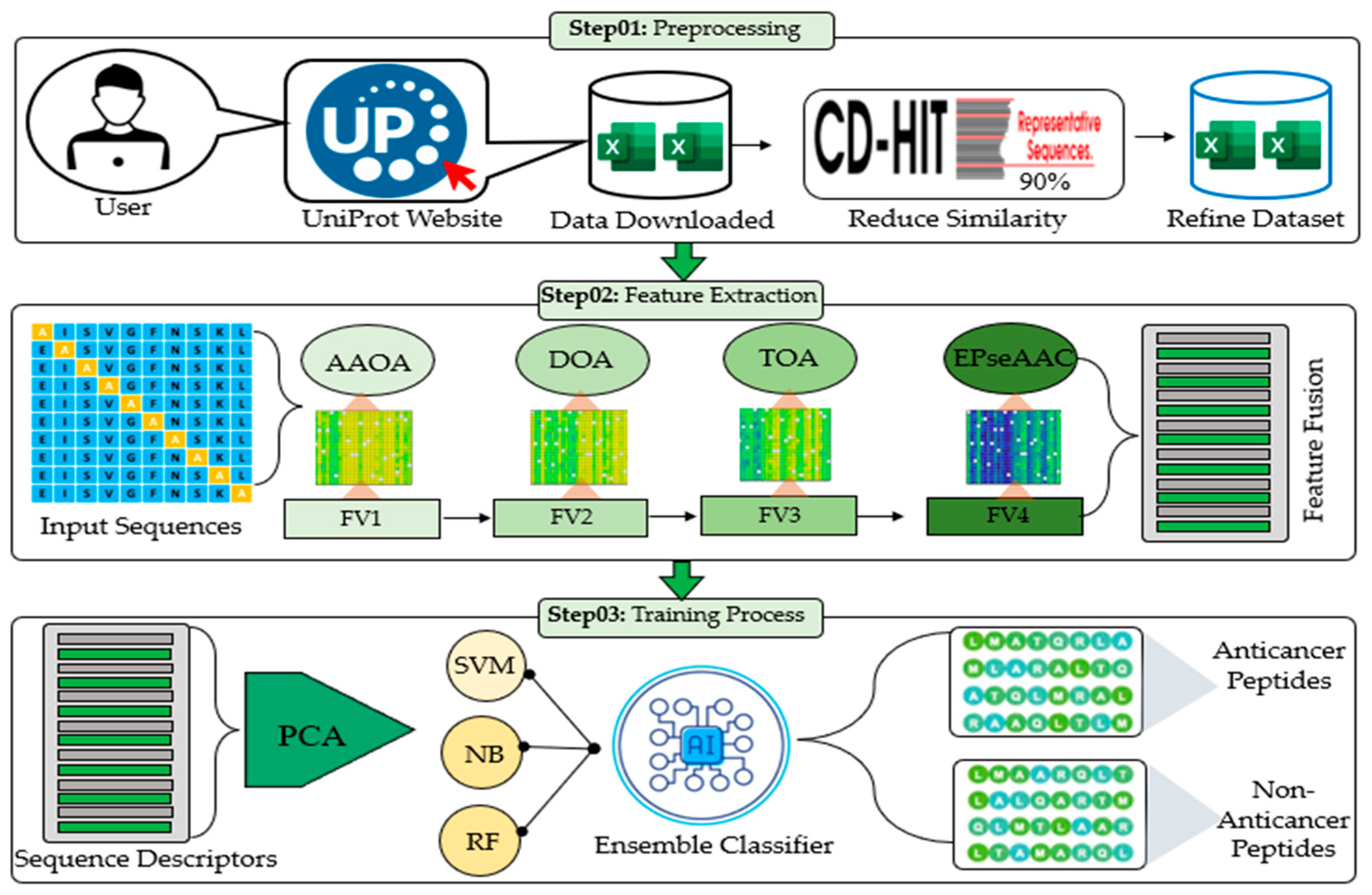
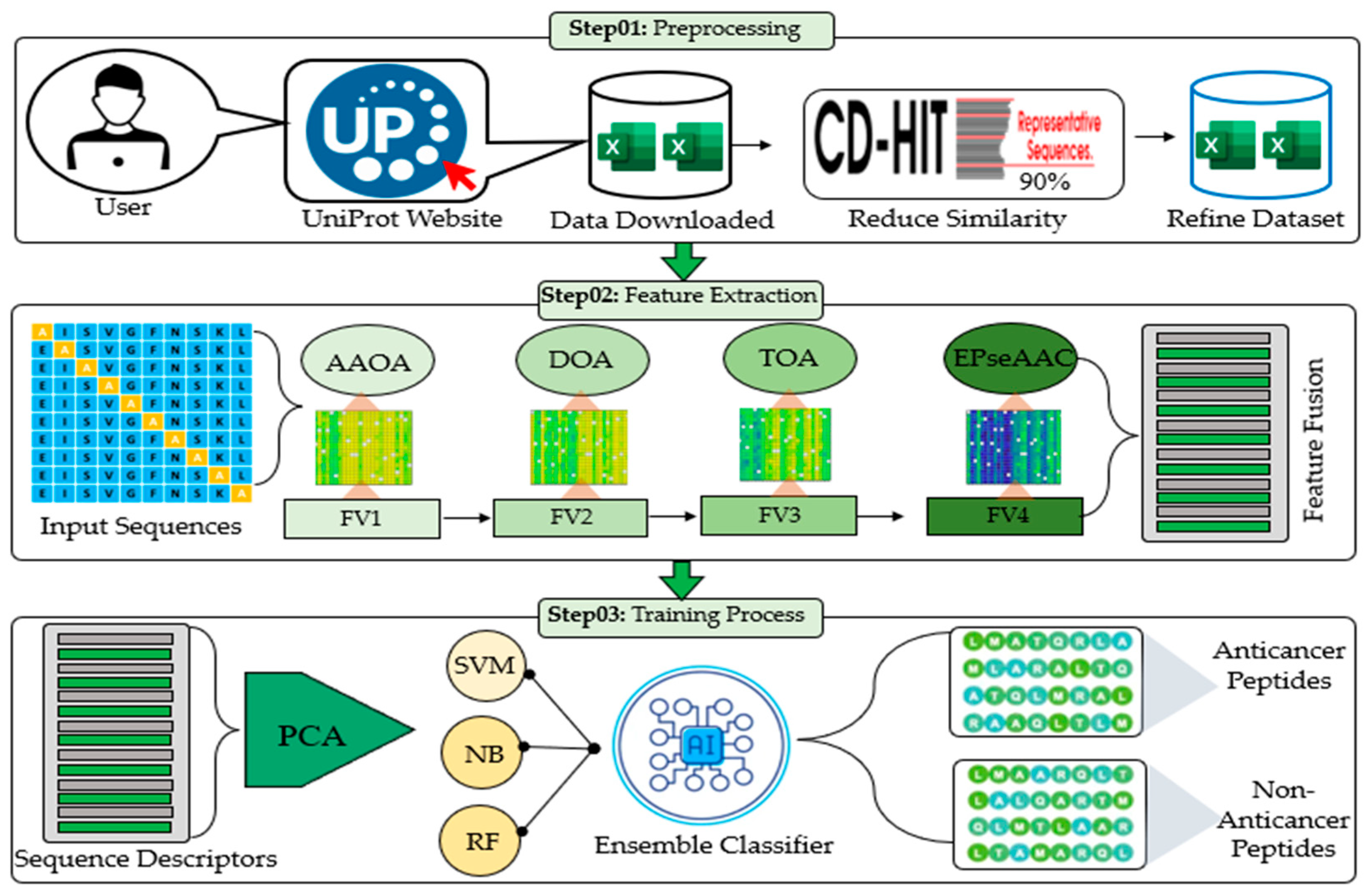
3. The Proposed Framework
| Algorithm 1: Pseudocode for the proposed framework for peptide classification |
| Input: Peptide Sequences (Šet_Train), (Šet_Test)(ƤŠeq)(Å)U(Ɐ) //Å and Ɐ represent anticancer and non-anticancer peptides Output: Peptide Sequences (Šet_Test) Y// represents the class label of the test dataset
Đ (ƤŠeq)(Å)U(Ɐ)//Đ represents the refined dataset for i = 1 to L-1Đ//L represents samples in the dataset Compare pattern (C) (ƤŠeq) Extract features(ƒ) (ƤŠeq) Save features (SF) Repeat (Л) Ϻ1, Ϻ2, Ϻ3, Ϻ4 //Л represents repeating step 2 for all methods end
Refine features (Яf1)
Equal sample (Æ) (Яf2)
(Яf2) + class label (CL) ensemble classifier (EC)
for j = 1 to L-1Đ//L represents samples in the test dataset Repeat step 2 feature extraction Output: Binary classification with class label end |
3.1. Dataset and Preprocessing
3.1.1. Dataset
3.1.2. Preprocessing Using the SMOTE
3.2. Computational Methods for Discriminative Features
3.2.1. Amino Acid Occurrence Analysis (AAOA)
3.2.2. Dipeptide Occurrence Analysis (DOA)
3.2.3. Tripeptide Occurrence Analysis (TOA)
3.2.4. Enhanced Physicochemical Property-Based Features
3.3. Ensemble Learning for Model Training
4. Experimental Analysis
4.1. Implementation Setup and System Specifications
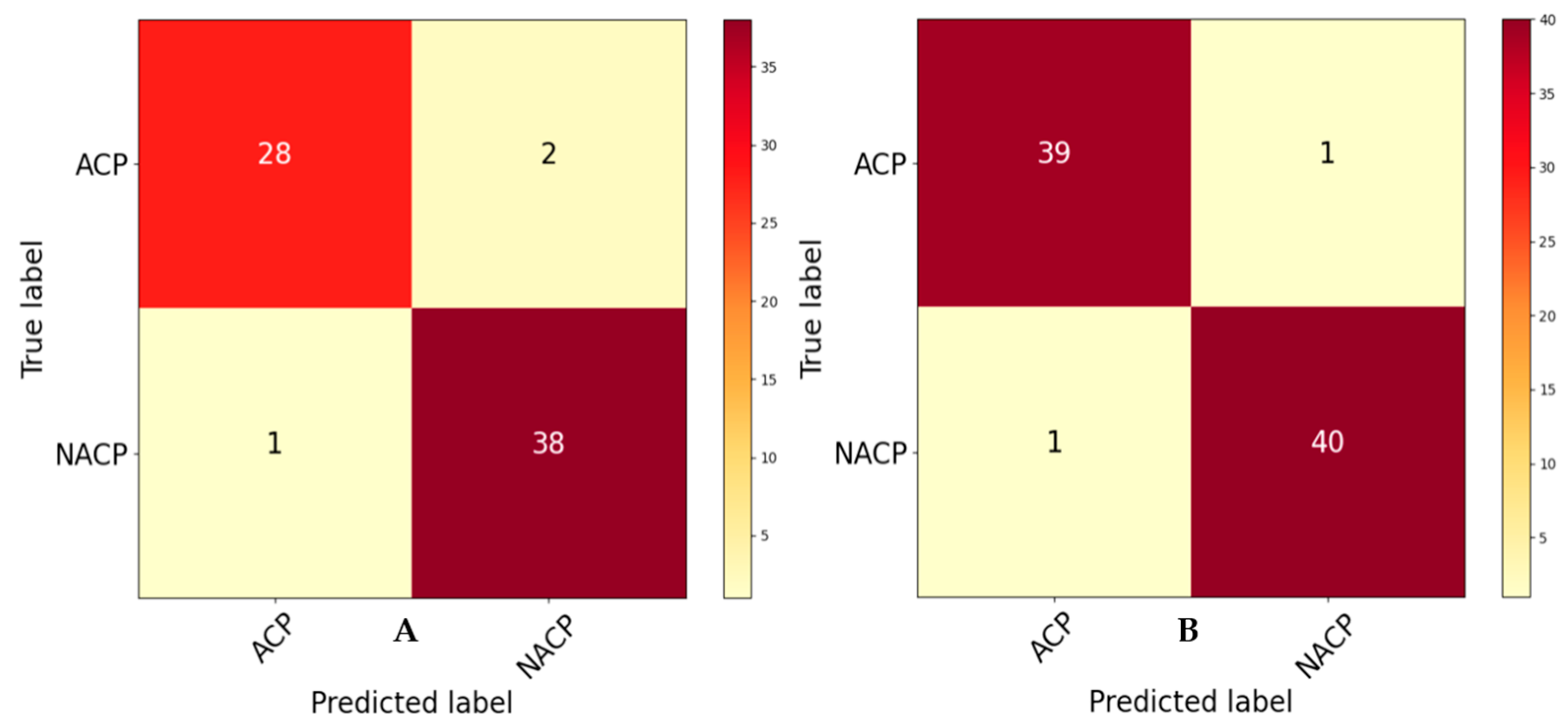
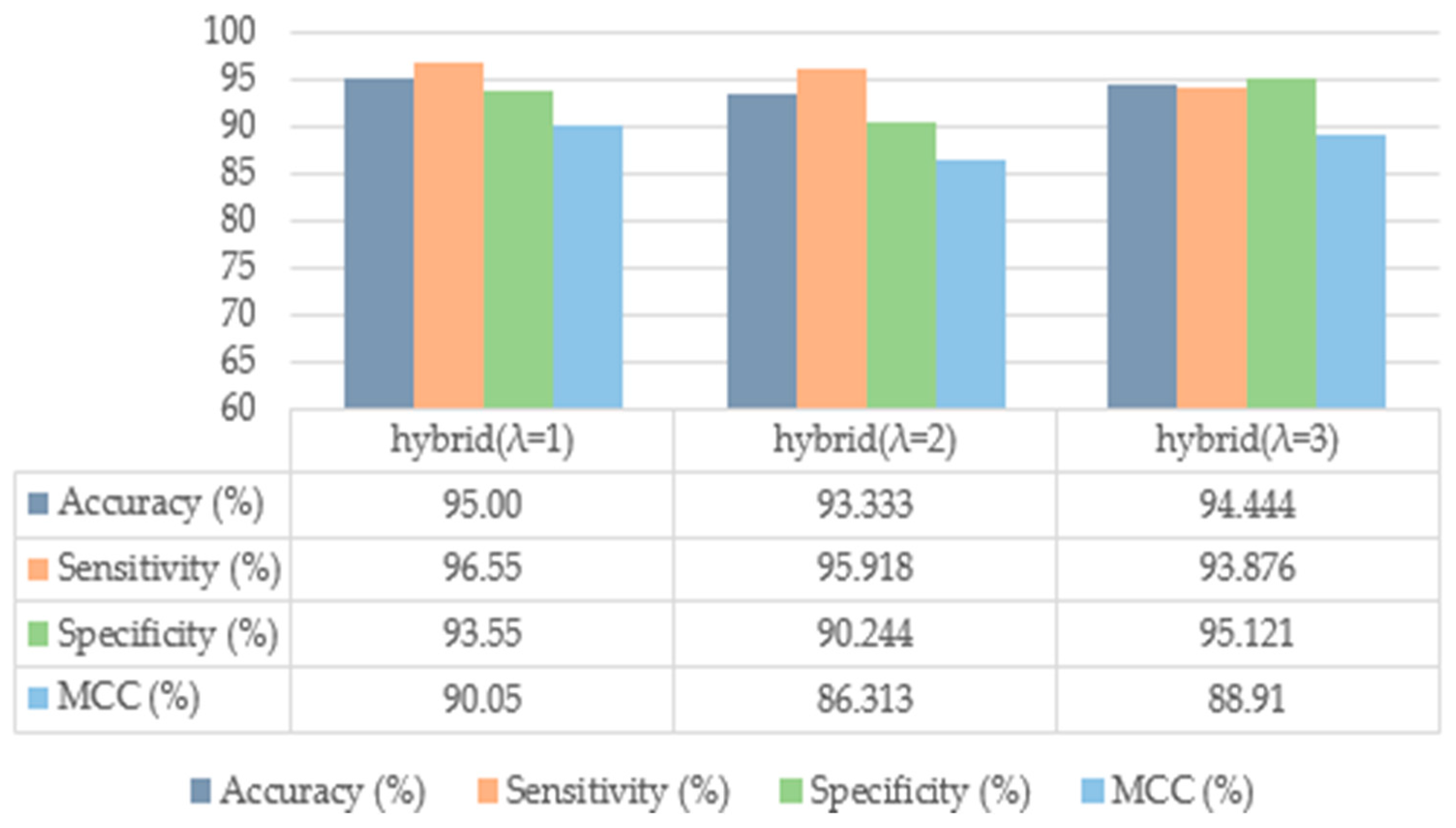
4.2. Ablation Study
4.2.1. Analysis of Benchmark Dataset
4.2.2. Analysis with Independent Datasets
4.3. Comparative Analysis
4.3.1. Performance Comparison with SOTA over Benchmark Dataset
4.3.2. Performance Comparison with SOTA using Independent Datasets
5. Conclusions and Future Research Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAOA | Amino acid occurrence analysis |
| ACC | Accuracy |
| ACP | Anticancer peptide |
| ACP-2DCNN | Anticancer peptide two-dimensional CNN |
| ACP-MHCNN | Anticancer peptide multi-headed CNN |
| AMDs | Advanced micro devices |
| cACP-2LFS | Classification of anticancer peptides with two-level feature selection |
| CCPM | Cervical cancer prediction model |
| CD-HIT | Cluster database at high identity with tolerance |
| cACP | Classifying anticancer peptides |
| CPUs | Central processing units |
| CNN | Convolutional neural network |
| DOA | Dipeptide occurrence analysis |
| DNA | Deoxyribonucleic acid |
| EPseAAC | Enhanced pseudo amino acid composition |
| MLACP | Machine learning anticancer peptide prediction |
| NB | Naive Bayes |
| NACP | Non-anticancer peptide |
| MCC | Mathews correlation coefficient |
| RF | Random forest |
| RTX | Ray tracing texel |
References
- Arora, T.; Grey, I. Health behaviour changes during COVID-19 and the potential consequences: A mini-review. J. Health Psychol. 2020, 25, 1155–1163. [Google Scholar] [CrossRef]
- Maki, O.; Alshaikhli, M.; Gunduz, M.; Naji, K.K.; Abdulwahed, M. Development of digitalization road map for healthcare facility management. IEEE Access 2022, 10, 14450–14462. [Google Scholar] [CrossRef]
- Kapoor, A.; Guha, S.; Das, M.K.; Goswami, K.C.; Yadav, R. Digital Healthcare: The Only Solution for Better Healthcare during COVID-19 Pandemic? Elsevier: Amsterdam, The Netherlands, 2020; Volume 72, pp. 61–64. [Google Scholar]
- Alshamrani, M. IoT and artificial intelligence implementations for remote healthcare monitoring systems: A survey. J. King Saud Univ. Comput. Inf. Sci. 2022, 34, 4687–4701. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
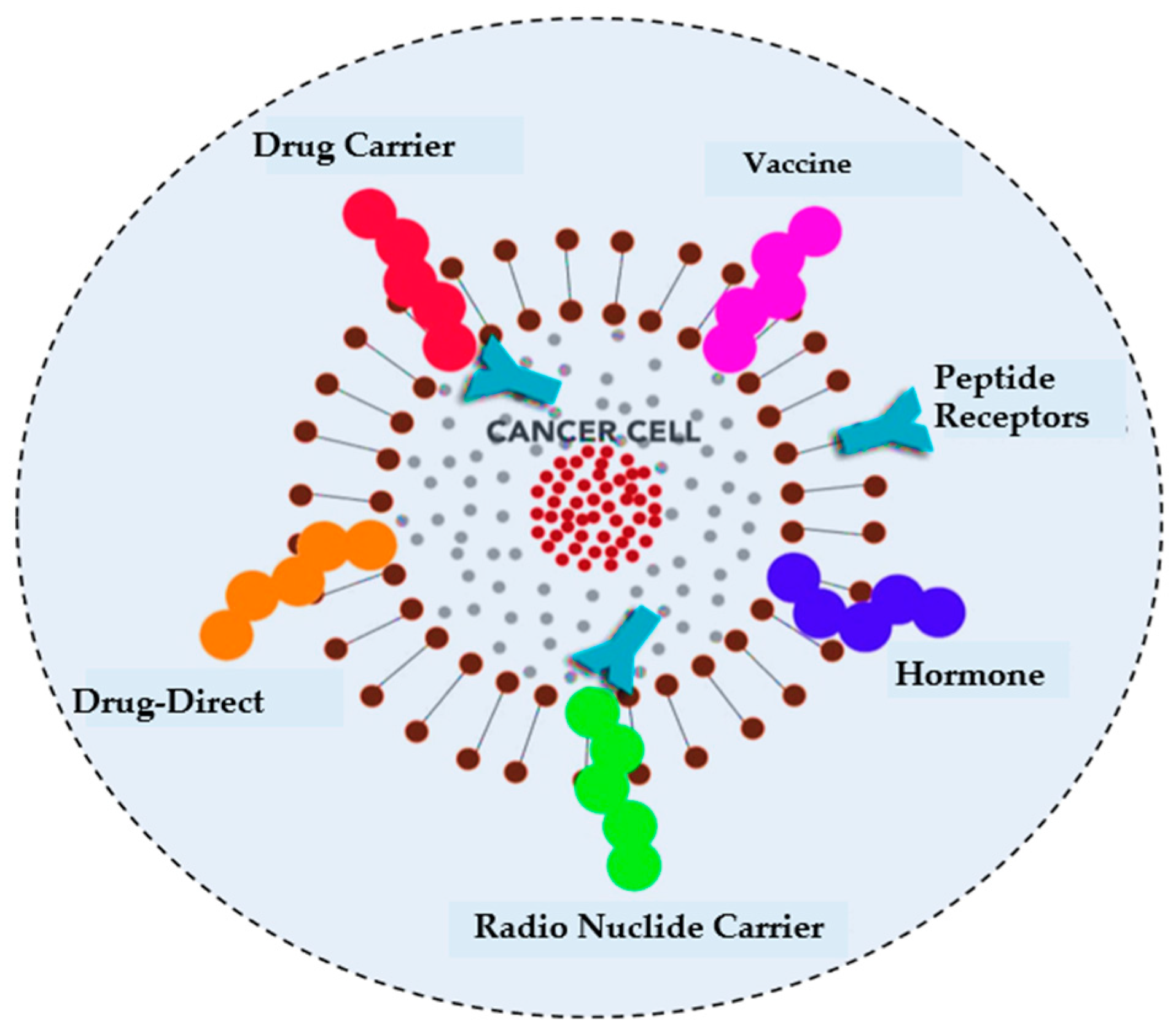
- Harris, F.; Dennison, S.R.; Singh, J.; Phoenix, D.A. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med. Res. Rev. 2013, 33, 190–234. [Google Scholar] [CrossRef] [PubMed]
- Karbalaeemohammad, S.; Naderi-Manesh, H. Two novel anticancer peptides from Aurein1. 2. Int. J. Pept. Res. Ther. 2011, 17, 159–164. [Google Scholar] [CrossRef]
- Ijaz, M.F.; Attique, M.; Son, Y. Data-driven cervical cancer prediction model with outlier detection and over-sampling methods. Sensors 2020, 20, 2809. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Islam, S.R.; Abdullah-Al-Wadud, M.; Islam, S.; Ali, F.; Park, K.S. Multiomics analysis reveals that GLS and GLS2 differentially modulate the clinical outcomes of cancer. J. Clin. Med. 2019, 8, 355. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2008, 1778, 357–375. [Google Scholar] [CrossRef]
- Mader, J.S.; Hoskin, D.W. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin. Investig. Drugs 2006, 15, 933–946. [Google Scholar] [CrossRef]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Feng, Q.; Yan, Q.; Hao, X.; Chen, Y. Alpha-helical cationic anticancer peptides: A promising candidate for novel anticancer drugs. Mini Rev. Med. Chem. 2015, 15, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Thundimadathil, J. Cancer treatment using peptides: Current therapies and future prospects. J. Amino Acids 2012, 2012, 967347. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A.; Raghava, G. In silico models for designing and discovering novel anticancer peptides. Sci. Rep. 2013, 3, 2984. [Google Scholar] [CrossRef] [PubMed]
- Hajisharifi, Z.; Piryaiee, M.; Beigi, M.M.; Behbahani, M.; Mohabatkar, H. Predicting anticancer peptides with Chou′ s pseudo amino acid composition and investigating their mutagenicity via Ames test. J. Theor. Biol. 2014, 341, 34–40. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Feng, P.; Lin, H.; Chou, K.-C. iACP: A sequence-based tool for identifying anticancer peptides. Oncotarget 2016, 7, 16895. [Google Scholar] [CrossRef]
- Akbar, S.; Hayat, M.; Iqbal, M.; Jan, M.A. iACP-GAEnsC: Evolutionary genetic algorithm based ensemble classification of anticancer peptides by utilizing hybrid feature space. Artif. Intell. Med. 2017, 79, 62–70. [Google Scholar] [CrossRef]
- Xu, L.; Liang, G.; Wang, L.; Liao, C. A novel hybrid sequence-based model for identifying anticancer peptides. Genes 2018, 9, 158. [Google Scholar] [CrossRef]
- Bansal, G.; Rajgopal, K.; Chamola, V.; Xiong, Z.; Niyato, D. Healthcare in metaverse: A survey on current metaverse applications in healthcare. IEEE Access 2022, 10, 119914–119946. [Google Scholar] [CrossRef]
- Tan, T.F.; Li, Y.; Lim, J.S.; Gunasekeran, D.V.; Teo, Z.L.; Ng, W.Y.; Ting, D.S. Metaverse and virtual health care in ophthalmology: Opportunities and challenges. Asia-Pac. J. Ophthalmol. 2022, 11, 237–246. [Google Scholar] [CrossRef]
- Ali, S.; Abdullah; Armand, T.P.T.; Athar, A.; Hussain, A.; Ali, M.; Yaseen, M.; Joo, M.-I.; Kim, H.-C. Metaverse in healthcare integrated with explainable ai and blockchain: Enabling immersiveness, ensuring trust, and providing patient data security. Sensors 2023, 23, 565. [Google Scholar] [CrossRef] [PubMed]
- Razdan, S.; Sharma, S. Internet of medical things (IoMT): Overview, emerging technologies, and case studies. IETE Tech. Rev. 2022, 39, 775–788. [Google Scholar] [CrossRef]
- Ge, L.; Liu, J.; Zhang, Y.; Dehmer, M. Identifying anticancer peptides by using a generalized chaos game representation. J. Math. Biol. 2019, 78, 441–463. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.-C.; You, Z.-H.; Zhou, X.; Cheng, L.; Li, X.; Jiang, T.-H.; Chen, Z.-H. ACP-DL: A deep learning long short-term memory model to predict anticancer peptides using high-efficiency feature representation. Mol. Ther. Nucleic Acids 2019, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-g.; Zhang, W.; Yang, X.; Li, C.; Chen, H. Acp-da: Improving the prediction of anticancer peptides using data augmentation. Front. Genet. 2021, 12, 698477. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Feng, G.; Jing, X.; Zhang, R.; Wang, P.; Wu, Q. Enacp: An ensemble learning model for identification of anticancer peptides. Front. Genet. 2020, 11, 760. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.-C. mACPpred: A support vector machine-based meta-predictor for identification of anticancer peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhagat, D.; Mahalwal, M.; Sharma, N.; Raghava, G.P. AntiCP 2.0: An updated model for predicting anticancer peptides. Brief. Bioinform. 2021, 22, bbaa153. [Google Scholar] [CrossRef]
- Akbar, S.; Hayat, M.; Tahir, M.; Khan, S.; Alarfaj, F.K. cACP-DeepGram: Classification of anticancer peptides via deep neural network and skip-gram-based word embedding model. Artif. Intell. Med. 2022, 131, 102349. [Google Scholar] [CrossRef]
- Akbar, S.; Rahman, A.U.; Hayat, M.; Sohail, M. cACP: Classifying anticancer peptides using discriminative intelligent model via Chou’s 5-step rules and general pseudo components. Chemom. Intell. Lab. Syst. 2020, 196, 103912. [Google Scholar] [CrossRef]
- Akbar, S.; Hayat, M.; Tahir, M.; Chong, K.T. cACP-2LFS: Classification of anticancer peptides using sequential discriminative model of KSAAP and two-level feature selection approach. IEEE Access 2020, 8, 131939–131948. [Google Scholar] [CrossRef]
- Ahmed, S.; Muhammod, R.; Khan, Z.H.; Adilina, S.; Sharma, A.; Shatabda, S.; Dehzangi, A. ACP-MHCNN: An accurate multi-headed deep-convolutional neural network to predict anticancer peptides. Sci. Rep. 2021, 11, 23676. [Google Scholar] [CrossRef]
- Ghulam, A.; Ali, F.; Sikander, R.; Ahmad, A.; Ahmed, A.; Patil, S. ACP-2DCNN: Deep learning-based model for improving prediction of anticancer peptides using two-dimensional convolutional neural network. Chemom. Intell. Lab. Syst. 2022, 226, 104589. [Google Scholar] [CrossRef]
- Park, H.W.; Pitti, T.; Madhavan, T.; Jeon, Y.-J.; Manavalan, B. MLACP 2.0: An updated machine learning tool for anticancer peptide prediction. Comput. Struct. Biotechnol. J. 2022, 20, 4473–4480. [Google Scholar]
- Chen, J.; Cheong, H.H.; Siu, S.W. xDeep-AcPEP: Deep learning method for anticancer peptide activity prediction based on convolutional neural network and multitask learning. J. Chem. Inf. Model. 2021, 61, 3789–3803. [Google Scholar] [CrossRef]
- Sun, M.; Yang, S.; Hu, X.; Zhou, Y. ACPNet: A deep learning network to identify anticancer peptides by hybrid sequence information. Molecules 2022, 27, 1544. [Google Scholar] [CrossRef] [PubMed]
- Onan, A.; Korukoğlu, S.; Bulut, H. A multiobjective weighted voting ensemble classifier based on differential evolution algorithm for text sentiment classification. Expert Syst. Appl. 2016, 62, 1–16. [Google Scholar] [CrossRef]
- Dimitriadou, E.; Weingessel, A.; Hornik, K. A cluster ensembles framework. In Design and Application of Hybrid Intelligent Systems; IOS Press: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Khan, S.U.; Haq, I.U.; Khan, Z.A.; Khan, N.; Lee, M.Y.; Baik, S.W. Atrous Convolutions and Residual GRU Based Architecture for Matching Power Demand with Supply. Sensors 2021, 21, 7191. [Google Scholar] [CrossRef]
- Khan, S.U.; Khan, N.; Ullah, F.U.M.; Kim, M.J.; Lee, M.Y.; Baik, S.W. Towards intelligent building energy management: AI-based framework for power consumption and generation forecasting. Energy Build. 2023, 279, 112705. [Google Scholar] [CrossRef]
- Hussain, A.; Khan, S.U.; Rida, I.; Khan, N.; Baik, S.W. Human Centric Attention with Deep Multiscale Feature Fusion Framework for Activity Recognition in Internet of Medical Things. Inf. Fusion 2023, 102211. [Google Scholar] [CrossRef]
- Hussain, A.; Khan, S.U.; Khan, N.; Shabaz, M.; Baik, S.W. AI-driven behavior biometrics framework for robust human activity recognition in surveillance systems. Eng. Appl. Artif. Intell. 2024, 127, 107218. [Google Scholar] [CrossRef]
- Hussain, A.; Amin, S.U.; Lee, H.; Khan, A.; Khan, N.F.; Seo, S. An Automated Chest X-Ray Image Analysis for Covid-19 and Pneumonia Diagnosis using Deep Ensemble Strategy. IEEE Access 2023, 11, 97207–97220. [Google Scholar] [CrossRef]
- Ekbal, A.; Saha, S. Weighted vote based classifier ensemble selection using genetic algorithm for named entity recognition. In Proceedings of the International Conference on Application of Natural Language to Information Systems, Cardiff, UK, 23–25 June 2010; pp. 256–267. [Google Scholar]
- Opitz, D.; Maclin, R. Popular ensemble methods: An empirical study. J. Artif. Intell. Res. 1999, 11, 169–198. [Google Scholar] [CrossRef]
- Hayat, M.; Khan, A.; Yeasin, M. Prediction of membrane proteins using split amino acid and ensemble classification. Amino Acids 2012, 42, 2447–2460. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Zhou, X.; Lin, H.; Chou, K.-C. iRNA (m6A)-PseDNC: Identifying N6-methyladenosine sites using pseudo dinucleotide composition. Anal. Biochem. 2018, 561, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Khan, S.U.; Khan, N.; Hussain, T.; Baik, S.W. An intelligent correlation learning system for person Re-identification. Eng. Appl. Artif. Intell. 2024, 128, 107213. [Google Scholar] [CrossRef]
- Dilshad, N.; Khan, S.U.; Alghamdi, N.S.; Taleb, T.; Song, J. Towards Efficient Fire Detection in IoT Environment: A Modified Attention Network and Large-Scale Dataset. IEEE Internet Things J. 2023. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Nguyen-Xuan, H.; Lee, J. A novel data-driven nonlinear solver for solid mechanics using time series forecasting. Finite Elem. Anal. Des. 2020, 171, 103377. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Lee, S.; Nguyen, P.-C.; Nguyen-Xuan, H.; Lee, J. Geometrically nonlinear postbuckling behavior of imperfect FG-CNTRC shells under axial compression using isogeometric analysis. Eur. J. Mech. A/Solids 2020, 84, 104066. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Khan, S.U.; Khan, N.; Hussain, T.; Muhammad, K.; Hijji, M.; Del Ser, J.; Baik, S.W. Visual Appearance and Soft Biometrics Fusion for Person Re-identification using Deep Learning. IEEE J. Sel. Top. Signal Process. 2023, 17, 575–586. [Google Scholar] [CrossRef]
- Chou, K.-C. Impacts of bioinformatics to medicinal chemistry. Med. Chem. 2015, 11, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Baik, R. MPPIF-Net: Identification of Plasmodium Falciparum Parasite Mitochondrial Proteins Using Deep Features with Multilayer Bi-directional LSTM. Processes 2020, 8, 725. [Google Scholar] [CrossRef]
- Cheng, X.; Xiao, X.; Chou, K.-C. pLoc-mGneg: Predict subcellular localization of Gram-negative bacterial proteins by deep gene ontology learning via general PseAAC. Genomics 2018, 110, 231–239. [Google Scholar] [CrossRef]
- Waris, M.; Ahmad, K.; Kabir, M.; Hayat, M. Identification of DNA binding proteins using evolutionary profiles position specific scoring matrix. Neurocomputing 2016, 199, 154–162. [Google Scholar] [CrossRef]
- Huang, T.; Chen, L.; Cai, Y.-D.; Chou, K.-C. Classification and analysis of regulatory pathways using graph property, biochemical and physicochemical property, and functional property. PLoS ONE 2011, 6, e25297. [Google Scholar] [CrossRef]
- Behbahani, M.; Mohabatkar, H.; Nosrati, M. Analysis and comparison of lignin peroxidases between fungi and bacteria using three different modes of Chou’s general pseudo amino acid composition. J. Theor. Biol. 2016, 411, 1–5. [Google Scholar] [CrossRef]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef]
- Chou, K.-C. An unprecedented revolution in medicinal chemistry driven by the progress of biological science. Curr. Top. Med. Chem. 2017, 17, 2337–2358. [Google Scholar] [CrossRef]
- Hajisharifi, Z.; Mohabatkar, H. In silico prediction of anticancer peptides by TRAINER tool. Mol. Biol. Res. Commun. 2013, 2, 39–45. [Google Scholar]
- Li, F.-M.; Wang, X.-Q. Identifying anticancer peptides by using improved hybrid compositions. Sci. Rep. 2016, 6, 33910. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, W.; Wang, D.; Wang, S.; Li, Q. Prediction of anticancer peptides using a low-dimensional feature model. Front. Bioeng. Biotechnol. 2020, 8, 892. [Google Scholar] [CrossRef] [PubMed]
- Fazal, E.; Ibrahim, M.S.; Park, S.; Naseem, I.; Wahab, A. Anticancer Peptides Classification Using Kernel Sparse Representation Classifier. IEEE Access 2023, 11, 17626–17637. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Total Samples | Training Samples | Test Samples |
|---|---|---|---|
| Benchmark [17] | 344 | 275 | 69 |
| Independent [17] | 300 | 240 | 60 |
| Method | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|
| Benchmark Dataset | ||||
| AAOA | 88.27 | 86.84 | 89.85 | 76.02 |
| DOA | 92.41 | 91.66 | 93.15 | 84.62 |
| TOA | 95.17 | 93.05 | 97.26 | 89.96 |
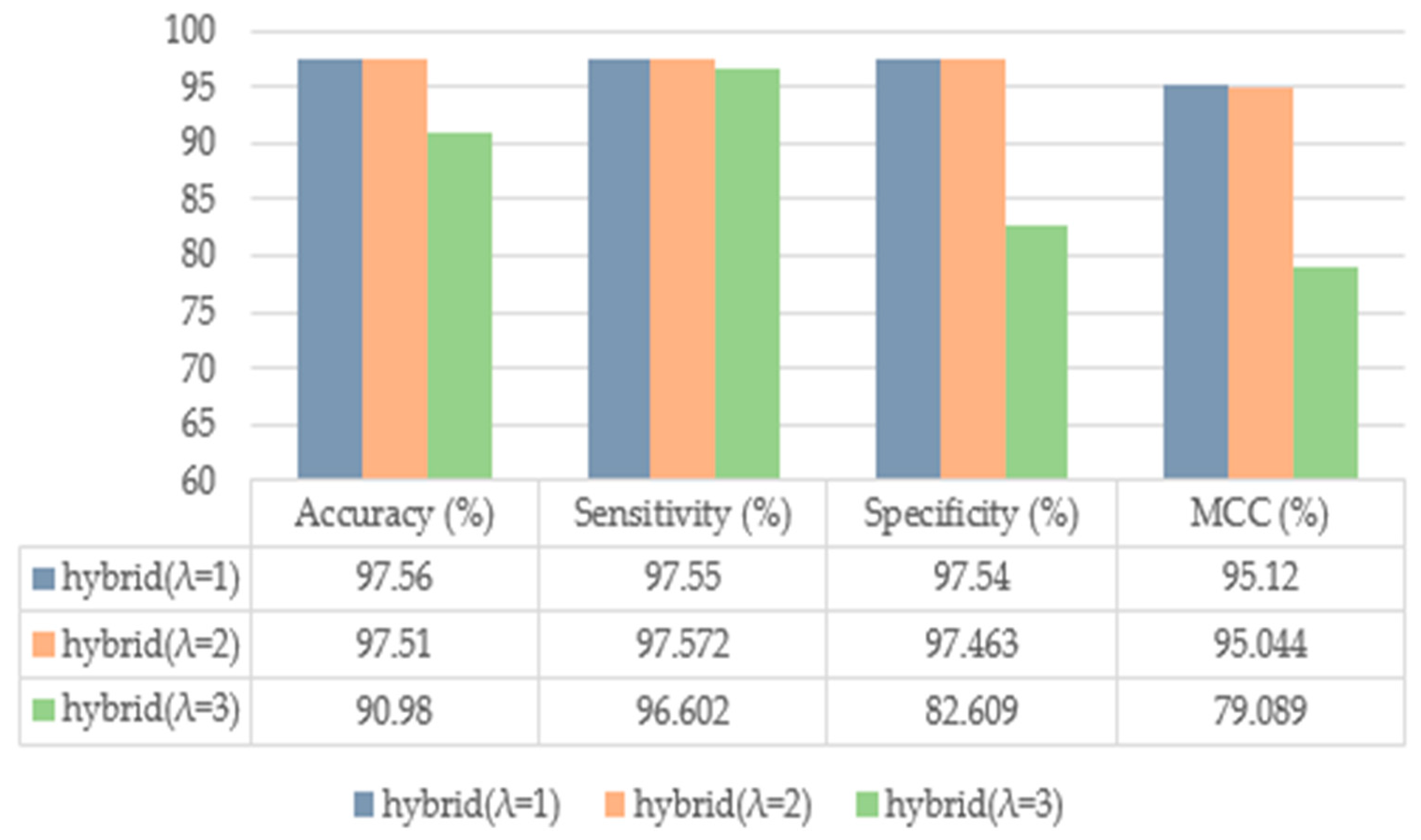
| EPseAAC (λ = 1) | 86.89 | 83.33 | 90.41 | 71.83 |
| EPseAAC (λ = 2) | 88.96 | 86.11 | 91.78 | 76.69 |
| EPseAAC (λ = 3) | 87.58 | 84.72 | 90.41 | 73.73 |
| AAOA + DOA | 93.79 | 91.66 | 95.89 | 87.11 |
| AAOA + EPseAAC (λ = 1) | 88.27 | 84.72 | 91.78 | 74.84 |
| AAOA + EPseAAC (λ = 2) | 83.57 | 76.92 s | 86.13 | 62.04 |
| AAOA + EPseAAC (λ = 3) | 88.27 | 84.72 | 91.78 | 74.84 |
| AAOA + TOA | 90.90 | 96.20 | 83.01 | 79.17 |
| DOA + EPseAAC (λ = 1) | 91.72 | 84.72 | 98.63 | 80.33 |
| DOA + EPseAAC (λ = 2) | 94.48 | 90.27 | 98.63 | 87.89 |
| DOA + EPseAAC (λ = 3) | 93.83 | 90.27 | 97.29 | 86.70 |
| DOA + TOA | 91.03 | 94.44 | 87.67 | 82.45 |
| TOA + EPseAAC (λ = 1) | 96.55 | 95.83 | 97.26 | 93.02 |
| TOA + EPseAAC (λ = 2) | 95.86 | 95.83 | 95.89 | 91.71 |
| TOA + EPseAAC (λ = 3) | 97.24 | 95.83 | 98.63 | 94.33 |
| AAOA + DOA + TOA | 94.48 | 98.61 | 90.41 | 89.10 |
| AAOA + DOA + EPseAAC (λ = 1) | 93.10 | 88.88 | 97.26 | 84.93 |
| AAOA + DOA + EPseAAC (λ = 2) | 92.41 | 90.27 | 94.52 | 84.24 |
| AAOA + DOA + EPseAAC (λ = 3) | 93.79 | 93.05 | 94.52 | 87.43 |
| AAOA+ TOA + EPseAAC (λ = 1) | 95.86 | 93.05 | 98.63 | 91.22 |
| AAOA + TOA + EPseAAC (λ = 2) | 94.48 | 93.05 | 95.89 | 88.07 |
| AAOA + TOA + EPseAAC (λ = 3) | 96.55 | 95.83 | 97.26 | 93.02 |
| DOA + TOA + EPseAAC (λ = 1) | 95.17 | 91.66 | 98.63 | 89.59 |
| DOA + TOA + EPseAAC (λ = 2) | 95.86 | 94.44 | 97.26 | 91.52 |
| DOA + TOA + EPseAAC (λ = 3) | 93.79 | 91.66 | 95.89 | 87.11 |
| Proposed (without SMOTE) | 95.65 | 95.55 | 95.00 | 91.16 |
| Proposed (with SMOTE) | 97.56 | 97.55 | 97.54 | 95.12 |
| Method | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|
| Independent Dataset | ||||
| AAOA | 88.88 | 93.87 | 82.92 | 77.14 |
| DOA | 91.11 | 95.91 | 85.36 | 81.46 |
| TOA | 84.44 | 77.55 | 92.68 | 64.55 |
| EPseAAC (λ = 1) | 89.13 | 92.15 | 85.36 | 77.84 |
| EPseAAC (λ = 2) | 86.66 | 89.79 | 82.92 | 73.13 |
| EPseAAC (λ = 3) | 86.06 | 89.79 | 82.92 | 73.13 |
| AAOA + DOA | 92.22 | 95.91 | 87.80 | 83.90 |
| AAOA + EPseAAC (λ = 1) | 90.00 | 93.87 | 85.36 | 79.55 |
| AAOA + EPseAAC (λ = 2) | 90.00 | 93.87 | 85.36 | 79.55 |
| AAOA + EPseAAC (λ = 3) | 88.88 | 91.83 | 85.36 | 77.53 |
| AAOA + TOA | 90.00 | 87.75 | 92.68 | 79.63 |
| DOA + EPseAAC (λ = 1) | 92.22 | 95.91 | 87.80 | 83.90 |
| DOA + EPseAAC (λ = 2) | 88.88 | 93.87 | 82.92 | 77.14 |
| DOA + EPseAAC (λ = 3) | 88.88 | 91.83 | 85.36 | 77.53 |
| DOA + TOA | 91.11 | 93.87 | 87.80 | 81.92 |
| TOA + EPseAAC (λ = 1) | 90.00 | 87.75 | 92.68 | 79.63 |
| TOA + EPseAAC (λ = 2) | 87.77 | 89.79 | 85.36 | 75.41 |
| TOA + EPseAAC (λ = 3) | 87.77 | 87.75 | 87.80 | 75.34 |
| AAOA + DOA + TOA | 92.22 | 93.87 | 90.24 | 84.27 |
| AAOA + DOA + EPseAAC (λ = 1) | 91.11 | 95.91 | 85.36 | 81.46 |
| AAOA + DOA + EPseAAC (λ = 2) | 88.88 | 91.83 | 85.36 | 77.53 |
| AAOA + DOA + EPseAAC (λ = 3) | 90.00 | 93.87 | 85.36 | 79.55 |
| AAOA+ TOA + EPseAAC (λ = 1) | 93.33 | 93.87 | 92.68 | 86.60 |
| AAOA + TOA + EPseAAC (λ = 2) | 88.88 | 91.83 | 85.36 | 77.53 |
| AAOA + TOA + EPseAAC (λ = 3) | 87.77 | 89.79 | 85.36 | 75.41 |
| DOA + TOA + EPseAAC (λ = 1) | 91.11 | 95.91 | 85.36 | 81.46 |
| DOA + TOA + EPseAAC (λ = 2) | 90.00 | 93.87 | 85.36 | 79.55 |
| DOA + TOA + EPseAAC (λ = 3) | 90.00 | 93.87 | 85.36 | 79.55 |
| Proposed (without SMOTE) | 93.75 | 92.00 | 94.87 | 86.87 |
| Proposed (with SMOTE) | 95.00 | 96.55 | 93.55 | 90.5 |
| Model/Year | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|
| SPAP [64] 2013 | 87.00 | 92.00 | 86.00 | 74.0 |
| LAK [16] 2014 | 92.68 | 89.70 | 85.18 | 78.0 |
| iACP [17] 2016 | 95.06 | 89.86 | 98.54 | 89.0 |
| IAP [65] 2016 | 93.61 | 89.86 | 96.12 | 86.0 |
| iACP-GAEnsC [18] 2017 | 96.45 | 95.36 | 97.57 | 91.0 |
| SAP [19] 2018 | 91.86 | 86.23 | 95.63 | 83.0 |
| LDFM [66] 2020 | 92.73 | 87.70 | 96.10 | 84.0 |
| ACP-KSRC [67] 2023 | 93.02 | 97.07 | 86.87 | 85.0 |
| Proposed (λ = 1) | 97.56 | 97.55 | 97.54 | 95.12 |
| Model/Year | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|
| NT5CT5 [15] 2013 | 92.65 | 74.67 | 94.44 | 61.0 |
| GCGR [24] 2018 | 96.36 | 69.33 | 99.07 | 76.0 |
| cACP [31] 2019 | 96.91 | 77.32 | 98.12 | 79.0 |
| ACP-MHCNN [33] 2021 | 91.0 | 97.6 | 84.2 | 82.0 |
| cACP-DeepGram [30] 2022 | 94.02 | 91.18 | 95.47 | 88.0 |
| Proposed (λ = 1) | 95.00 | 96.55 | 93.55 | 90.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danish, S.; Khan, A.; Dang, L.M.; Alonazi, M.; Alanazi, S.; Song, H.-K.; Moon, H. Metaverse Applications in Bioinformatics: A Machine Learning Framework for the Discrimination of Anti-Cancer Peptides. Information 2024, 15, 48. https://doi.org/10.3390/info15010048
Danish S, Khan A, Dang LM, Alonazi M, Alanazi S, Song H-K, Moon H. Metaverse Applications in Bioinformatics: A Machine Learning Framework for the Discrimination of Anti-Cancer Peptides. Information. 2024; 15(1):48. https://doi.org/10.3390/info15010048
Chicago/Turabian StyleDanish, Sufyan, Asfandyar Khan, L. Minh Dang, Mohammed Alonazi, Sultan Alanazi, Hyoung-Kyu Song, and Hyeonjoon Moon. 2024. "Metaverse Applications in Bioinformatics: A Machine Learning Framework for the Discrimination of Anti-Cancer Peptides" Information 15, no. 1: 48. https://doi.org/10.3390/info15010048
APA StyleDanish, S., Khan, A., Dang, L. M., Alonazi, M., Alanazi, S., Song, H.-K., & Moon, H. (2024). Metaverse Applications in Bioinformatics: A Machine Learning Framework for the Discrimination of Anti-Cancer Peptides. Information, 15(1), 48. https://doi.org/10.3390/info15010048