
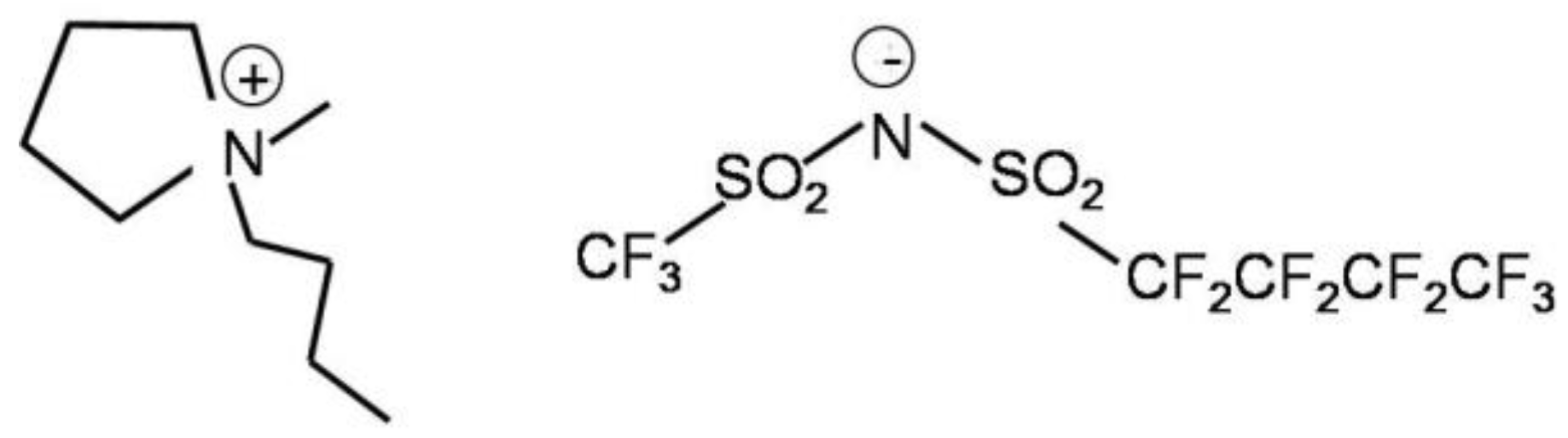
A Study of the Conformers of the (Nonafluorobutanesulfonyl)imide Ion by Means of Infrared Spectroscopy and Density Functional Theory (DFT) Calculations

 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
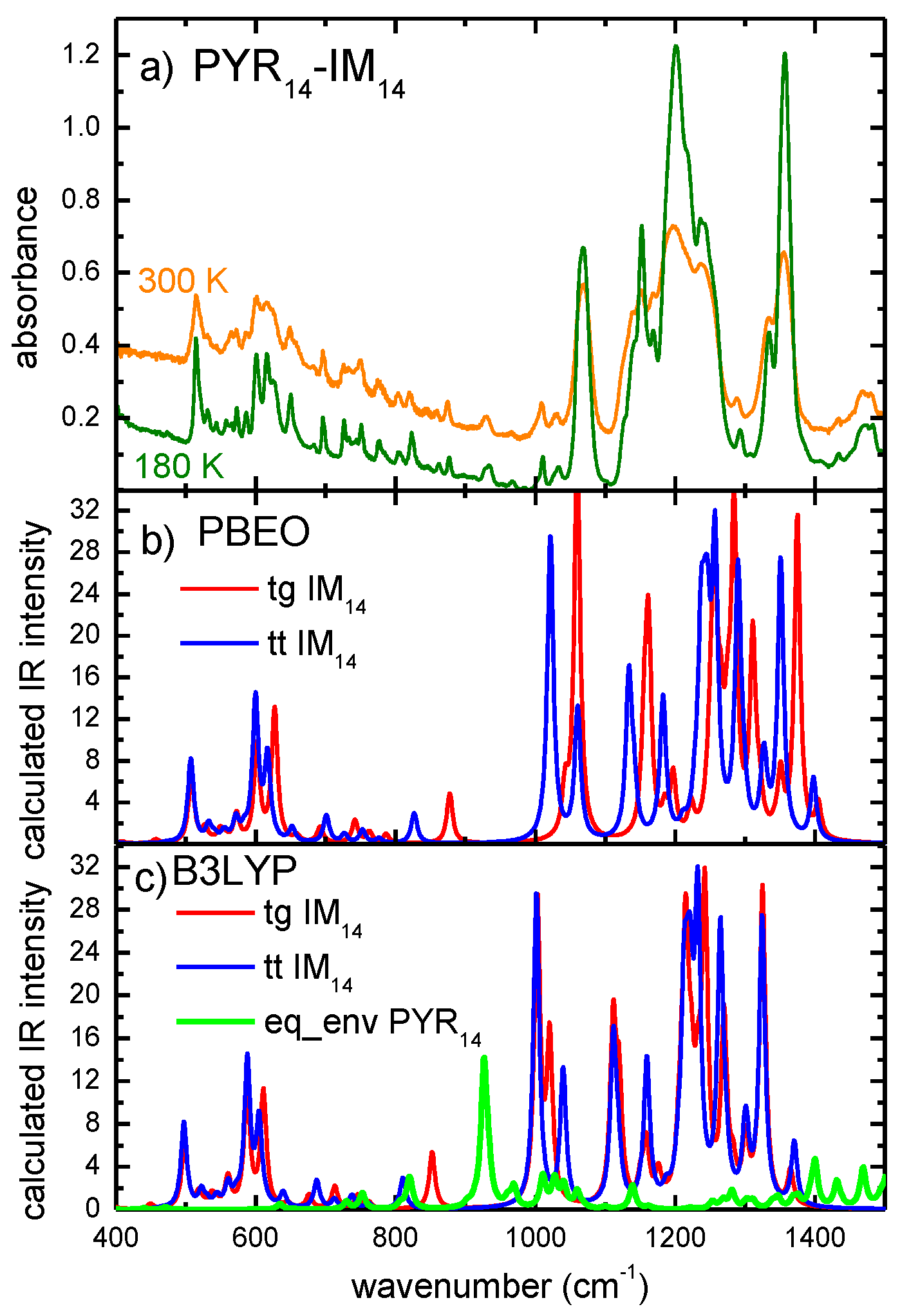
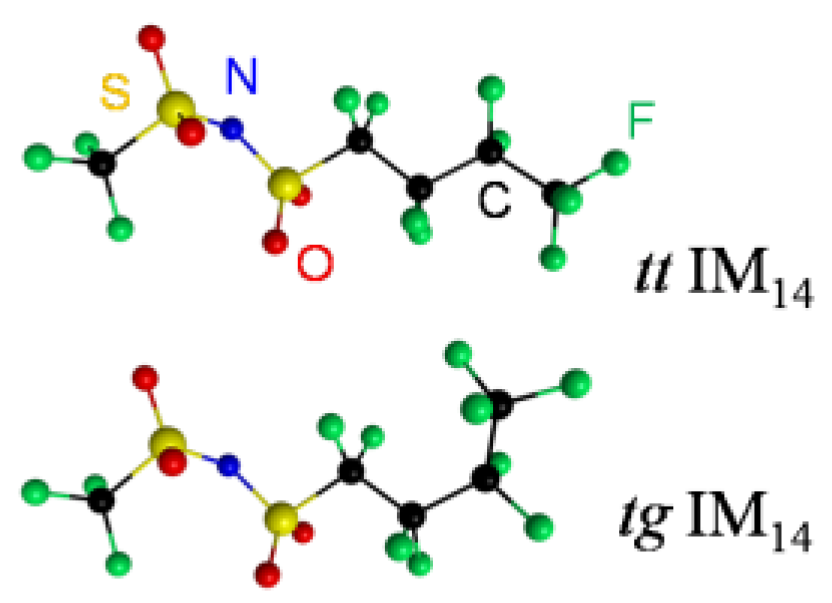
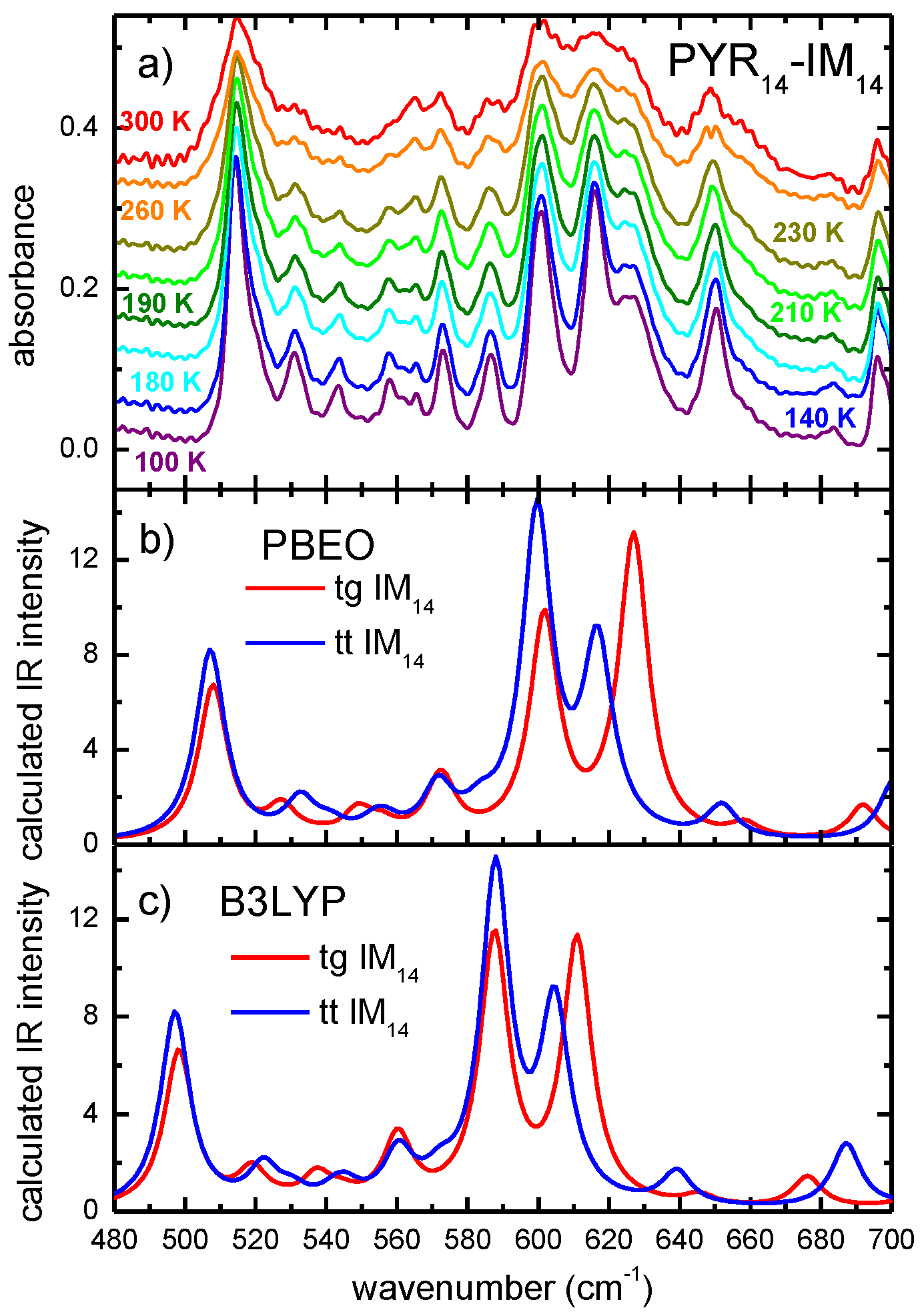
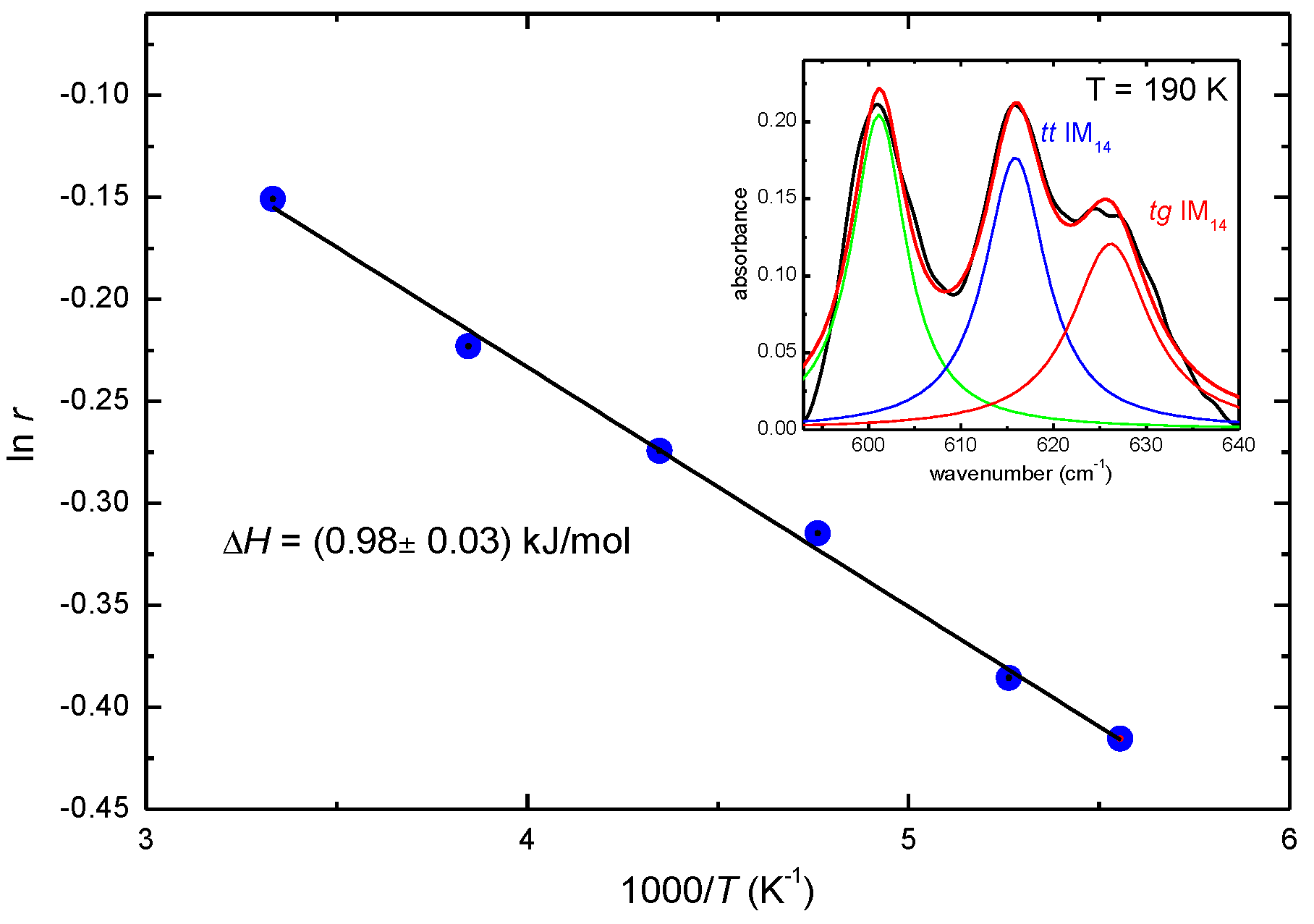
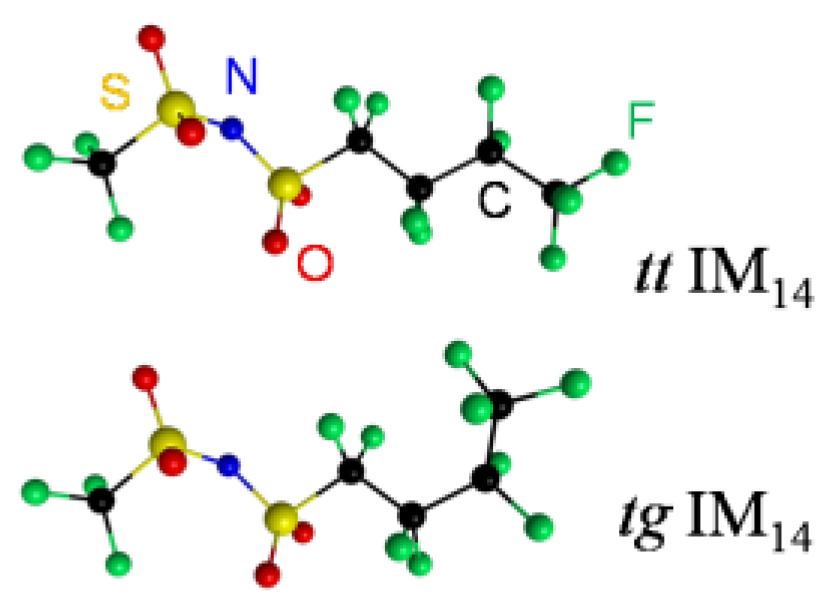
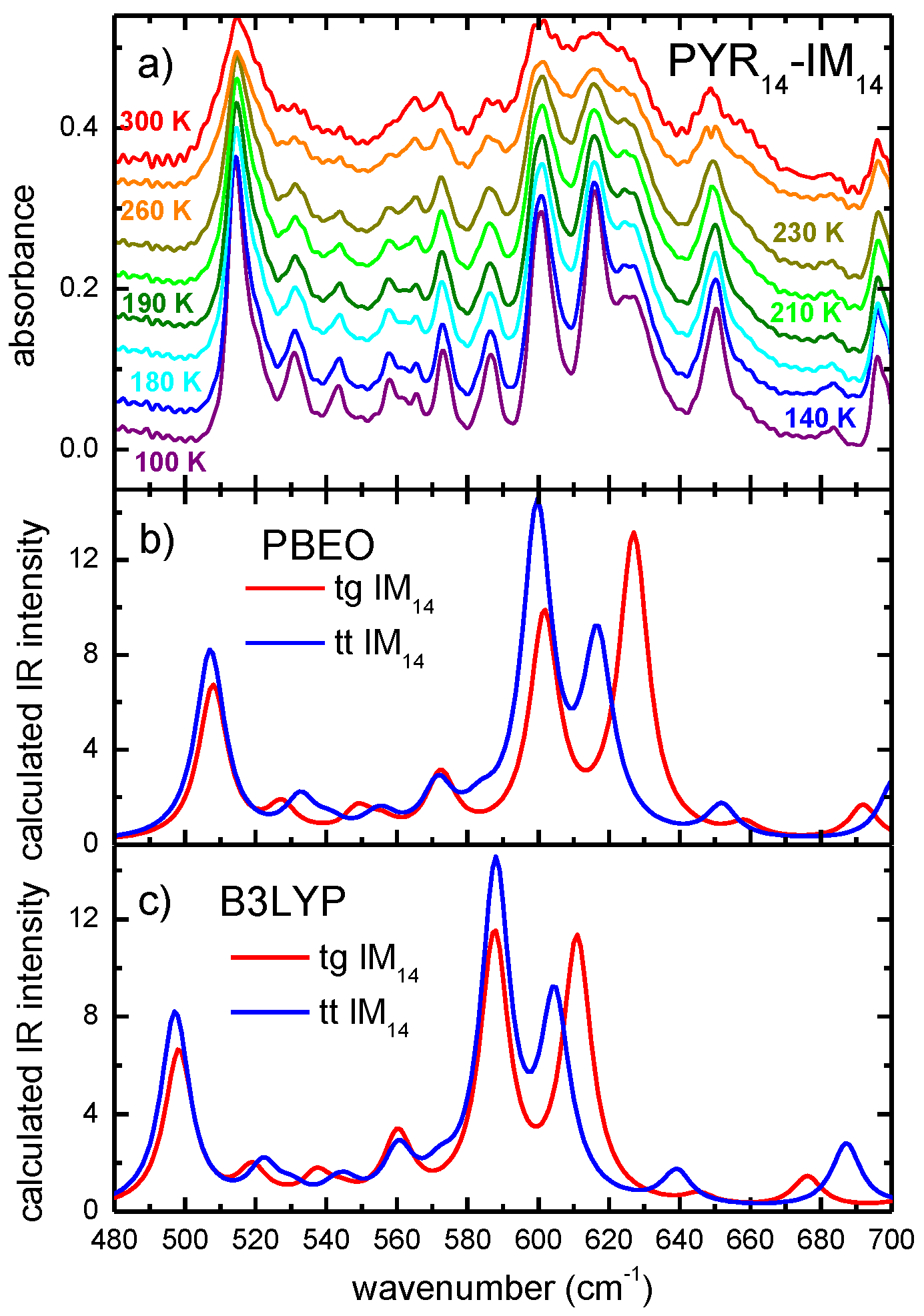
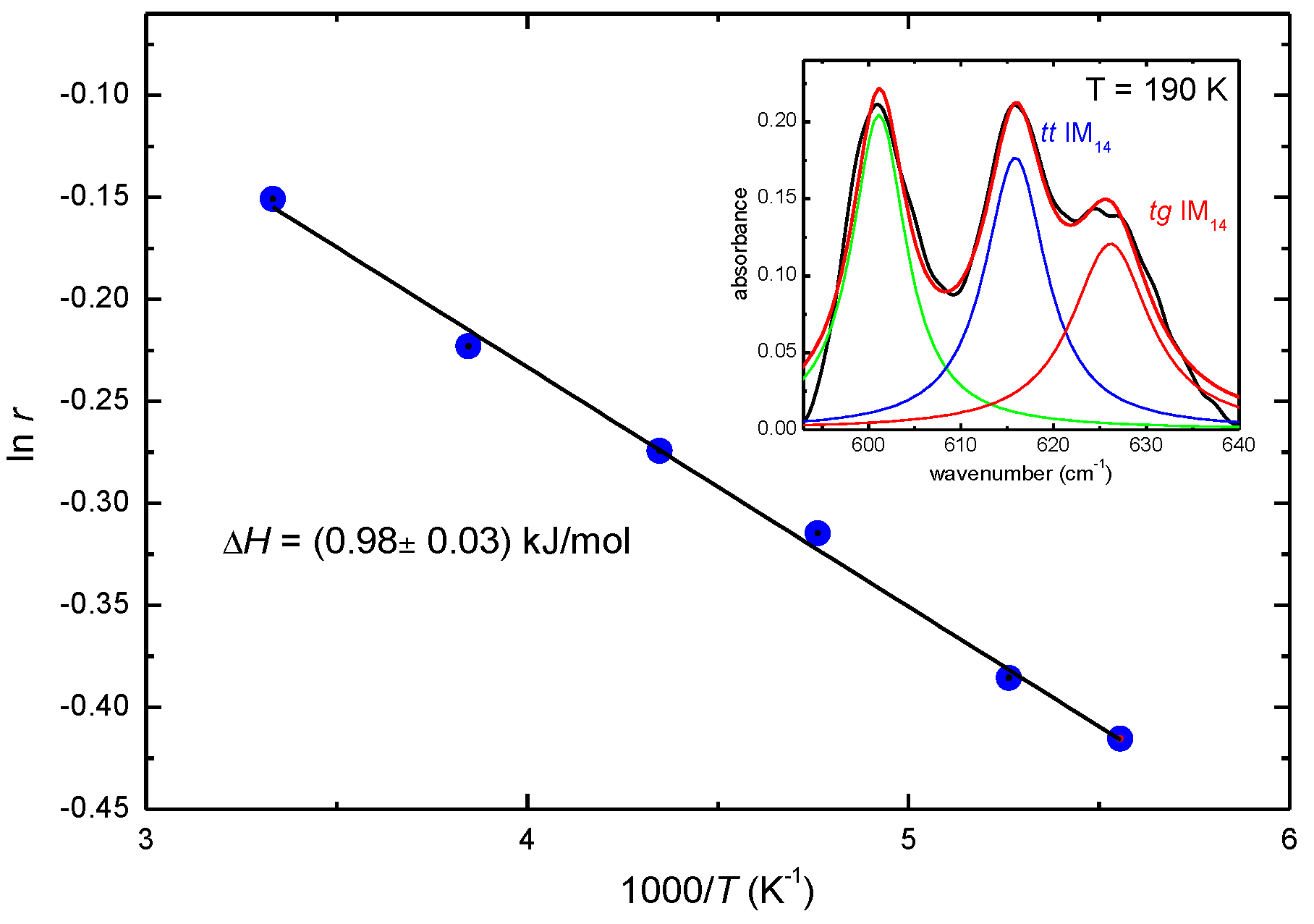
2. Results and Discussion
3. Materials and Methods
3.1. Experimental
3.2. Computational
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Welton, T. Ionic liquids in catalysis. Coord. Chem. Rev. 2004, 248, 2459–2477. [Google Scholar] [CrossRef]
- Navarra, M.A. Ionic liquids as safe electrolyte components for Li-metal and Li-ion batteries. Mat. Res. Soc. Bull. 2013, 38, 548–553. [Google Scholar] [CrossRef]
- Armand, M.; Endres, F.; MacFarlane, D.R.; Ohno, H.; Scrosati, B. Ionic-liquid materials for the electrochemical challenges of the future. Nat. Mater. 2009, 8, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Matic, A.; Scrosati, B. Ionic liquids for energy applications. MRS Bull. 2013, 38, 533–537. [Google Scholar] [CrossRef]
- Kunze, M.; Jeong, S.; Paillard, E.; Winter, M.; Passerini, S. Melting behavior of pyrrolidinium-based ionic liquids and their binary mixtures. J. Phys. Chem. 2010, 114, 12364–12369. [Google Scholar] [CrossRef]
- Palumbo, O.; Vitucci, F.M.; Trequattrini, F.; Paolone, A. A study of the conformers of the N,N-diethyl-N-methyl-N-propylammonium ion by means of infrared spectroscopy and DFT calculations. Vib. Spec. 2015, 80, 11–16. [Google Scholar] [CrossRef]
- Herstedt, M.; Smirnov, M.; Johansson, P.; Chami, M.; Grondin, J.; Servant, L.; Lassègues, J.C. Spectroscopic characterization of the conformational states of the bis(trifluoromethanesulfonyl)imide anion (TFSI−). J. Raman Spectrosc. 2005, 36, 762–770. [Google Scholar] [CrossRef]
- Fujii, K.; Takamuku, T.; Kanzaki, R.; Umebayashi, Y.; Ishiguro, S.-I. Conformational equilibrium of bis(trifluoromethanesulfonyl)imide anion of a room-temperature ionic liquid: Raman spectroscopic study and DFT calculations. J. Phys. Chem. 2006, 110, 8179–8183. [Google Scholar] [CrossRef] [PubMed]
- Canongia Lopes, J.N.; Shimizu, K.; Pádua, A.A.H.; Umebayashi, Y.; Fukuda, S.; Fujii, K.; Ishiguro, S.-I. A tale of two ions: The conformational landscapes of bis(trifluoromethanesulfonyl) amide and N,N-dialkylpyrrolidinium. J. Phys. Chem. B 2008, 112, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Vitucci, F.M.; Trequattrini, F.; Palumbo, O.; Brubach, J.-B.; Roy, P.; Paolone, A. Infrared spectra of bis(trifluoromethanesulfonyl)imide based ionic liquids: Experiments and DFT simulations. Vib. Spec. 2014, 74, 81–87. [Google Scholar] [CrossRef]
- Vitucci, F.M.; Trequattrini, F.; Palumbo, O.; Brubach, J.-B.; Roy, P.; Navarra, M.A.; Panero, S.; Paolone, A. Stabilization of different conformers of bis(trifluoromethanesulfonyl)imide anion in ammonium based ionic liquids at low temperatures. J. Phys. Chem. 2014, 118, 8758–8764. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, A.; Matic, A.; Johansson, P.; Jacobsson, P.; Börjesson, L.; Fernicola, A.; Panero, S.; Scrosati, B.; Ohno, H. Conformational evolution of TFSI- in protic and aprotic ionic liquids. J. Raman Spectrosc. 2011, 42, 522–528. [Google Scholar] [CrossRef]
- Fujii, K.; Seki, S.; Fukuda, S.; Kanzaki, R.; Takamuku, T.; Umebayashi, Y.; Ichiguro, S.-I. Anion conformation of low-viscosity room temperature ionic liquid 1-ethyl-3methylimidazolium bis(fluorosulfonyl)imide. J. Phys. Chem. 2007, 111, 12829–12833. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, T.; Fujii, K.; Kanzaki, R.; Chiba, K.; Yamamoto, H.; Umebayashi, Y.; Ishiguro, S.-I. Conformational structure of room temperature ionic liquid N-butyl-N-methyl-pyrrolidiniumbis(trifluoromethanesulfonyl)imide—Raman spectroscopic study and DFT calculations. J. Mol. Liquids 2007, 131–132, 216–224. [Google Scholar] [CrossRef]
- Gatto, S.; Palumbo, O.; Caramazza, S.; Trequattrini, F.; Postorino, P.; Appetecchi, G.B.; Paolone, A. The infrared spectrum of bis(fluorosulfonyl)imide revisited: attractive performances of the PBE0/6–31G∗∗ model. Vib. Spec. 2016, 82, 16–21. [Google Scholar] [CrossRef]
- Palumbo, O.; Trequattrini, F.; Vitucci, F.M.; Navarra, M.A.; Panero, S.; Paolone, A. An infrared spectroscopy study of the conformational evolution of the bis(trifluoromethanesulfonyl)imide ion in the liquid and in the glass state. Adv. Condens. Matter Phys. 2015, 2015, 176067. [Google Scholar] [CrossRef]
- Trequattrini, F.; Palumbo, O.; Gatto, S.; Appetecchi, G.B.; Paolone, A. A computational and experimental study of the conformers of pyrrolidinium ionic liquid cations containing an ethoxy group in the alkyl side chain. Adv. Chem. 2016, 2016, 1–9. [Google Scholar] [CrossRef]
- Capitani, F.; Gatto, S.; Postorino, P.; Palumbo, O.; Trequattrini, F.; Deutsch, M.; Brubach, J.-B.; Roy, P.; Paolone, A. The complex dance of the two conformers of bis(trifluoromethanesulfonyl)imide as a function of pressure and temperature. J. Phys. Chem. 2016, 120, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Capitani, F.; Trequattrini, F.; Palumbo, O.; Paolone, A.; Postorino, P. Phase transitions of PYR14-TFSI as a function of pressure and temperature: The competition between smaller volume and lower energy conformer. J. Phys. Chem. 2016, 120, 2921–2928. [Google Scholar] [CrossRef] [PubMed]
- Sobczyk, L.; Chudoba, D.; Tolstoy, P.M.; Filarowski, A. Some brief notes on theoretical and experimental investigations of intramolecular hydrogen bonding. Molecules 2016, 21, 1657–1676. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J.; Holfter, H.; Klapötke, T.M. Preparation and X-ray crystal structure of caesiumimidodisulphuryl fluoride Cs[N(SO2F)2]. J. Fluor. Chem. 1996, 78, 51–53. [Google Scholar] [CrossRef]
- Matsumoto, K.; Oka, T.; Nohira, T.; Hagiwara, R. Polymorphism of alkali bis(fluorosulfonyl)amides (M[N(SO2F)2], M = Na, K, and Cs). Inorg. Chem. 2013, 52, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Beran, M.; Príhoda, J.; Žák, Z.; Cerník, M. A new route to the syntheses of alkali metal bis(fluorosulfuryl)imides: Crystal structure of LiN(SO2F)2. Polyhedron 2006, 25, 1292–1298. [Google Scholar] [CrossRef]
- Ishida, H. Conformations of pyrrolidinium ion studied by molecular orbital calculations. Z. Naturforsch. 2000, 55a, 665–666. [Google Scholar]
- Lassègues, J.C.; Grondin, J.; Holomb, R.; Johansson, P. Raman and AB initio study of the conformational isomerism in the 1-ethyl-3-methyl-imidazolium bis(trifluoromethanesulfonyl)imide ionic liquid. J. Raman Spectrosc. 2007, 3, 551–558. [Google Scholar] [CrossRef]
- Ribeiro, M.C. High viscosity of imidazolium ionic liquids with the hydrogen sulfate anion: A raman spectroscopy study. J. Phys. Chem. 2012, 116, 7281–7290. [Google Scholar] [CrossRef] [PubMed]
- Vitucci, F.M.; Palumbo, O.; Trequattrini, F.; Brubach, J.-B.; Roy, P.; Meschini, I.; Croce, F.; Paolone, A. Interaction of 1-butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide with an electrospun PVdF membrane: Temperature dependence of the concentration of the anion conformers. J. Chem. Phys. 2015, 143, 143. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, O.; Trequattrini, F.; Vitucci, F.M.; Paolone, A. Relaxation dynamics and phase transitions in ionic liquids: viscoelastic properties from the liquid to the solid state. J. Phys. Chem. 2015, 119, 12905–12911. [Google Scholar] [CrossRef]
- Montanino, M.; Alessandrini, F.; Passerini, S.; Appetecchi, G.B. Water-based synthesis of hydrophobic ionic liquids for high-energyelectrochemical devices. Electrochim. Acta 2013, 96, 124–133. [Google Scholar] [CrossRef]
- Jeremias, S.; Carewska, M.; Conte, L.; Passerini, S.; Appetecchi, G.B. Asymmetry effect of novel per(fluoroalkylsulfonyl)imide anions in pyrrolidinium ionic liquids. RSC Adv. 2013, 3, 17755–17761. [Google Scholar] [CrossRef]
- Castiglione, F.; Raos, G.; Appetecchi, G.B.; Montanino, M.; Passerini, S.; Moreno, M.; Famulari, A.; Mele, A. Blending ionic liquids: How physico-chemical properties change. Phys. Chem. Chem. Phys. 2010, 12, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Russina, O.; Lo Celso, F.; Di Michiele, M.; Passerini, S.; Appetecchi, G.B.; Castiglione, F.; Mele, A.; Caminiti, R.; Triolo, A. Mesoscopic structural organization in triphilic room temperature ionic liquids. Faraday Discuss. 2013, 167, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; GuidiCestelli, M.; Nucara, A.; Marcouille, O.; Calvani, P.; Giura, P.; Paolone, A.; Mathis, Y.-L.; Gerschel, A. Spectral distribution of infrared synchrotron radiation by an insertion device and its edges: A comparison between experimental and simulated spectra. Phys. Rev. Lett. 2000, 84, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Brubach, J.-B.; Calvani, P.; De Marzi, G.; Filabozzi, A.; Gerschel, A.; Giura, P.; Lupi, S.; Marcouille, O.; Mermet, A.; et al. Infrared synchrotron radiation: From the production to the spectroscopic and microscopic applications. Nucl. Instrum. Methods Phys. Res. Sect. 2001, 426, 467–468. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J. A Guide to Molecular Mechanics and Quantum Chemical Calculations; Wavefunction, Inc.: Irvine, CA, USA, 2003. [Google Scholar]
- Granovsky, A.A. Firefly version 8.0. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 21 February 2017).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Bode, B.M.; Gordon, M.S. MacMolPlt: A graphical user interface for GAMESS. J. Mol. Graphics and Modeling 1999, 16, 133–138. [Google Scholar] [CrossRef]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palumbo, O.; Trequattrini, F.; Appetecchi, G.B.; Paolone, A. A Study of the Conformers of the (Nonafluorobutanesulfonyl)imide Ion by Means of Infrared Spectroscopy and Density Functional Theory (DFT) Calculations. Challenges 2017, 8, 7. https://doi.org/10.3390/challe8010007
Palumbo O, Trequattrini F, Appetecchi GB, Paolone A. A Study of the Conformers of the (Nonafluorobutanesulfonyl)imide Ion by Means of Infrared Spectroscopy and Density Functional Theory (DFT) Calculations. Challenges. 2017; 8(1):7. https://doi.org/10.3390/challe8010007
Chicago/Turabian StylePalumbo, Oriele, Francesco Trequattrini, Giovanni Battista Appetecchi, and Annalisa Paolone. 2017. "A Study of the Conformers of the (Nonafluorobutanesulfonyl)imide Ion by Means of Infrared Spectroscopy and Density Functional Theory (DFT) Calculations" Challenges 8, no. 1: 7. https://doi.org/10.3390/challe8010007
APA StylePalumbo, O., Trequattrini, F., Appetecchi, G. B., & Paolone, A. (2017). A Study of the Conformers of the (Nonafluorobutanesulfonyl)imide Ion by Means of Infrared Spectroscopy and Density Functional Theory (DFT) Calculations. Challenges, 8(1), 7. https://doi.org/10.3390/challe8010007