
About the Purification Route of Ionic Liquid Precursors



Abstract
:1. Introduction
- (i)
- Preparation of an intermediate product (hereafter, precursor) using amine and alkyl bromide as the reagents;
- (ii)
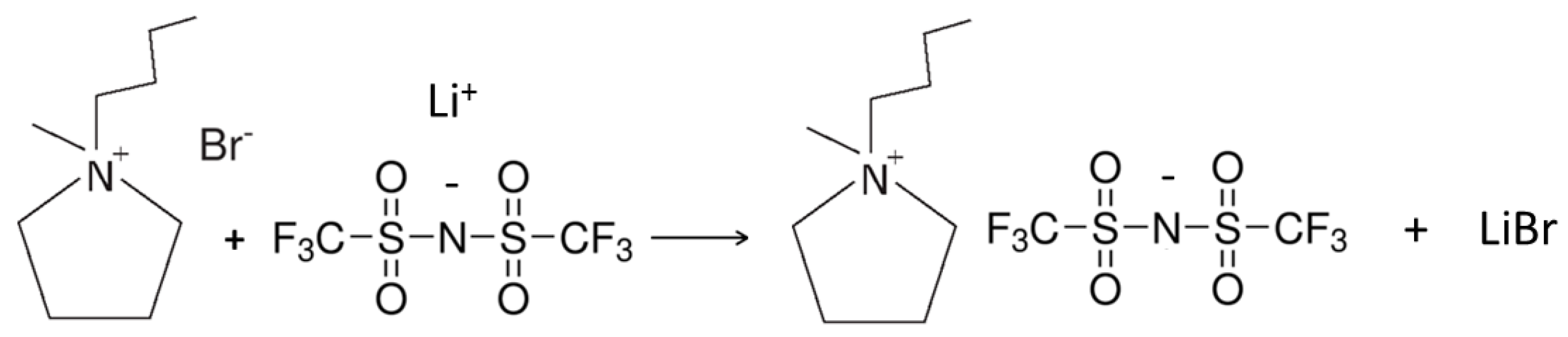
- The precursor is reacted with proper lithium or sodium salt (metathesis reaction) to obtain the ionic liquid.
2. Results and Discussion
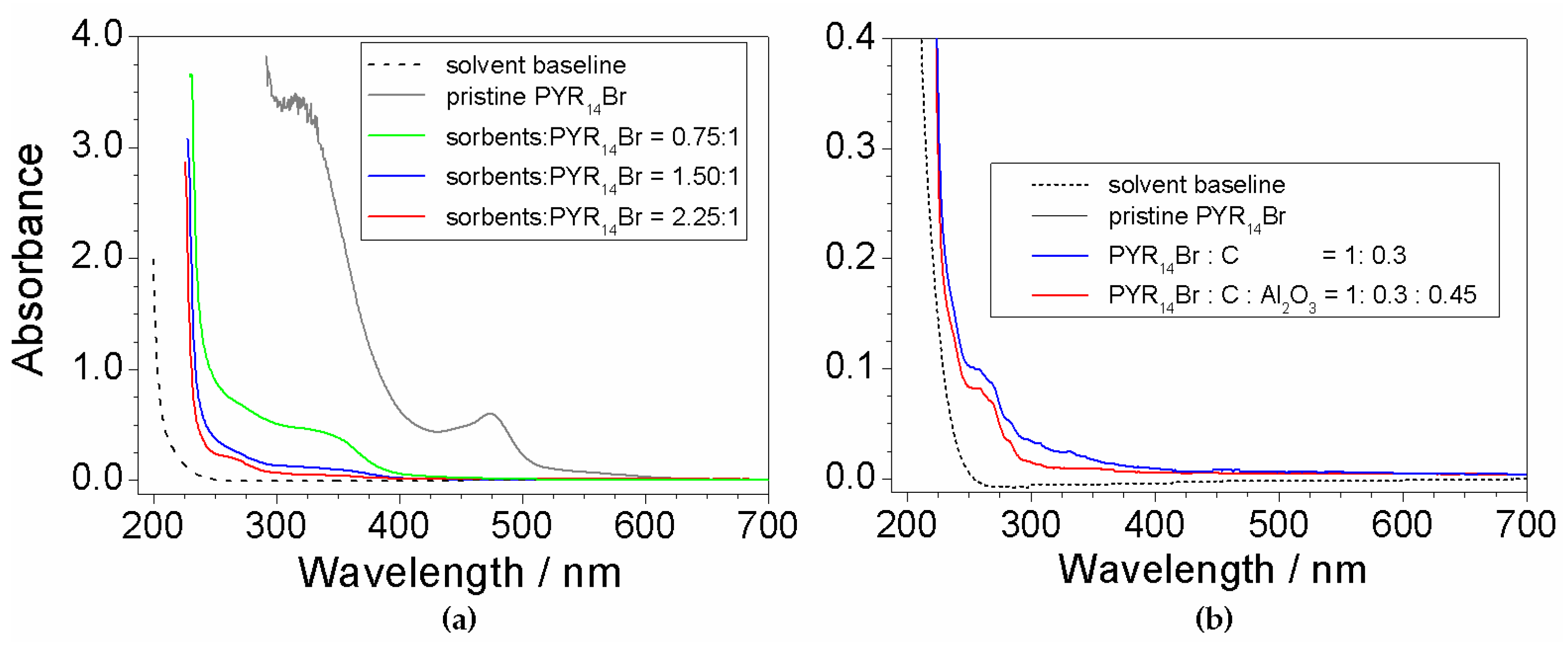
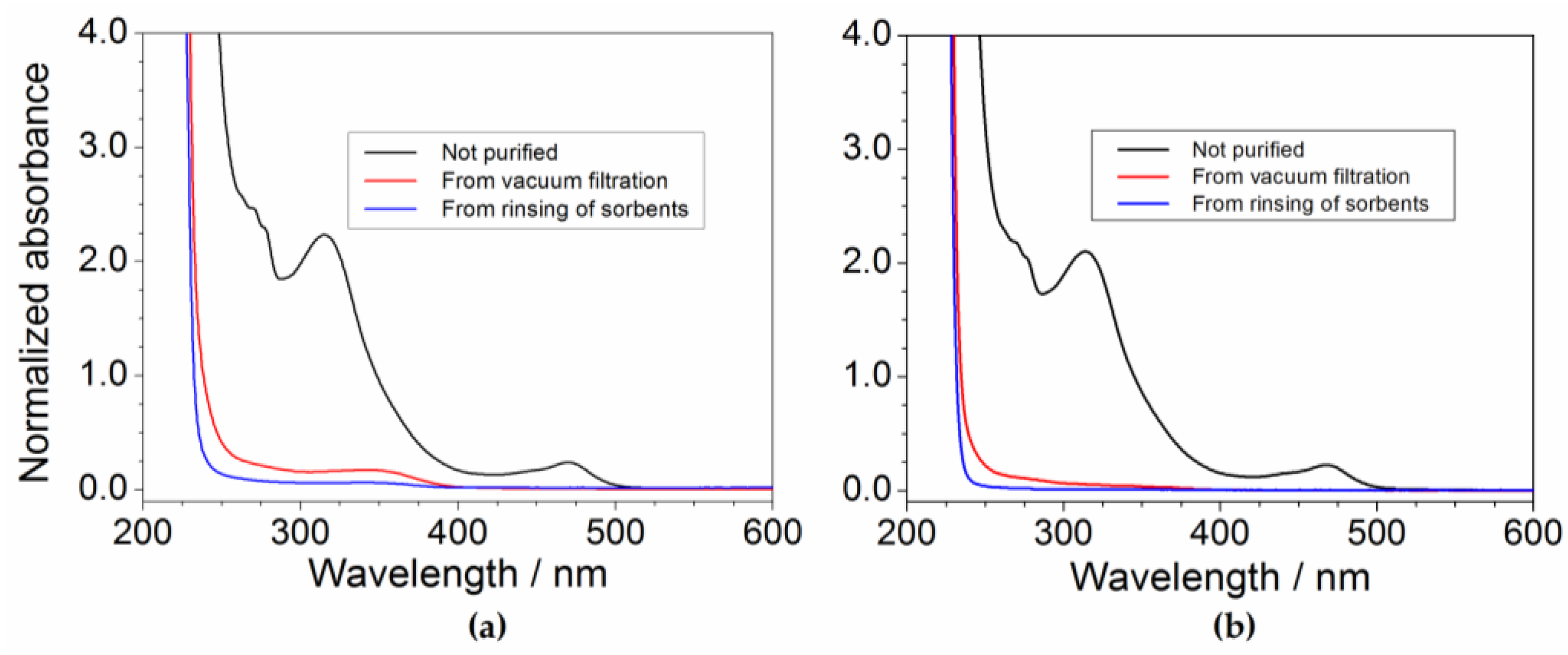
2.1. Preliminary Treatment of Sorbents
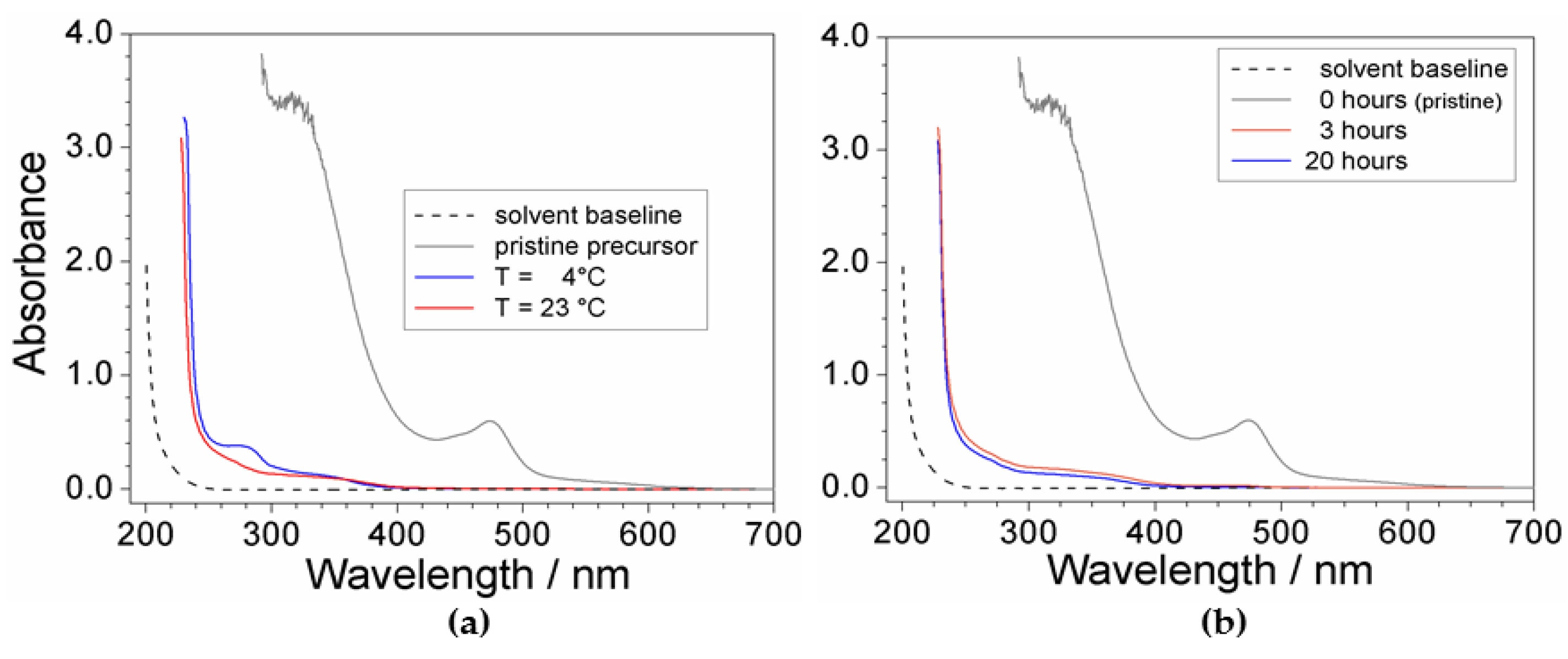
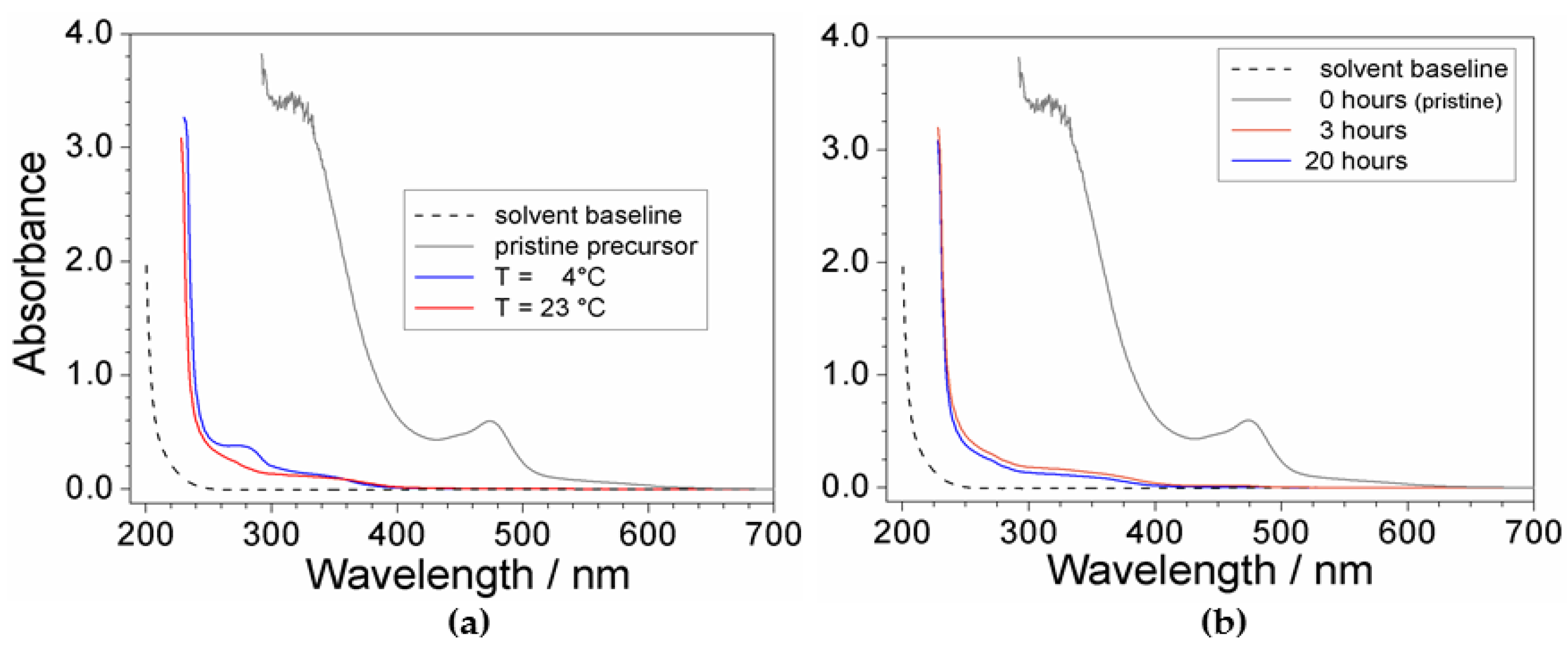
2.2. Effect of Processing Conditions
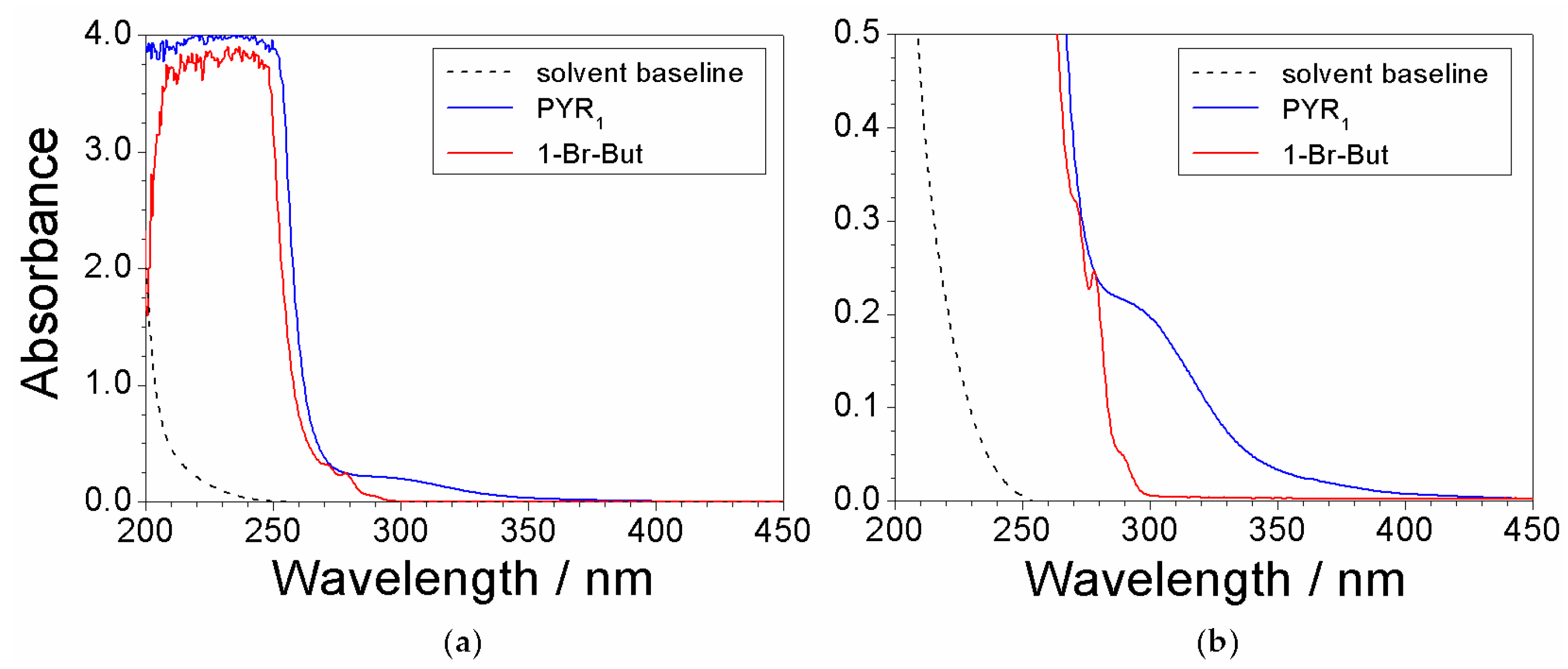
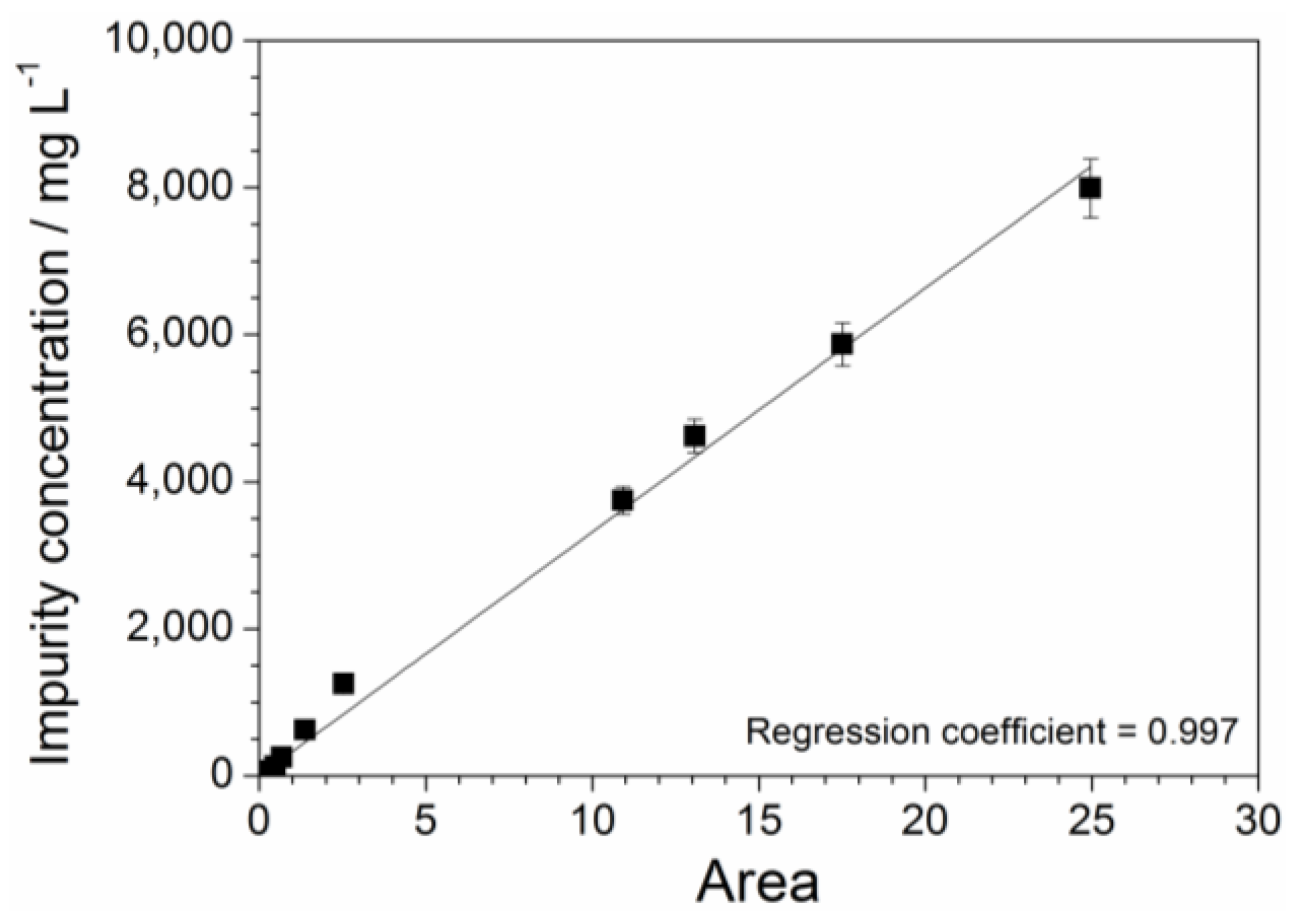
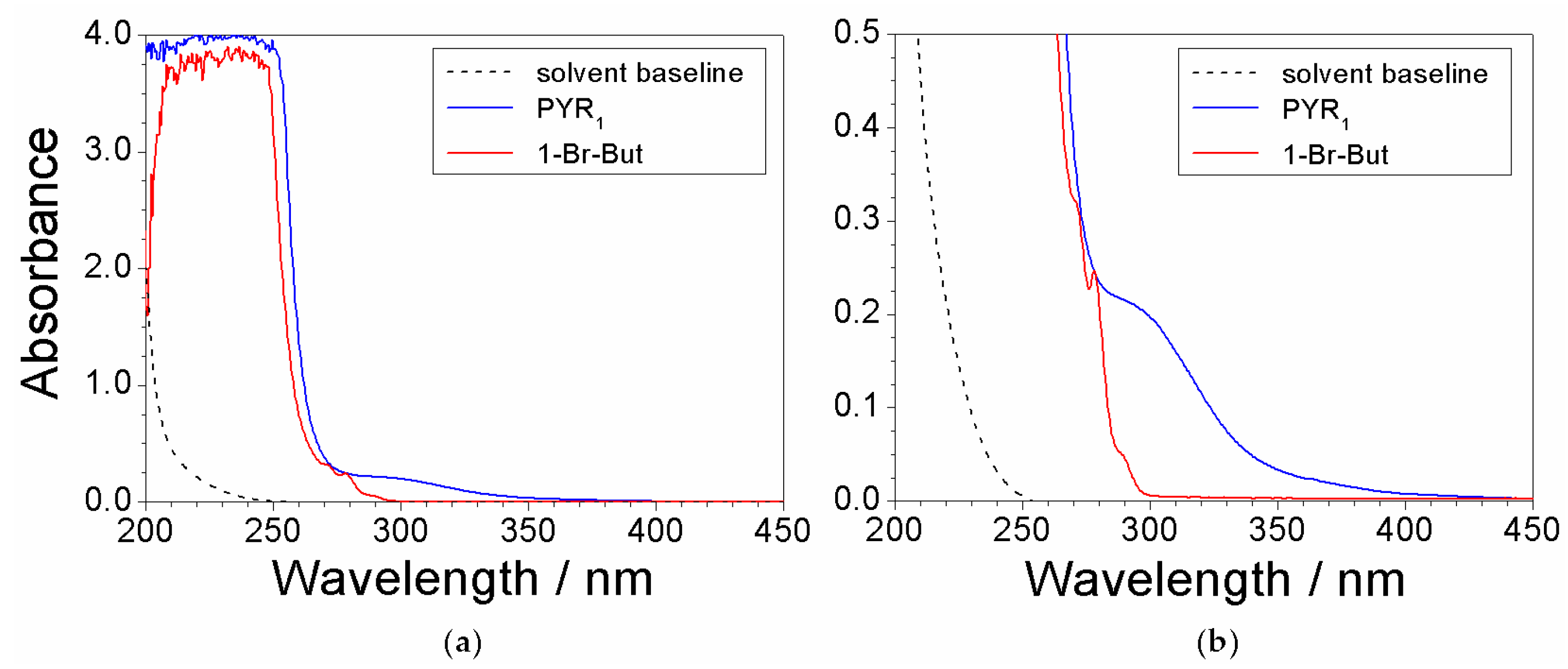
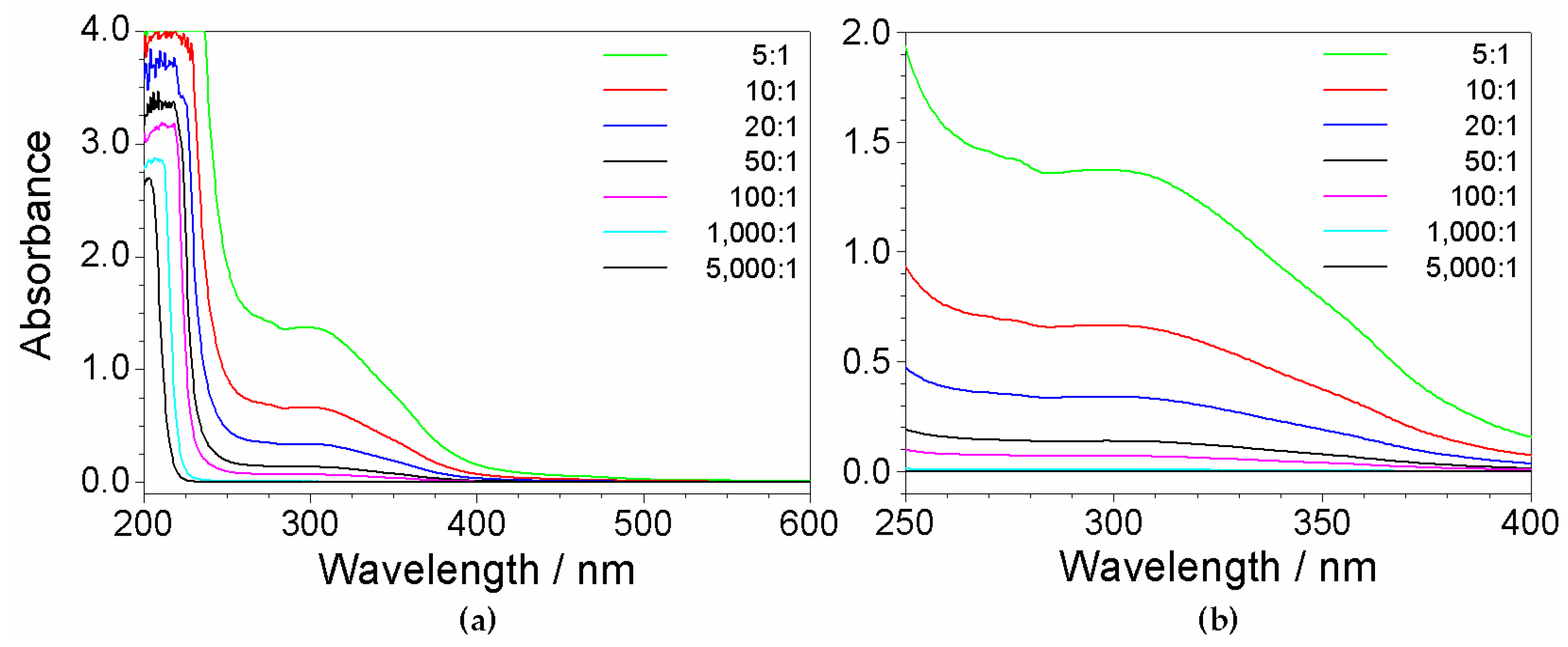
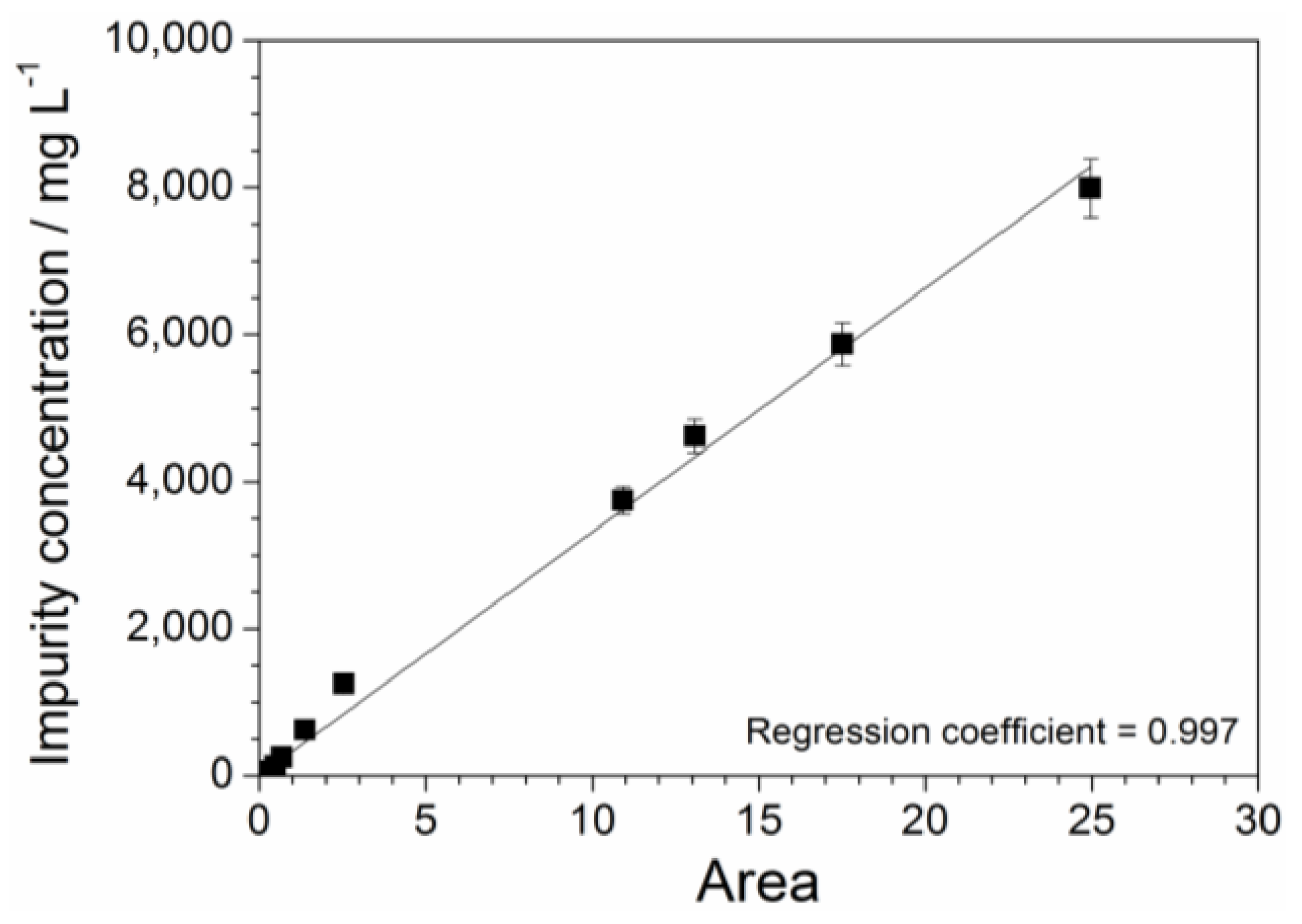
2.3. Absorbance Dependence on the Impurity Content
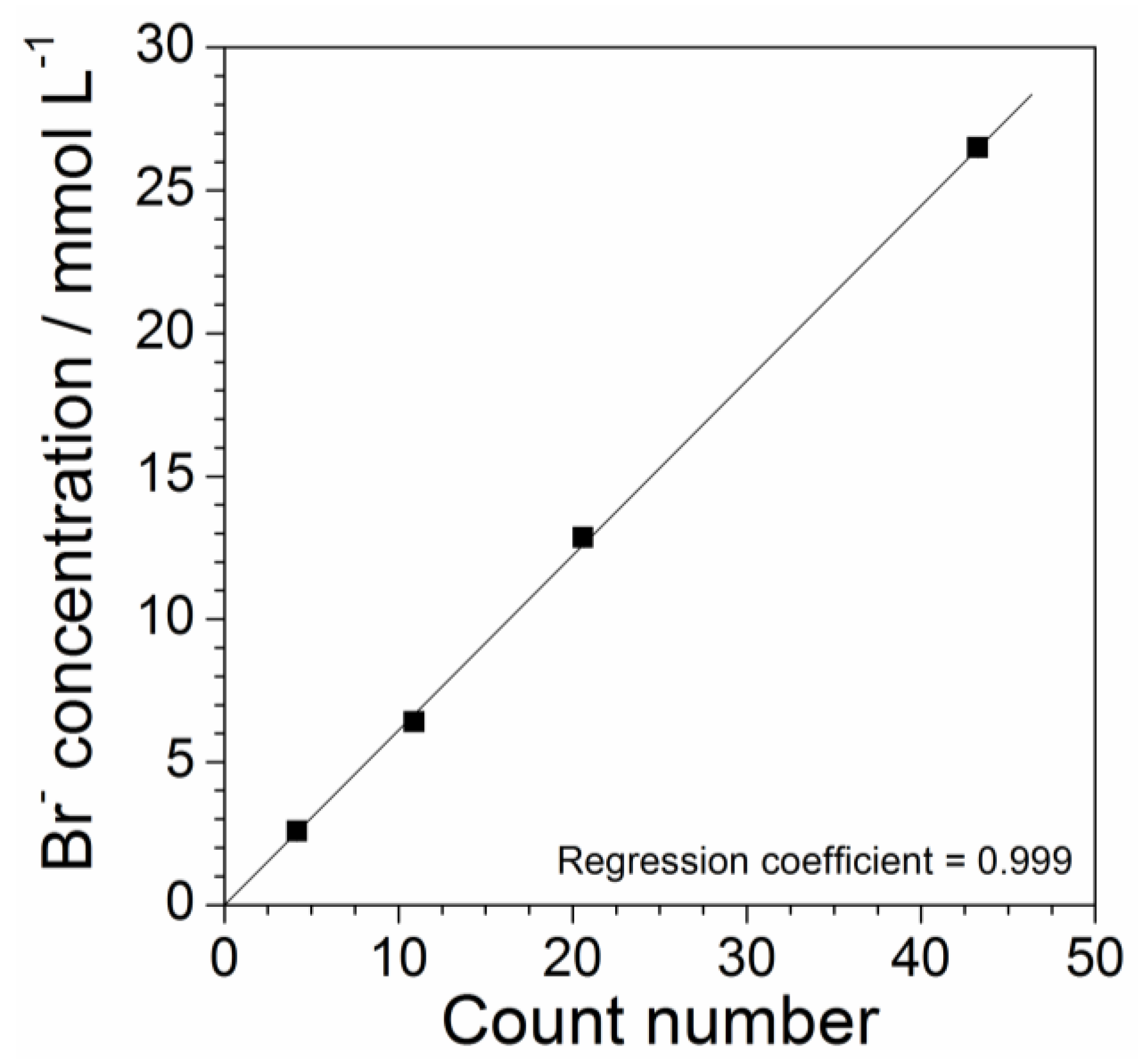
2.4. Determination of Precursor Content in Aqueous Phase
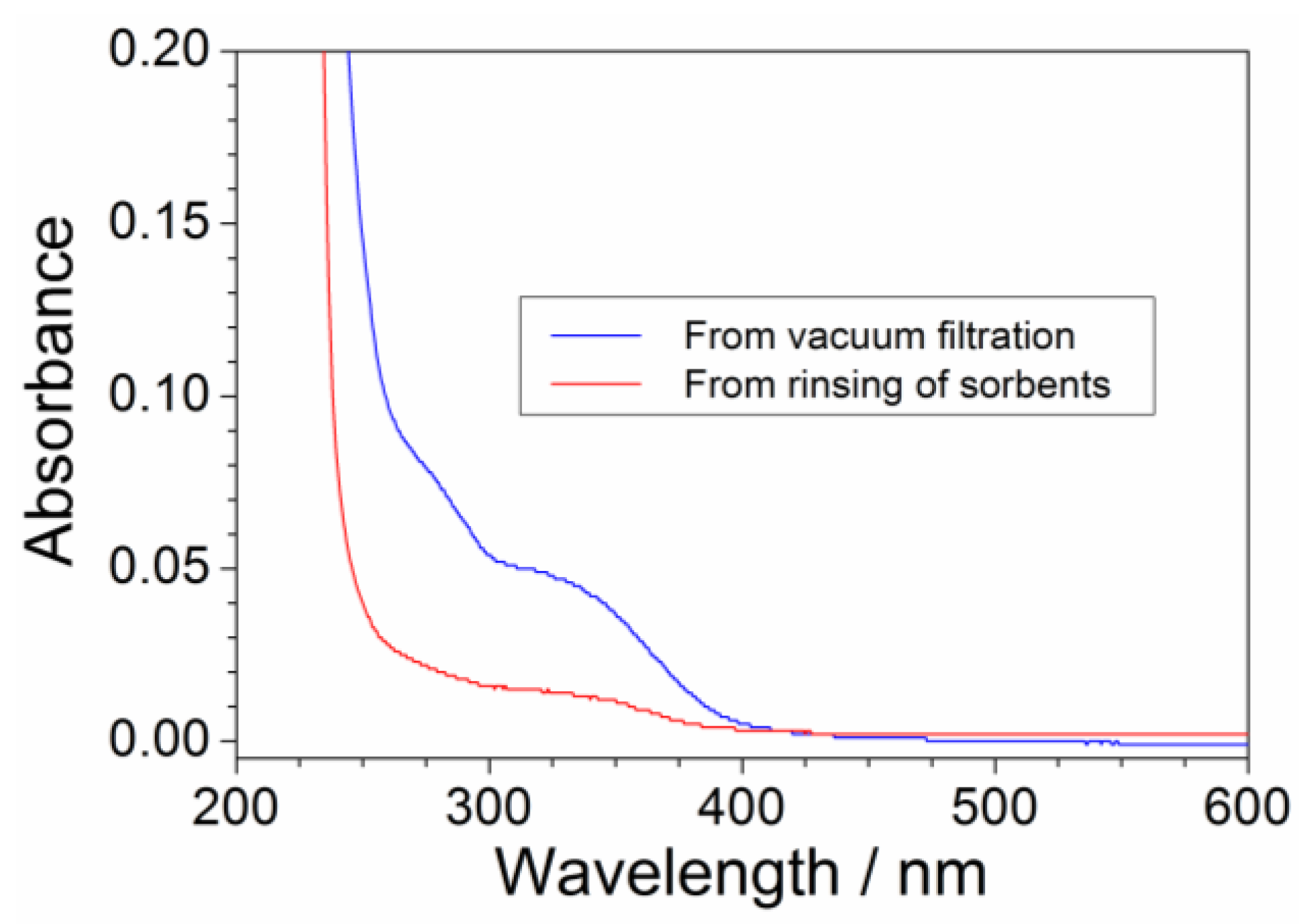
2.5. Efficiency of the Precursor Recover Process
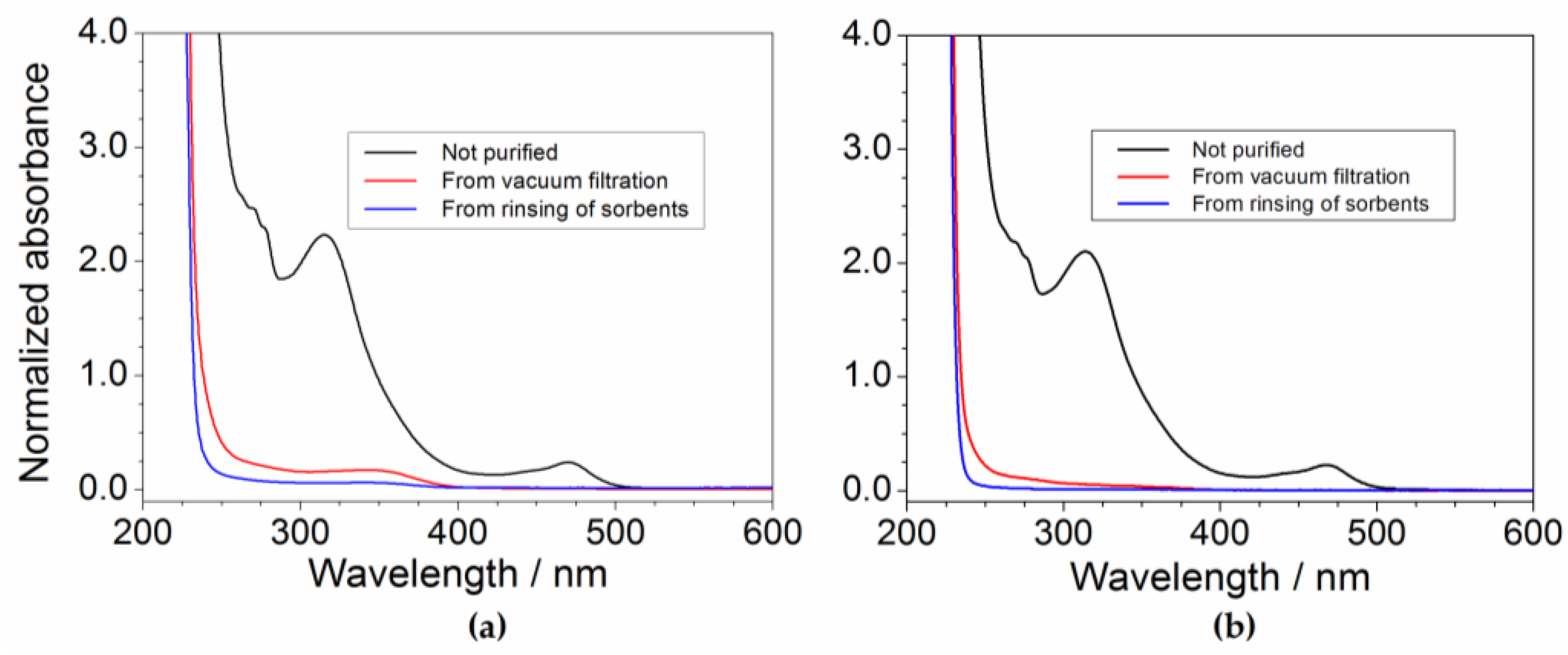
2.6. Determination of the Impurity Content in Purified Precursor
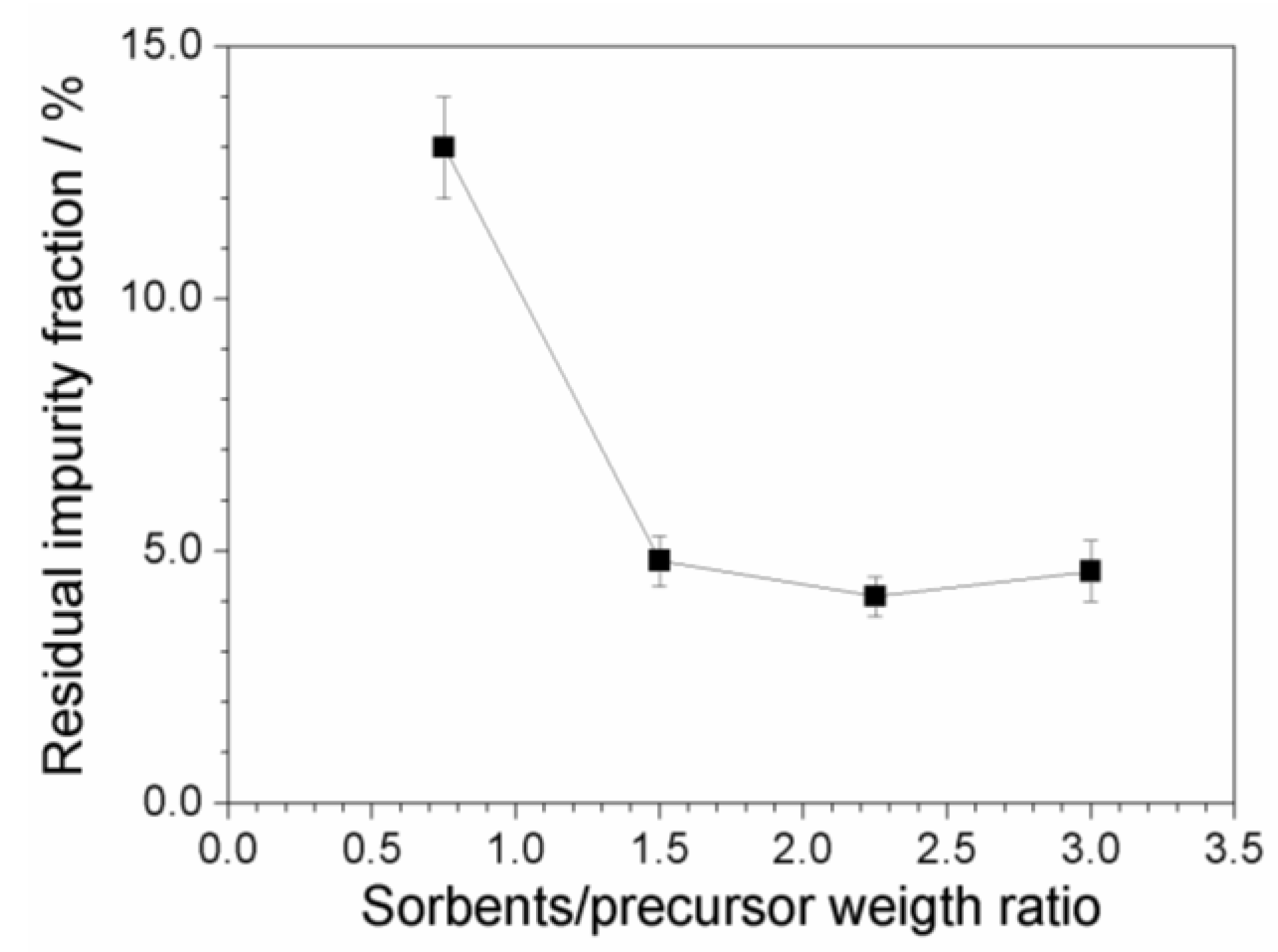
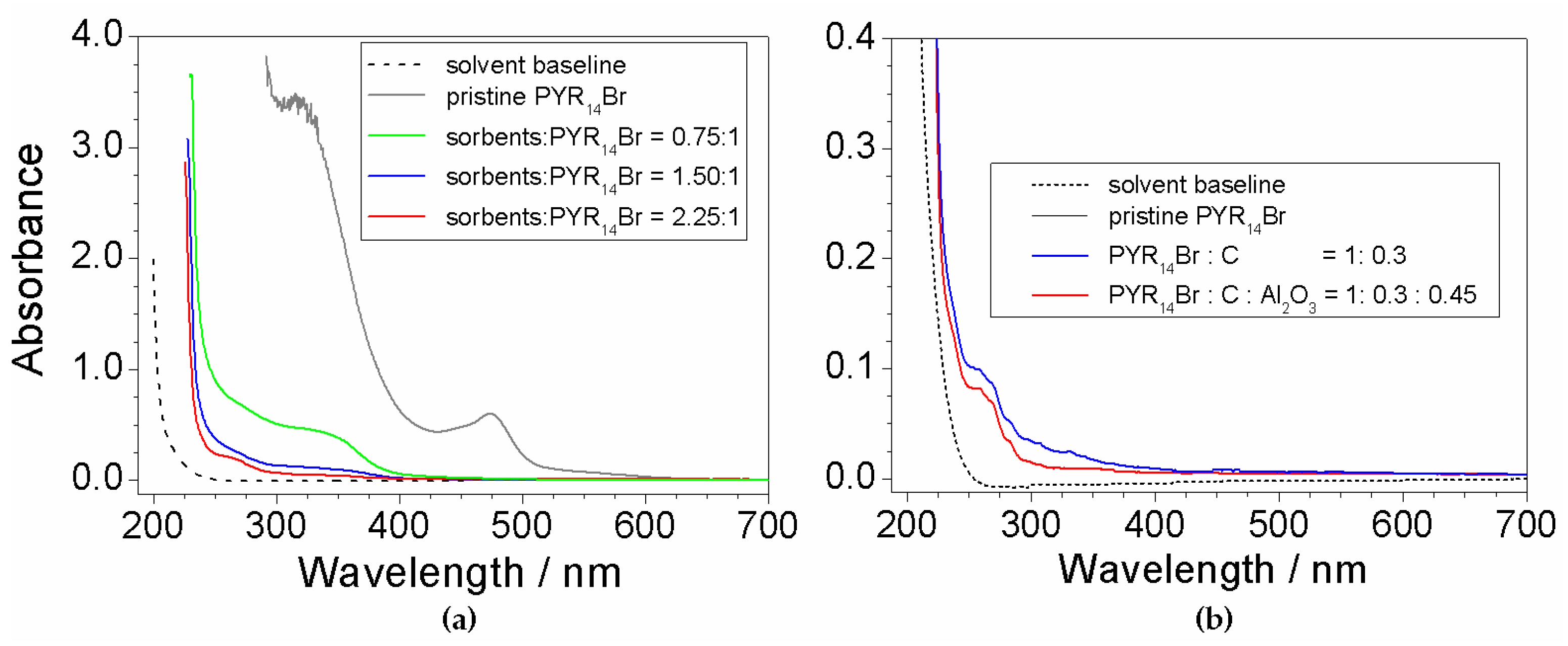
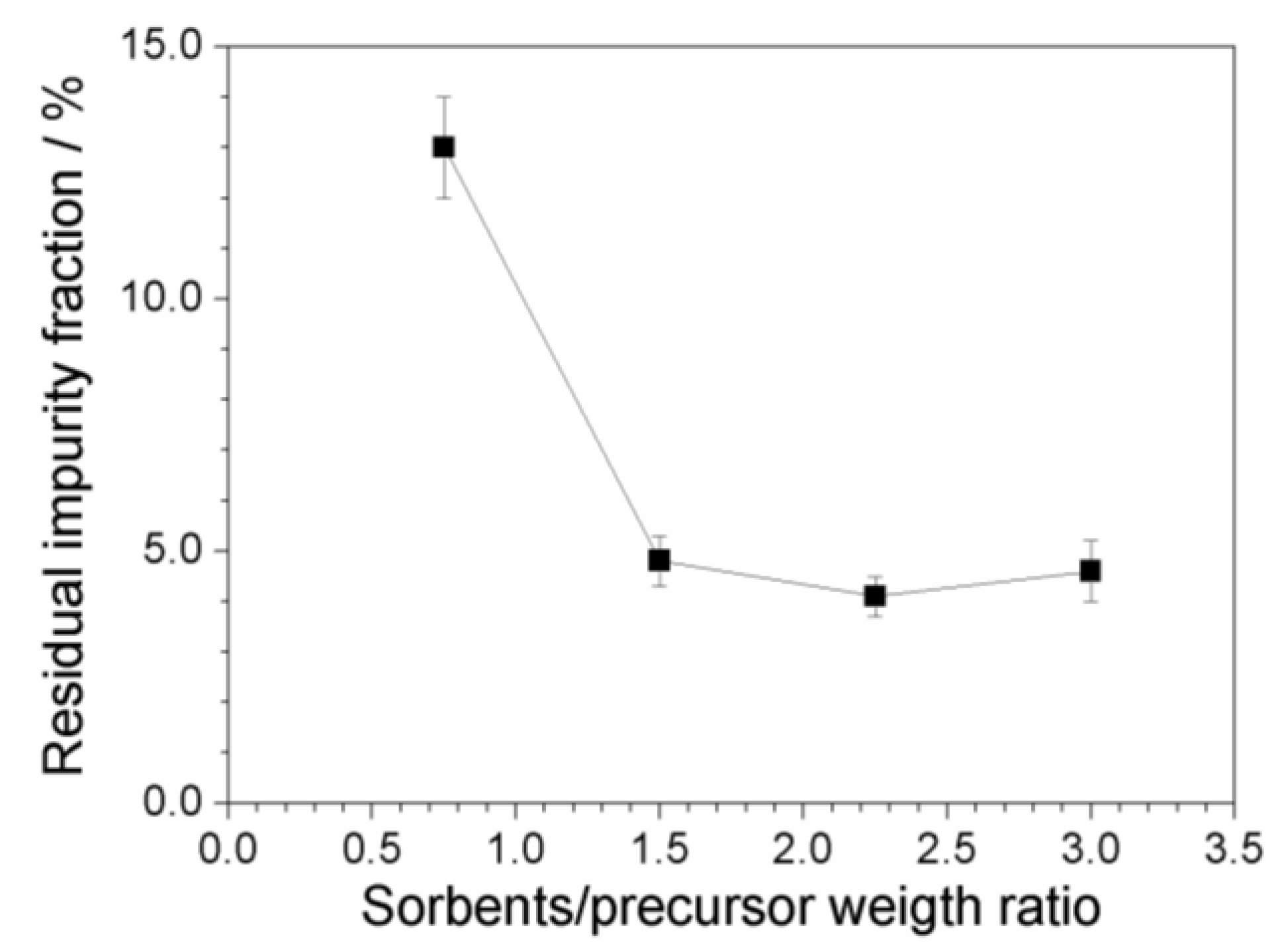
2.7. Investigation on Sorbents:Precursor Ratio
3. Materials and Methods
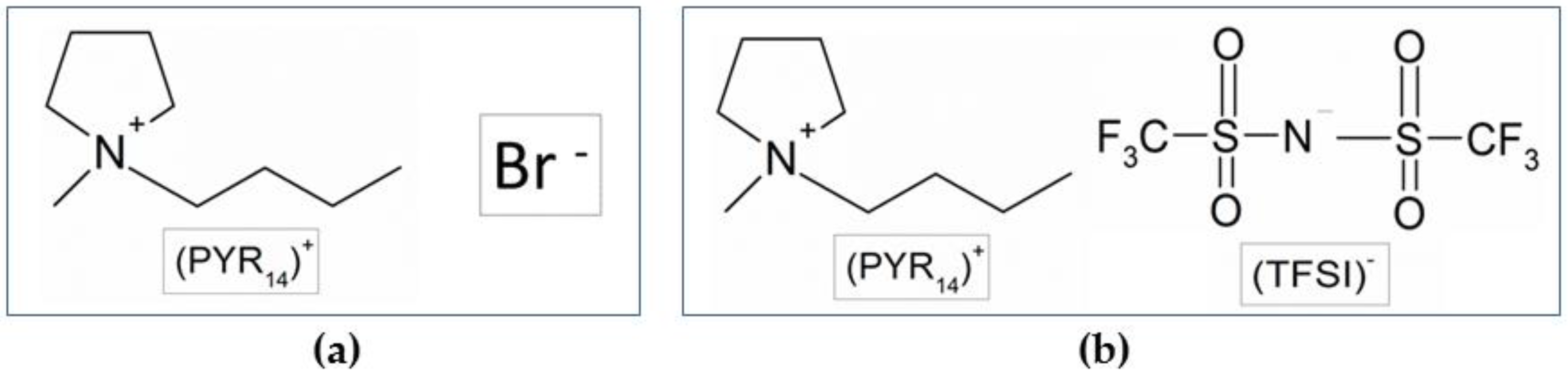
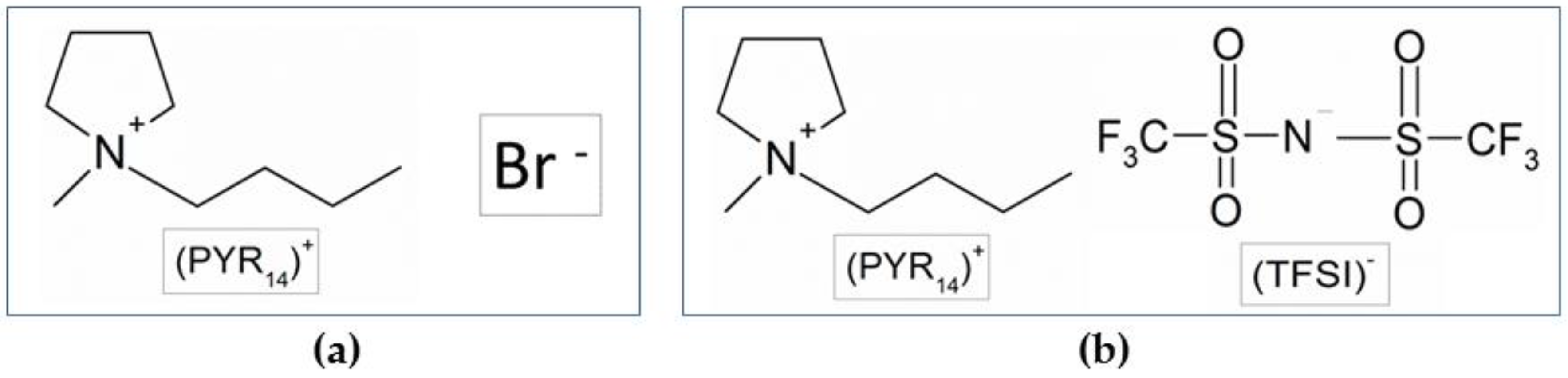
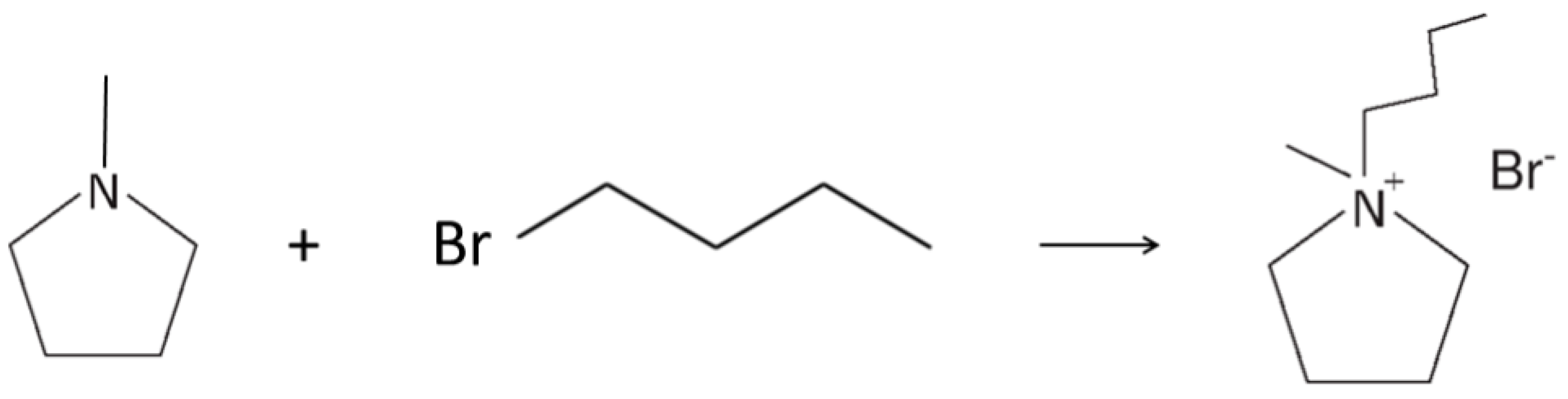
3.1. Preparation of the PYR14Br Precursor
3.2. Cleaning of Sorbent Materials
3.3. Purification of Precursor
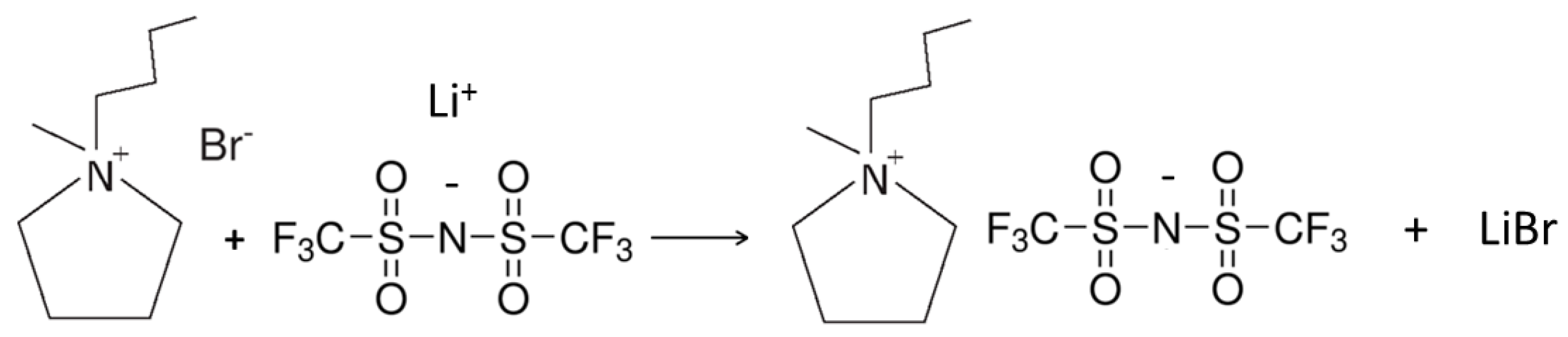
3.4. Preparation of the PYR14TFSI Ionic Liquid
3.5. Analytical Methods
3.6. Electrochemical Measurements
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rogers, J.R.D.; Seddon, K.R. Ionic Liquids: Industrial Application to Green Chemistry; ACS Symposium Series 818; American Chemical Society: Washington, DC, USA, 2002. [Google Scholar]
- Chiappe, C.; Pieraccini, D. Ionic liquids: Solvent properties and organic reactivity. J. Phys. Org. Chem. 2005, 18, 275–297. [Google Scholar] [CrossRef]
- Ohno, H. (Ed.) Electrochemical Aspects of Ionic Liquids; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Wasserscheid, P.; Keim, W. Ionic liquids—New “solutions” for transition metal catalysis. Angew. Chem. Int. Ed. 2000, 39, 3772–3789. [Google Scholar] [CrossRef]
- Earle, M.J.; Seddon, K.R. Ionic liquids. Green solvents for the future. Pure Appl. Chem. 2000, 72, 1391–1398. [Google Scholar] [CrossRef]
- Anderson, J.L.; Ding, J.; Welton, T.; Armstrong, D.W. Characterizing ionic liquids on the basis of multiple solvation interactions. J. Am. Chem. Soc. 2002, 124, 14247–14254. [Google Scholar] [CrossRef] [PubMed]
- Dupont, J.; de Souza, R.F.; Suarez, P.A.Z. Ionic liquid (molten salt) phase organometallic catalysis. Chem. Rev. 2002, 102, 3667–3692. [Google Scholar] [CrossRef] [PubMed]
- Pereiro, A.B.; Rodríguez, A. An ionic liquid proposed as solvent in aromatic hydrocarbon separation by liquid extraction. AIChE J. 2010, 56, 381–386. [Google Scholar] [CrossRef]
- Westerholt, A.; Liebert, V.; Gmehling, J. Influence of ionic liquids on the separation factor of three standard separation problems. Fluid Phase Equilibria 2009, 280, 56–60. [Google Scholar] [CrossRef]
- Domańska, U.; Redhi, G.G.; Marciniak, A. Activity coefficients at infinite dilution measurements for organic solutes and water in the ionic liquid 1-butyl-1-methylpyrrolidinium trifluoromethanesulfonate using GLC. Fluid Phase Equilibria 2009, 278, 97–102. [Google Scholar] [CrossRef]
- Marciniak, A.; Karczemna, E. Influence of cation structure on binary liquid–liquid equilibria for systems containing ionic liquids based on trifluoromethanesulfonate anion with hydrocarbons. J. Phys. Chem. B 2010, 114, 5470–5474. [Google Scholar] [CrossRef] [PubMed]
- Domańska, U.; Królikowski, M.; Ślesińska, K. Phase equilibria study of the binary systems (ionic liquid + thiophene): Desulphurization process. J. Chem. Thermodyn. 2009, 41, 1303–1311. [Google Scholar] [CrossRef]
- Lombardo, M.; Easwar, S.; Pasi, F.; Trombini, C.; Dhavale, D.D. Protonated arginine and lysine as catalysts for the direct asymmetric aldol reaction in ionic liquids. Tetrahedron 2008, 64, 9203–9207. [Google Scholar] [CrossRef]
- Huang, J.-F.; Baker, G.A.; Luo, H.; Hong, K.; Li, Q.F.; Bjerrum, N.J.; Dai, S. Brønsted acidic room temperature ionic liquids derived from N,N-dimethylformamide and similar protophilic amides. Green Chem. 2006, 8, 599–602. [Google Scholar] [CrossRef]
- Dzyuba, S.V.; Kollar, K.D.; Sabnis, S.S. Synthesis of imidazolium room-temperature ionic liquids. Exploring green chemistry and click chemistry paradigms in undergraduate organic chemistry laboratory. J. Chem. Educ. 2009, 86, 856–858. [Google Scholar] [CrossRef]
- Shen, X.-D.; Shi, L.-L.; Long, Z.; Zhou, X.-B.; Liang, D.-Q. Experimental study on the kinetic effect of N-butyl-N-methylpyrrolidinium bromide on CO2 hydrate. J. Mol. Liq. 2016, 223, 672–677. [Google Scholar] [CrossRef]
- Bhatt, A.I.; May, I.; Volkovich, V.A.; Hetherington, M.E.; Lewin, B.; Thied, R.C.; Ertok, N. Group 15 quaternary alkyl bistriflimides: Ionic liquids with potential application in electropositive metal deposition and as supporting electrolytes. J. Chem. Soc. Dalton Trans. 2002, 4532–4534. [Google Scholar] [CrossRef]
- Panozzo, S.; Armand, M.; Stephan, O. Light-emitting electrochemical cells using a molten delocalized salt. Appl. Phys. Lett. 2002, 80, 679–681. [Google Scholar] [CrossRef]
- Wang, P.; Zakeeruddin, S.M.; Exnar, I.; Gratzel, M. High efficiency dye-sensitized nanocrystalline solar cells based on ionic liquid polymer gel electrolyte. Chem. Commun. 2002, 2972–2973. [Google Scholar] [CrossRef]
- Fuller, J.; Breda, A.C.; Carlin, R.T. Ionic liquid-polymer gel electrolytes from hydrophilic and hydrophobic ionic liquids. J. Electroanal. Chem. 1998, 459, 29–34. [Google Scholar] [CrossRef]
- Nakagawa, H.; Izuchi, S.; Kunawa, K.; Nukuda, T.; Aihara, Y. Liquid and polymer gel electrolytes for lithium batteries composed of room-temperature molten salt doped by lithium salt. J. Electrochem. Soc. 2003, 150, A695–A700. [Google Scholar] [CrossRef]
- Noda, A.; Susan, M.A.B.H.; Kudo, K.; Mitsushima, S.; Hayamizu, K.; Watanabe, M. Brønsted acid-base ionic liquids as proton-conducting non aqueous electrolytes. J. Phys. Chem. B 2003, 107, 4024–4033. [Google Scholar] [CrossRef]
- Biso, M.; Mastragostino, M.; Montanino, M.; Passerini, S.; Soavi, F. Electropolymerization of poly(3-methylthiophene) in pyrrolidinium-based ionic liquids for hybrid supercapacitors. Electrochim. Acta 2008, 53, 7967–7971. [Google Scholar] [CrossRef]
- Bielawski, C.W.; Ruoff, R.S.; Agnihotri, D.K.; Dreryer, D.R.; Stoller, M.D.; Zhu, Y. Ionic Liquids for Use in Ultracapacitor and Graphene-Based Ultracapacitor. U.S. Patent, US 2011/0080689 A1, 7 April 2011. [Google Scholar]
- Appetecchi, G.B.; Montanino, M.; Passerini, S. Ionic liquid-based electrolytes for high-energy lithium batteries. In Ionic Liquids Science and Applications; ACS Symposium Series 1117; Visser, A.E., Bridges, N.J., Rogers, R.D., Eds.; Oxford University Press, Inc.: New York, NY, USA; American Chemical Society: Washington, DC, USA, 2013. [Google Scholar]
- Passerini, S.; Montanino, M.; Appetecchi, G.B. Lithium polymer batteries based on ionic liquids. In Polymers for Energy Storage and Conversion; Mittal, V., Ed.; John Wiley: Hoboken, NJ, USA; Scriverner Publishing: Salem, MA, USA, 2013. [Google Scholar]
- Li, L.; Wang, J.; Yang, P.; Guo, S.; Wang, H.; Yang, X.; Ma, X.; Yang, S.; Wu, B. Preparation and characterization of gel polymer electrolytes containing N-butyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide ionic liquid for lithium ion batteries. Electrochim. Acta 2013, 88, 147–156. [Google Scholar] [CrossRef]
- Scrosati, B.; Garche, J. Lithium batteries: Status, prospects and future. J. Power Sources 2010, 195, 2419–2430. [Google Scholar] [CrossRef]
- Spotnitz, R.; Franklin, J. Abuse behavior of high-power, lithium-ion cells. J. Power Sources 2003, 113, 81–100. [Google Scholar] [CrossRef]
- Yang, H.; Amiruddin, S.; Bang, H.J.; Sun, Y.K.; Prakash, J. A review of li-ion cell chemistries and their potential use as hybrid electric vehicles. J. Ind. Eng. Chem. 2006, 12, 12–38. [Google Scholar]
- Abraham, D.P.; Roth, E.P.; Kostecky, R.; McCarthy, K.; MacLaren, S.; Doughty, D.H. Diagnostic examination of thermally abused high-power lithium-ion cells. J. Power Sources 2006, 161, 648–657. [Google Scholar] [CrossRef]
- Bandhauer, T.M.; Garimella, S.; Fuller, T.F. A critical review of thermal issues in lithium-ion batteries. J. Electrochem. Soc. 2011, 158, R1–R25. [Google Scholar] [CrossRef]
- Appetecchi, G.B.; Scaccia, S.; Tizzani, C.; Alessandrini, F.; Passerini, S. Synthesis of hydrophobic ionic liquids for electrochemical applications. J. Electrochem. Soc. 2006, 153, A1685–A1691. [Google Scholar] [CrossRef]
- Passerini, S.; Appetecchi, G.B. Toward more environmentally friendly routes to high purity ionic liquids. MRS Bull. 2013, 38, 540–547. [Google Scholar] [CrossRef]
- Earle, M.J.; Gordon, C.M.; Plechkova, N.V.; Seddon, K.R.; Welton, W. Decolorization of ionic liquids for Spectroscopy. Anal. Chem. 2007, 79, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Clare, B.R.; Bayley, P.M.; Best, A.S.; Forsyth, M.; MacFarlane, D.R. Purification or contamination? The effect of sorbents on ionic liquids. Chem. Commun. 2008, 2689–2691. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.W.; Gourisankar, S.P.; Montchamp, J.-L.; Dzyuba, S.V. Silver-free synthesis of nitrate-containing room-temperature ionic liquids. New J. Chem. 2011, 35, 909–914. [Google Scholar] [CrossRef]
- Montanino, M.; Alessandrini, F.; Passerini, S.; Appetecchi, G.B. Water-based synthesis of hydrophobic ionic liquids for high-energy electrochemical devices. Electrochim. Acta 2013, 96, 124–133. [Google Scholar] [CrossRef]
- Henderson, W.A.; Passerini, S. Phase Behavior of Ionic Liquid− LiX Mixtures: Pyrrolidinium Cations and TFSI-Anions. Chem. Mater. 2004, 16, 2881–2885. [Google Scholar] [CrossRef]
- Randström, S.; Appetecchi, G.B.; Lagergren, C.; Moreno, A.; Passerini, S. The influence of air and its components on the cathodic stability of N-butyl-N-methylpyrrolidinium bis(trifluoromethansulfonyl) imide. Electrochim. Acta 2007, 53, 1837–1842. [Google Scholar] [CrossRef]
- Appetecchi, G.B.; Montanino, M.; Carewska, M.; Moreno, M.; Alessandrini, F.; Passerini, S. Chemical-physical properties of bis(perfluoroalkylsulfonyl)imide anion-based ionic liquids. Electrochim. Acta 2011, 56, 1300–1307. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dilution Ratio of the Aqueous Phase | Impurity Concentration of the Aqueous Phase (mg L−1) | Impurity Content with Respect to the Precursor Mass (ppm) |
|---|---|---|
| 0:1 * | 12,490 ± 70 | 16,020 ± 80 |
| 5:1 | 2500 ± 20 | 3200 ± 20 |
| 10:1 | 1249 ± 6 | 1602 ± 8 |
| 20:1 | 625 ± 3 | 801 ± 4 |
| 50:1 | 250 ± 2 | 320 ± 2 |
| 100:1 | 125 ± 1 | 160 ± 1 |
| 200:1 | 63 ± 1 | 80 ± 1 |
| 500:1 | 25 ± 1 | 32 ± 1 |
| 1000:1 | 13 ± 1 | 16 ± 1 |
| 5000:1 | 3 ± 1 | 3 ± 1 |
| PYR14Br:C:Al2O3 Weight Ratio | Recovered Fraction of PYR14Br (wt%) | |
|---|---|---|
| From Vacuum Filtration | From Sorbent Rinsing | |
| 1:0.30:0.45 | 91 ± 2 | 9 ± 2 |
| 1:0.60:0.90 | 83 ± 2 | 17 ± 2 |
| 1:0.90:1.35 | 77 ± 2 | 23 ± 2 |
| 1:1.20:1.80 | 69 ± 2 | 31 ± 2 |
| Batch | PYR14Br | C | Al2O3 |
|---|---|---|---|
| I | 1 | 0.30 | 0.45 |
| II | 1 | 0.60 | 0.90 |
| III | 1 | 0.90 | 1.35 |
| IV | 1 | 1.20 | 1.80 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Francesco, M.; Simonetti, E.; Gorgi, G.; Appetecchi, G.B. About the Purification Route of Ionic Liquid Precursors. Challenges 2017, 8, 11. https://doi.org/10.3390/challe8010011
De Francesco M, Simonetti E, Gorgi G, Appetecchi GB. About the Purification Route of Ionic Liquid Precursors. Challenges. 2017; 8(1):11. https://doi.org/10.3390/challe8010011
Chicago/Turabian StyleDe Francesco, Massimo, Elisabetta Simonetti, Gianremo Gorgi, and Giovanni Battista Appetecchi. 2017. "About the Purification Route of Ionic Liquid Precursors" Challenges 8, no. 1: 11. https://doi.org/10.3390/challe8010011
APA StyleDe Francesco, M., Simonetti, E., Gorgi, G., & Appetecchi, G. B. (2017). About the Purification Route of Ionic Liquid Precursors. Challenges, 8(1), 11. https://doi.org/10.3390/challe8010011