


Explainabilty Comparison between Random Forests and Neural Networks—Case Study of Amino Acid Volume Prediction



Abstract
1. Introduction
2. Background
2.1. Protein Data Bank
HEADER OXYGEN TRANSPORT 09-MAY-09 3HEN TITLE FERRIC HORSE HEART MYOGLOBIN; H64V/V67R MUTANT COMPND MOL_ID: 1; COMPND 2 MOLECULE: MYOGLOBIN; SOURCE 2 ORGANISM_SCIENTIFIC: EQUUS CABALLUS; SOURCE 3 ORGANISM_COMMON: DOMESTIC HORSE, EQUINE; SEQRES 1 A 153 GLY LEU SER ASP GLY GLU TRP GLN GLN VAL LEU ASN VAL SEQRES 2 A 153 TRP GLY LYS VAL GLU ALA ASP ILE ALA GLY HIS GLY GLN SEQRES 3 A 153 GLU VAL LEU ILE ARG LEU PHE THR GLY HIS PRO GLU THR SEQRES 4 A 153 LEU GLU LYS PHE ASP LYS PHE LYS HIS LEU LYS THR GLU SEQRES 5 A 153 ALA GLU MET LYS ALA SER GLU ASP LEU LYS LYS VAL GLY SEQRES 6 A 153 THR ARG VAL LEU THR ALA LEU GLY GLY ILE LEU LYS LYS SEQRES 7 A 153 LYS GLY HIS HIS GLU ALA GLU LEU LYS PRO LEU ALA GLN SEQRES 8 A 153 SER HIS ALA THR LYS HIS LYS ILE PRO ILE LYS TYR LEU SEQRES 9 A 153 GLU PHE ILE SER ASP ALA ILE ILE HIS VAL LEU HIS SER SEQRES 10 A 153 LYS HIS PRO GLY ASP PHE GLY ALA ASP ALA GLN GLY ALA SEQRES 11 A 153 MET THR LYS ALA LEU GLU LEU PHE ARG ASN ASP ILE ALA SEQRES 12 A 153 ALA LYS TYR LYS GLU LEU GLY PHE GLN GLY ATOM 1 N GLY A 1 -1.476 41.015 -11.482 1.00 40.53 N ATOM 2 CA GLY A 1 -2.113 40.213 -12.574 1.00 40.50 C ATOM 3 C GLY A 1 -1.163 40.052 -13.757 1.00 38.97 C ATOM 4 O GLY A 1 -0.026 40.555 -13.734 1.00 40.91~O
2.2. Random Forest and MLP
2.3. The Eli-5 Tool
3. Description of the Methodology
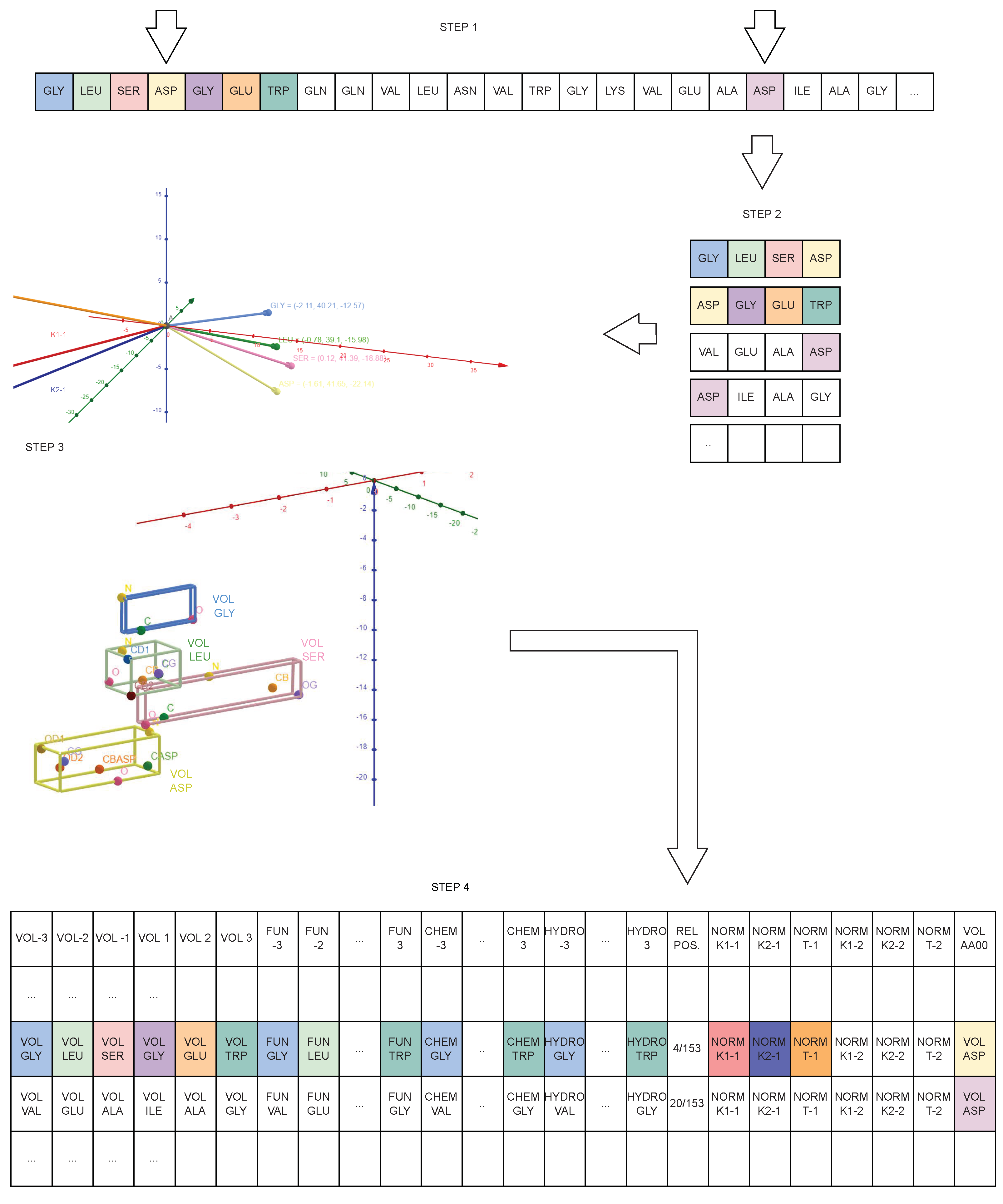
4. From PDB to Geometrical Data
4.1. Data Preparation Process
4.2. Protein Geometrical Modelling
- Three distances , ;
- The angles , determined by the vectors joining the centres;
- The angle corresponding to the two curvature vectors.
4.3. DBR2 Scheme Definition
4.4. DBR2 Instance Population
5. Building the ML Models
5.1. Experiment Description
- Volume of the amino acid located in position −3, −2, −1, 1, 2, 3 from AA0;
- Functional class of the amino acid located in position −3, −2, −1, 1, 2, 3 from AA0;
- Chemical class of the amino acid located in position −3, −2, −1, 1, 2, 3 from AA0;
- Hydrophilic class of the amino acid located in position −3, −2, −1, 1, 2, 3 from AA0;
- Relative position of AA0 in the chain;
- Norm of the 1st and 2nd curvature of the quadruplet in which AA0 is in position 1;
- Norm of the 1st and 2nd curvature of the quadruplet in which AA0 is in position 4;
- Norm of the torsion of the quadruplet in which AA0 is in position 1;
- Norm of the torsion of the quadruplet in which AA0 is in position 4;
- Volume of AA0
5.1.1. Lysine Analysis
5.1.2. All Aminos, Separated
5.1.3. All Aminos
5.2. Analyzing Data with RF
- Table that contains features about 3 amino acids preceding and following LYS;
- Table that contains features about 6 amino acids preceding and following LYS;
- Table that contains features about 9 amino acids preceding and following LYS;
- Table that contains features about 3 amino acids preceding and following each residue;
- The number of estimators: Random Forest trees may vary from 50 to 500, with step 10;
- The depth of the tree: the maximum number of children from root to node further leaf, can range from 5 to 12. The value None was also attached to the list, which means default maximum depth was not chosen.
LYS-3: {'max_depth': None, 'n_estimators': 300}
Model Performance
Average Error: 9.1795 degrees.
Accuracy = 81.75
LYS-6 : {'max_depth': None, 'n_estimators': 300}
Model Performance
Average Error: 8.6356 degrees.
Accuracy = 82.04
LYS-9: {'max_depth': None, 'n_estimators': 300}
Model Performance
Average Error: 8.5398 degrees.
Accuracy = 83.39
LYS-3 {'max_depth': None, 'n_estimators': 300}:
Average Error: 9.1833 degrees.
Accuracy = 81.72%.
Means absolute error 9.183323451548308
Means squared error 186.71606785645264
Root means squared error 13.664408800107402
Minimum max depth of tree = 30
16737 nodes 25068 edges 8420 iter 13.43~sec
LYS-6 {'max_depth': None, 'n_estimators': 300}:
Average Error: 8.5886 degrees.
Accuracy = 82.15%.
Means absolute error 8.588595380071665
Means squared error 166.42968189426642
Root means squared error 12.900762841563534
Minimum max depth of tree = 32
16789 nodes 25137 edges 8155 iter 13.78~sec
LYS-9 {'max_depth': None, 'n_estimators': 300}:
Average Error: 8.5366 degrees.
Accuracy = 83.39%.
Means absolute error 8.536609032254626
Means squared error 165.80542920374762
Root means squared error 12.876545701536093
Minimum max depth of tree = 31
14937 nodes 22372 edges 7057 iter 8.21 sec
5.3. Analyzing Data with MLP
- The size of the hidden layers, from 100 to 1000 with step 300;
- The activation function, deleting from the grid those that did not carry, in any case, to the algorithm convergence;
- The solver, choosing the one compatible with the activation functions defined;
- The maximum number of iterations, from 100 to 1000 with step 200.
LYS-3
Parameters:
{'activation': logistic, 'hidden_layer_sizes': 700,
'max_iter':900, 'solver': 'adam'}
Accuracy = 79.22
LYS-6
Parameters:
{'activation': logistic, 'hidden_layer_sizes': 700,
'max_iter':900, 'solver': 'adam'}
Accuracy = 80.35
LYS-9
Parameters:
{'activation': logistic, 'hidden_layer_sizes': 700,
'max_iter':900, 'solver': 'adam'}
Accuracy = 81.27
6. Explainability Analysis and Discussion
6.1. Lysine Analysis
6.2. All Aminos, Separated
- When the amino acid is considered in its configuration with three previous and subsequent residues, the variable that most affects the prediction is always one of the volumes of the surrounding residues, except in the case of aspartic acid in which the chemical property of a residue emerges previously. The following variables also apply for the greater part of the volumes.
- Even considering the information on the six residues prior to and after the AA0, the volumes are the features that mostly influence the prediction. However, in the case of Aspartic Acid and Glutamine, their relative position in the chain acquires the role of main Features Importance (FI).
- Finally, considering a round of the AA0 of 9 amino acid ray, the analysis reports, again, as first FI, one of the volumes of the surrounding amino acids, for almost all residues. In the case of aspartic acid and cysteine, in fact, the relative position is confirmed as the main FI and in the case of isoleucine the functional characteristic is of one of the following residues.
- When the amino acid is considered in its configuration with three previous and subsequent residues, the variable that most affects the prediction is always one of the volumes of the surrounding residues, with an exception made for cystein which shows the highest score in prediction for the geometric feature torsion of the first quadruplet. The following FIs are also, for the most part, volumes, but features related to the geometry still emerge.
- Even when considering the information on the six residues prior to and after the AA0, the volumes are the most influential features of the prediction. This is the case even with the greater impact as compared to the previous case. However, in the case of proline and serine, torsion of a quadruplet in the chain acquires the role of the main FI.
- Finally, considering the surroundings of the AA0 of the 9 amino acid ray, the analysis confirms the pattern established by the previous cases, reporting, again, as first in FI, one of the volumes of the surrounding amino acids, for almost all residues, without any exceptions. The geometrical features seem to appear as the third or following, in the classification of FI.
- On the one hand, the role of the relative position of the AA0 stands out, as the main feature, showing its fundamental role in this type of analysis;
- On the other hand, contrary to what might be expected, the main FI that emerges when excluding volumes is not the characteristics of the amino acids (chemical, functional or hydrophobic), but the geometric ones, formulated ad hoc for the problem in examination. This has enhanced and supported the validity and correctness of the model created.
- Clear predominance of the geometric features related only to the torsion parameters, which decreases slightly only in the analysis of feature 9 residues in the surroundings.
- On the other hand, contrary to what might be expected—in complete disagreement with the same analysis conducted with RF—the other features that emerge when excluding volumes are not the other geometric characteristics—i.e., curvatures—but both relative position and the amino acid properties (chemical, functional or hydrophobic).
6.3. All Aminos
TOT-3 {'max_depth': None, 'n_estimators': 200}:
Average Error: 4.0070 degrees.
Accuracy = 86.91%.
Means absolute error: 4.010753088384853
Means squared error: 62.97177931847423
Root means squared error: 7.935475998229358
Max_depth min = 42
129749 nodes 194571 edges 70263 iter 1524.40 sec
TOT-3: {'activation': 'logistic',
'hidden_layer_sizes': 700, 'max_iter': 1000, 'solver': 'adam'}
Average Error: 5.8446 degrees.
Accuracy = 73.19
TOT NO POS 3 {'max_depth': None, 'n_estimators': 200}:
Average Error: 4.0616 degrees.
Accuracy = 86.43%.
Means absolute error: 4.07168620428757
Means squared error: 64.37940508449833
Root means squared error: 8.023677777958081
Max_depth min = 42
129765 nodes 194598 edges 69420 iter 1516.01 sec
TOT NO POS 3: {'activation': 'logistic',
'hidden_layer_sizes': 700,'max_iter': 1000, 'solver': 'adam'}
Average Error: 6.3096 degrees.
Accuracy = 67.55
- On one hand both, the models confirm the predominance of relative positions and volumes, so the main features seem to be well detached by the different approach.
- On the other hand, analysis with RF seems to underline the strength of the mathematical model; instead, analysis with MLP emphasizes the role of the torsion in determining the target variable and a quite significant impact of chemical properties.
7. Related Works
- A comparative analysis of different Natural Language Processing (NLP) models in sentiment analysis of single domain Twitter messages [28]. In this paper, some ML models are analyzed and a comparison is made against classification accuracy and explainability capabilities.
- A study on the explainability in Deep Neural Network (DNN)s for image-based mammography analysis is reported in [29]. The paper makes a contribution with the introduction of an explainability method (named oriented, modified integrated gradients, OMIG) and its application to image analysis.
- The authors in [30] carry out a concrete analysis concerning the explainability of glaucoma prediction by merging the information coming from two different sources (tomography images and medical data).
8. Conclusions and Future Developments
- The members of the chosen protein family—i.e., myoglobin—are very similar in their sequences (85–99% identity): this similarity implies that trained predictors exhibit high performance that is not met in reality; Otherwise, it was decided to perform this kind of analysis—aware of such similarity—because all the proteins belonging to the same family have similar functions. This similitude is intentional and allows one to study how geometrical features can vary within a similar primary structure. Moreover, it is clear that this has no impact on the results: the most important features are not directly related to the primary structure but to an amino acid’s properties and its relative position in the chain (there could be multiple within the same structure).
- The present work is based on the mathematical model reported in [10], which has its limited scope and constraining hypotheses: in particular, it does not explicitly consider gaps in the protein structure and does not use sophisticated bioinformatics methods for the evaluation of the protein structure.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations/Code
| 3HEN | Ferric Horse Heart Myoglobin; H64V/V67R Mutant |
| AA0 | Amino acid 0 |
| AI | Artificial Intelligence |
| CSV | Comma Separated Value |
| DBR2 | Rosy and Roberta Database |
| DNN | Deep Neural Network |
| FI | Features Importance |
| LYS | Lysine |
| MAE | mean absolute error |
| ML | Machine Learning |
| MLP | Multilayer Perceptron Neural Network |
| MSE | mean squared error |
| NLP | Natural Language Processing |
| NN | Neural Network |
| PDB | Protein Data Bank |
| RF | Random Forest |
| RMSE | root mean squared error |
| V&V | Verification & Validation |
| XAI | eXplainable Artificial Intelligence |
References
- Aceto, G.; Persico, V.; Pescapé, A. Industry 4.0 and Health: Internet of Things, Big Data and Cloud Computing for Healthcare 4.0. J. Ind. Inf. Integr. 2020, 18, 100129. [Google Scholar] [CrossRef]
- Allam, Z.; Dhunny, Z.A. On big data, Artificial Intelligence and smart cities. Cities 2019, 89, 80–91. [Google Scholar] [CrossRef]
- El Sallab, A.; Abdou, M.; Perot, E.; Yogamani, S. Deep reinforcement learning framework for autonomous driving. Electron. Imaging 2017, 2017, 70–76. [Google Scholar] [CrossRef]
- Murdoch, T.B.; Detsky, A.S. The inevitable application of big data to health care. JAMA 2013, 309, 1351–1352. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Tay, F.E. Support vector machine with adaptive parameters in financial time series forecasting. IEEE Trans. Neural Netw. 2003, 14, 1506–1518. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, F.; Marulli, F.; Mercaldo, F.; Marrone, S.; Santone, A. Enhanced Privacy and Data Protection using Natural Language Processing and Artificial Intelligence. In Proceedings of the 2020 International Joint Conference on Neural Networks (IJCNN), Glasgow, UK, 19–24 July 2020; pp. 1–8. [Google Scholar]
- Antão, L.; Pinto, R.; Reis, J.; Gonçalves, G. Requirements for testing and validating the industrial internet of things. In Proceedings of the 2018 IEEE International Conference on Software Testing, Verification and Validation Workshops (ICSTW), Vasteras, Sweden, 9–13 April 2018; pp. 110–115. [Google Scholar] [CrossRef]
- Zhou, J.; Gandomi, A.H.; Chen, F.; Holzinger, A. Evaluating the quality of Machine Learning explanations: A survey on methods and metrics. Electronics 2021, 10, 593. [Google Scholar] [CrossRef]
- Barredo Arrieta, A.; Díaz-Rodríguez, N.; Del Ser, J.; Bennetot, A.; Tabik, S.; Barbado, A.; Garcia, S.; Gil-Lopez, S.; Molina, D.; Benjamins, R.; et al. Explainable Artificial Intelligence (XAI): Concepts, taxonomies, opportunities and challenges toward responsible AI. Inf. Fusion 2020, 58, 82–115. [Google Scholar] [CrossRef]
- Vitale, F. On statistically meaningful geometric properties of digital three-dimensional structures of proteins. Math. Comput. Model. 2008, 48, 141–160. [Google Scholar] [CrossRef]
- Vitale, F. A topology for the space of protein chains and a notion of local statistical stability for their three-dimensional structures. Math. Comput. Model. 2008, 48, 610–620. [Google Scholar] [CrossRef]
- On a 3D-matrix representation of the tertiary structure of a protein. Math. Comput. Model. 2006, 43, 1434–1464. [CrossRef]
- Gilpin, L.H.; Bau, D.; Yuan, B.Z.; Bajwa, A.; Specter, M.; Kagal, L. Explaining explanations: An overview of interpretability of machine learning. In Proceedings of the 2018 IEEE 5th International Conference on Data Science and Advanced Analytics (DSAA), Turin, Italy, 1–3 October 2018; pp. 80–89. [Google Scholar] [CrossRef]
- Miller, T. Explanation in Artificial Intelligence: Insights from the social sciences. Artif. Intell. 2019, 267, 1–38. [Google Scholar] [CrossRef]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Weissig, H.; Shindyalov, I.; Bourne, P. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Heinecke, J.; Tan, H.; Ford, P.C.; Richter-Addo, G.B. The Distal Pocket Histidine Residue in Horse Heart Myoglobin Directs the O-Binding Mode of Nitrite to the Heme Iron. J. Am. Chem. Soc. 2009, 131, 18119–18128. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Nassif, A.B.; Ho, D.; Capretz, L.F. Towards an early software estimation using log-linear regression and a multilayer perceptron model. J. Syst. Softw. 2013, 86, 144–160. [Google Scholar] [CrossRef]
- Kuzlu, M.; Cali, U.; Sharma, V.; Güler, Ö. Gaining insight into solar photovoltaic power generation forecasting utilizing explainable Artificial Intelligence tools. IEEE Access 2020, 8, 187814–187823. [Google Scholar] [CrossRef]
- Sarp, S.; Knzlu, M.; Cali, U.; Elma, O.; Guler, O. An interpretable solar photovoltaic power generation forecasting approach using an explainable Artificial Intelligence tool. In Proceedings of the 2021 IEEE Power & Energy Society Innovative Smart Grid Technologies Conference (ISGT), Washington, DC, USA, 16–18 February 2021. [Google Scholar] [CrossRef]
- Vij, A.; Nanjundan, P. Comparing Strategies for Post-Hoc Explanations in Machine Learning Models. Lect. Notes Data Eng. Commun. Technol. 2022, 68, 585–592. [Google Scholar] [CrossRef]
- Cock, P.J.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. BioPython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; Te Beek, T.A.H.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- Xu, F.; Uszkoreit, H.; Du, Y.; Fan, W.; Zhao, D.; Zhu, J. Explainable AI: A Brief Survey on History, Research Areas, Approaches and Challenges. In Proceedings of the CCF International Conference on Natural Language Processing and Chinese Computing, Dunhuang, China, 9–14 October 2019; Volume 11839 LNAI, pp. 563–574. [Google Scholar] [CrossRef]
- De Mulder, W.; Valcke, P. The need for a numeric measure of explainability. In Proceedings of the 2021 IEEE International Conference on Big Data, Orlando, FL, USA, 15–18 December 2021; pp. 2712–2720. [Google Scholar] [CrossRef]
- Bedué, P.; Fritzsche, A. Can we trust AI? An empirical investigation of trust requirements and guide to successful AI adoption. J. Enterp. Inf. Manag. 2022, 35, 530–549. [Google Scholar] [CrossRef]
- London, A.J. Artificial Intelligence and Black-Box Medical Decisions: Accuracy versus Explainability. Hastings Cent. Rep. 2019, 49, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Fiok, K.; Karwowski, W.; Gutierrez, E.; Wilamowski, M. Analysis of sentiment in tweets addressed to a single domain-specific Twitter account: Comparison of model performance and explainability of predictions. Expert Syst. Appl. 2021, 186, 115771. [Google Scholar] [CrossRef]
- Amanova, N.; Martin, J.; Elster, C. Explainability for deep learning in mammography image quality assessment. Mach. Learn. Sci. Technol. 2022, 3, 025015. [Google Scholar] [CrossRef]
- Kamal, M.S.; Dey, N.; Chowdhury, L.; Hasan, S.I.; Santosh, K. Explainable AI for Glaucoma Prediction Analysis to Understand Risk Factors in Treatment Planning. IEEE Trans. Instrum. Meas. 2022, 71, 1–9. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acids | Features about 3 Amino Acids Preceding and Following AA0 | Features about 6 Amino Acids Preceding and Following AA0 | Features about 9 Amino Acids Preceding and Following AA0 | |||
|---|---|---|---|---|---|---|
| Average Error | Accuracy | Average Error | Accuracy | Average Error | Accuracy | |
| GLY | 0.5385 | 75.54% | 0.5151 | 70.97% | 0.4468 | 78.68% |
| ALA | 0.7402 | 93.28% | 0.6254 | 93.97% | 0.6009 | 94.01% |
| VAL | 1.8636 | 92.94% | 1.4999 | 94.18% | 1.5162 | 93.98% |
| LEU | 2.8407 | 93.65% | 2.5713 | 94.23% | 2.4643 | 94.42% |
| ILE | 2.6804 | 93.02% | 2.4010 | 93.74% | 2.2002 | 94.25% |
| MET | 3.8676 | 92.29% | 2.8892 | 94.59% | 3.0405 | 94.29% |
| SER | 1.0704 | 93.76% | 1.0899 | 93.05% | 1.0885 | 93.25% |
| PRO | 1.6259 | 91.33% | 1.4459 | 92.46% | 1.3758 | 92.71% |
| THR | 1.6654 | 93.65% | 1.4738 | 94.31% | 1.4648 | 94.33% |
| CYS | 4.3423 | 71.34% | 3.4113 | 77.39% | 1.6509 | 93.66% |
| ASN | 2.4039 | 93.87% | 2.0666 | 94.81% | 1.8991 | 94.69% |
| GLN | 5.9150 | 88.48% | 4.4296 | 91.30% | 5.1124 | 91.34% |
| PHE | 4.6112 | 93.14% | 3.5674 | 94.76% | 3.4181 | 94.65% |
| TYR | 5.1439 | 94.54% | 4.0024 | 95.63% | 4.6987 | 95.48% |
| TRP | 4.8437 | 95.60% | 4.3107 | 95.98% | 4.2929 | 95.80% |
| LYS | 9.1833 | 81.72% | 8.5886 | 82.15% | 8.5366 | 83.39% |
| HIS | 3.3231 | 93.25% | 3.1346 | 93.60% | 2.8392 | 94.33% |
| ARG | 10.5045 | 69.72% | 10.1136 | 68.02% | 10.1957 | 68.46% |
| ASP | 2.8097 | 92.15% | 2.5156 | 93.04% | 2.5252 | 93.00% |
| GLU | 5.2578 | 88.68% | 5.1794 | 88.16% | 4.9968 | 89.58% |
| Amino Acids | Features about 3 Amino Acids Preceding and Following AA0 | Features about 6 Amino Acids Preceding and Following AA0 | Features about 9 Amino Acids Preceding and Following AA0 | |||
|---|---|---|---|---|---|---|
| Average Error | Accuracy | Average Error | Accuracy | Average Error | Accuracy | |
| GLY | 0.4813 | 74.08% | 1.5946 | 30.03% | 1.6121 | 10.36% |
| ALA | 0.6569 | 93.62% | 2.1218 | 78.64% | 1.8956 | 80.09% |
| VAL | 1.7685 | 93.14% | 4.8140 | 80.44% | 4.1212 | 83.52% |
| LEU | 3.2014 | 92.66% | 6.8586 | 85.08% | 6.7312 | 84.92% |
| ILE | 2.6705 | 92.96% | 6.8473 | 82.38% | 6.8182 | 82.39% |
| MET | 4.1665 | 92.59% | 7.8958 | 84.33% | 7.6145 | 86.10% |
| SER | 1.0998 | 93.36% | 2.7085 | 83.28% | 2.8237 | 82.77% |
| PRO | 1.5146 | 92.02% | 3.9080 | 79.57% | 3.7529 | 81.58% |
| THR | 1.7111 | 93.39% | 3.8131 | 85.29% | 3.7133 | 85.47% |
| CYS | 1.2494 | 95.18% | 3.4752 | 80.31% | 2.0410 | 91.85% |
| ASN | 2.1876 | 93.87% | 4.1435 | 89.31% | 3.9476 | 89.00% |
| GLN | 5.6628 | 90.25% | 11.1820 | 77.89% | 10.7624 | 81.91% |
| PHE | 3.8403 | 94.11% | 11.4726 | 83.08% | 12.1458 | 81.15% |
| TYR | 7.1454 | 93.21% | 12.9676 | 86.32% | 14.0738 | 85.72% |
| TRP | 5.5169 | 94.84% | 15.7977 | 85.43% | 8.6417 | 91.98% |
| LYS | 9.4384 | 81.81% | 16.5806 | 67.33% | 15.6154 | 70.29% |
| HIS | 3.3458 | 93.27% | 8.5671 | 83.00% | 7.5599 | 85.21% |
| ARG | 10.2835 | 71.13% | 17.4146 | 60.54% | 15.9363 | 61.93% |
| ASP | 2.7899 | 92.36% | 5.6960 | 84.28% | 5.1691 | 85.76% |
| GLU | 5.4416 | 88.77% | 10.4980 | 78.21% | 9.7335 | 79.30% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Fazio, R.; Di Giovannantonio, R.; Bellini, E.; Marrone, S. Explainabilty Comparison between Random Forests and Neural Networks—Case Study of Amino Acid Volume Prediction. Information 2023, 14, 21. https://doi.org/10.3390/info14010021
De Fazio R, Di Giovannantonio R, Bellini E, Marrone S. Explainabilty Comparison between Random Forests and Neural Networks—Case Study of Amino Acid Volume Prediction. Information. 2023; 14(1):21. https://doi.org/10.3390/info14010021
Chicago/Turabian StyleDe Fazio, Roberta, Rosy Di Giovannantonio, Emanuele Bellini, and Stefano Marrone. 2023. "Explainabilty Comparison between Random Forests and Neural Networks—Case Study of Amino Acid Volume Prediction" Information 14, no. 1: 21. https://doi.org/10.3390/info14010021
APA StyleDe Fazio, R., Di Giovannantonio, R., Bellini, E., & Marrone, S. (2023). Explainabilty Comparison between Random Forests and Neural Networks—Case Study of Amino Acid Volume Prediction. Information, 14(1), 21. https://doi.org/10.3390/info14010021