Abstract
Indane and phenylpropyl derivatives are interesting precursors for the synthesis of bioactive compounds, including those with antifungal or anti-inflammatory properties. In light of the increasing interest in the biocatalytic potential of marine-derived fungi, a study was conducted in which the substrates indene (1), indanone (2), 5-chloroindanone (2a), 1-phenylpropyl acetate (3), and 1-(4′-chlorophenyl)propyl acetate (3a) were biotransformed by the marine sediment-derived fungal strains Purpureocillium lilacinum BC17-2 and Emericellopsis maritima BC17. Fermentations led to the isolation of sixteen derivatives, which exhibited noteworthy stereoselectivities. The absolute configurations of the optically active indane and phenylpropyl derivatives isolated were determined through electronic circular dichroism and optical rotation dispersion computational calculations. Furthermore, given the known biocatalytic potential of the phytopathogenic fungus Botrytis cinerea to modify the structures of certain antifungal phenylpropyl derivatives, substrates 3 and 3a were also subjected to biotransformation by the strain B. cinerea UCA992. The antifungal activities of the biotransformation products (R)-5, (S)-6, syn-(1S,2R)-7, anti-(1R,2R)-7, (R)-8, (R)-9, threo-(1R,2R)-11, and erythro-(1R,2S)-11 were evaluated against B. cinerea UCA992 using a resazurin-based microdilution method.
1. Introduction
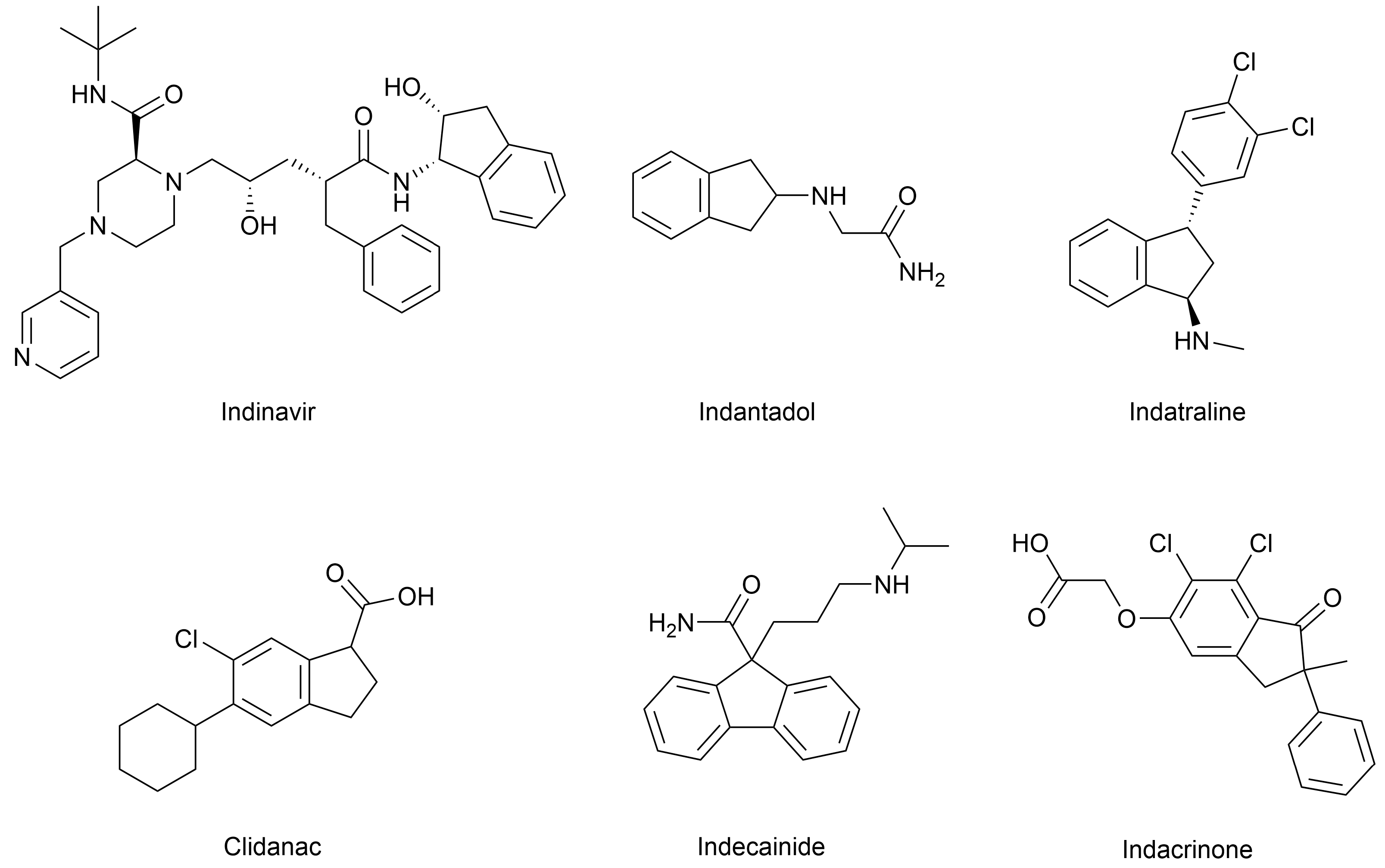
Optically active indane and phenylpropyl derivatives are interesting precursors for the synthesis of biologically active compounds. In particular, indane derivatives play a crucial role in the development of pharmaceutical compounds [1,2,3]. This relevance is largely due to the unique physicochemical properties of the indane (2,3-dihydro-1H-indene) ring system, which combines a rigid bicyclic structure with both aromatic and aliphatic characteristics. Such a framework offers considerable versatility for chemical modification, as it allows the introduction of substituents in multiple spatial orientations—up to six on the saturated ring and four on the aromatic moiety—thus offering ample opportunities for structural optimization in drug design [4]. For instance, indane-1,3-diones have demonstrated significant potential as α-glucosidase and urease inhibitors [5,6], while (1S,2R)-1-aminoindan-2-ol has been utilized in the synthesis of indinavir, a highly effective HIV protease inhibitor used in antiretroviral therapy [7,8] (Figure 1). Other indane-based compounds used in the clinic include indantadol, a potent monoamine oxidase (MAO) inhibitor; the amine uptake inhibitor indatraline; the anti-inflammatory agent clidanac; the antiarrhythmic drug indecainide; and the diuretic indacrinone, among others [4] (Figure 1).
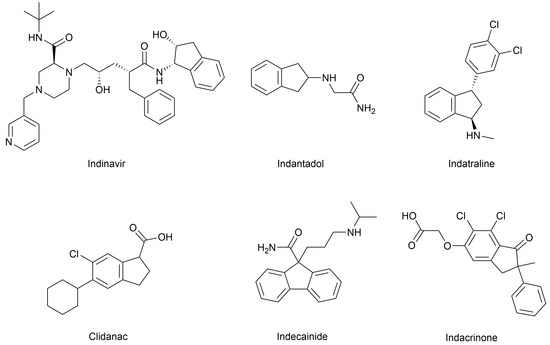
Figure 1.
Representative examples of biologically active indane-containing compounds reported in the literature [4].
Furthermore, recent studies have highlighted the high cytotoxic, antiproliferative, antioxidant, anti-inflammatory, and anti-metastatic activities of hybrid compounds containing the 2-arylideneindane-1,3-dione group [9].
Similarly, optically active phenylpropyl derivatives, such as chlorophenylpropanols, have shown remarkable antifungal properties against the phytopathogenic fungi Botrytis cinerea and Colletotrichum gloeosporoides [10,11], in addition to their application as fragrance ingredients [12].
In recent years, biocatalysis has gained significant recognition as a valuable tool in the chemical synthesis of innovative drug derivatives, agrochemicals, and fragrances [13]. This biological method, which involves the use of microorganisms or isolated enzymes, enables the catalysis of regio- and stereoselective reactions with substantial yields, providing an alternative to conventional synthetic methods. Moreover, this approach aligns closely with the principles of green chemistry, as it operates under mild and cost-effective conditions, such as aqueous media and physiological pH and temperature [14]. Marine microorganisms, such as yeasts, fungi, and bacteria, have been identified as a rich source of novel enzymes that may exhibit characteristics such as high salt tolerance, barophilicity, hyperthermostability, and cold adaptability, making them promising and valuable biocatalysts [15,16,17]. The recent isolation of strains P. lilacinum BC17-2 and E. maritima BC17 from sediments of the Bay of Cádiz provides a valuable opportunity to explore their biotechnological potential, a field that has been scarcely evaluated. To date, studies have primarily focused on investigating the potential of the Emericellopsis genus to biodegrade lignocellulose and aflatoxin B1 [18,19,20]. Furthermore, recent reports have highlighted the ability of P. lilacinum to biodegrade plastics and solid waste from the leather industry [21,22,23,24]. Additionally, our research group has recently demonstrated the ability of both strains to biotransform thiochroman derivatives with notable results [25].
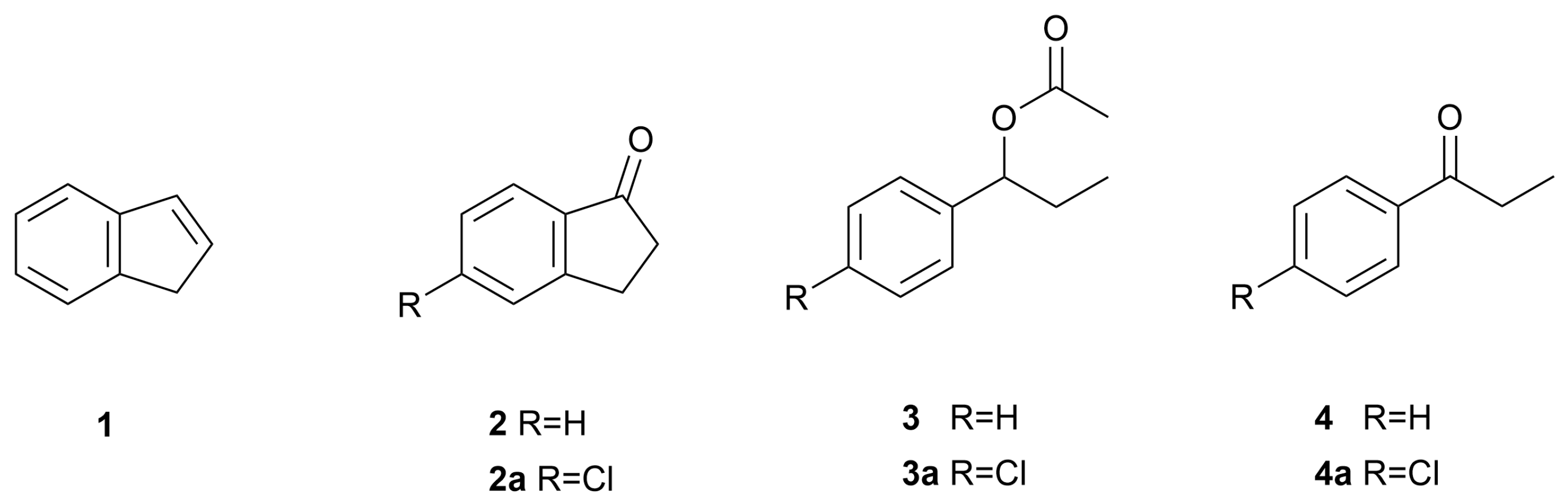
As part of our ongoing research on the biocatalytic potential of both strains, this study reports the biotransformation of the substrates indene (1), indanone (2), 5-chloroindanone (2a), (±)-1-phenylpropyl acetate (3), and (±)-1-(4′-chlorophenyl)propyl acetate (3a) by the marine-derived fungi P. lilacinum BC17-2 and E. maritima BC17 (Figure 2). Given the known biocatalytic potential of B. cinerea to transform the structures of certain antifungal phenylpropyl derivatives [26], substrates 3 and 3a were also subjected to biotransformation by the strain B. cinerea UCA992. Moreover, considering the documented antifungal activity of chlorinated derivatives of these xenobiotics against B. cinerea [11,27], the antifungal activity of the non-chlorinated biotransformation products (R)-5, (S)-6, syn-(1S,2R)-7, anti-(1R,2R)-7, (R)-8, (R)-9, threo-(1R,2R)-11, and erythro-(1R,2S)-11 was evaluated against B. cinerea UCA992 using the resazurin-based microdilution method.
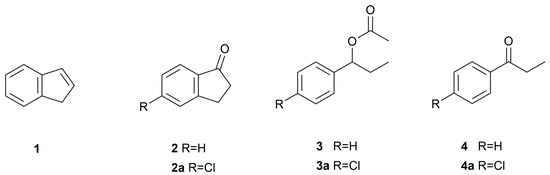
Figure 2.
Precursor and xenobiotic substrates subjected to biotransformation by the marine-derived strains P. lilacinum BC17-2 and E. maritima BC17.
2. Materials and Methods
2.1. General Experimental Procedures
Optical rotations were determined using a JASCO P-2000 polarimeter (JASCO Corporation, Tokyo, Japan). 1H and 13C NMR measurements were performed on Agilent 400, 500, and 600 MHz (Agilent Technologies, Santa Clara, CA, USA) and Bruker 400, 500, and 700 MHz NMR spectrometers (Bruker Corporation, Billerica, MA, USA) using SiMe4 as the internal reference. Chemical shifts are expressed in ppm (δ) and referenced to CDCl3 (Eurisotop, Saint-Aubiu, France, δH 7.25, δC 77.0) and CD3OD (Eurisotop, Saint-Aubiu, France, δH 3.30, δC 49.0). TLC was performed on Merck Kiesegel 60 F254, 0.2 mm thick (Darmstadt, Germany). Silica gel (60–200 µm) (Merck Group, Darmstadt, Germany) was used for column chromatography. Purifications by means of HPLC were performed on a Hitachi HPLC system equipped with a UV-VIS detector (Hitachi-Primaide 1410) and a differential refractometer detector (Hitachi-Chromaster RI-5450) (Hitachi High-Technologies Corporation, Tokyo, Japan), employing an ACE 5 Sil column (4.6 × 250 mm, 5 µm) (Advanced Chromatography Technologies, Aberdeen, Scotland, UK) with isocratic elution of n-hexane/ethyl acetate and chloroform/methanol mixtures as eluents. Diastereoisomeric (de) and enantiomeric excesses (ee) were determined on a Hitachi Chromaster HPLC system equipped with a Diode Array Detector (Hitachi-Chromaster 5430) and a column oven (Hitachi-Chromaster 5310) (Hitachi High-Technologies Corporation, Tokyo, Japan) using Chiralcel OD (4.6 × 250 mm, 10 µm) (Daicel Corporation, Osaka, Japan) and Chiralcel IB N-5 (4.6 × 250 mm, 5 µm) (Daicel Corporation, Osaka, Japan) chiral columns, with n-hexane/i-PrOH mixtures as eluent. Antifungal assays were performed using a Multiskan GO microplate spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
2.2. Reagents and Solvents
All solvents used were of HPLC grade (Panreac, Barcelona, Spain). Indanone (2), 5-chloroindanone (2a), propiophenone (4), and 4′-chloropropiophenone (4a) were provided by Sigma-Aldrich (Darmstadt, Germany), while indene (1) and NaBH4 were obtained from Thermo Fisher Scientific Chemicals (Waltham, MA, USA). Microbial growth media were provided by Condalab S.L. (Madrid, Spain).
2.3. Microorganisms
The marine-derived fungal strains Purpureocillium lilacinum BC17-2 and Emericellopsis maritima BC17 were isolated from sediments collected in the inner Cádiz Bay (Spain). The Botrytis cinerea UCA992 strain was isolated from Domecq vineyard grapes in Jerez de la Frontera, Cádiz, Spain. The fungal strains were previously isolated and identified as pure cultures using both morphological and molecular techniques, including ITS, 28S rRNA, and β-tubulin gene sequencing [25,28]. All fungal strains used in this study are preserved in the Mycological Herbarium Collection of the University of Cádiz. Spore suspensions of these strains are maintained as viable at −40 °C in 80% glycerol.
2.4. Synthesis of (±)-1-Phenylpropyl Acetate (3) and (±)-1-(4′-Chlorophenyl)propyl Acetate (3a)
Compounds propiophenone (4) (2 g, 0.015 mol) and 4′-chloropropiophenone (4a) (2 g, 0.012 mol) were treated with NaBH4 (1 g, 0.026 mol) in methylene chloride/methanol 1:1 (200 mL). The reaction mixtures were stirred for 24 h at room temperature. Upon completion, the solvents were removed under reduced pressure, and the resulting crudes were neutralized with 10% aqueous HCl and extracted with ethyl acetate. The organic layers were dried over anhydrous Na2SO4 and the solvents were evaporated using a rotary evaporator. The reaction crudes were chromatographed on a silica gel column, using hexane-ethyl acetate mixtures as eluent, yielding (±)-1-phenylpropan-1-ol (9) (1940 mg, 0.015 mol, 95% yield) and (±)-1-(4′-chlorophenyl)propan-1-ol (9a) (1980 mg, 0.012 mol, 97% yield), respectively. The 1H NMR spectra of the obtained products were consistent with those previously reported in the literature [11,29].
Compounds 9 (1940 mg, 0.015 mol) and 9a (1980 mg, 0.012 mol) were dissolved in a catalytic amount of dry pyridine, and acetic anhydride (50 mL) was added dropwise. The reaction mixtures were stirred for 24 h. The solvents were removed, and the crude reaction products were chromatographed to afford (±)-1-phenylpropyl acetate (3) (1649 mg, 9.3 mmol, 65% yield) and (±)-1-(4′-chlorophenyl)propyl acetate (3a) (1850 mg, 8.7 mmol, 75% yield), respectively. The 1H NMR spectra of the obtained compounds were consistent with those previously reported in the bibliography [11,30].
2.5. General Biotransformation Method
P. lilacinum BC17-2 and E. maritima BC17 were cultured either under static conditions in 1 L Roux bottles or shaking at 220 rpm in 500 mL Erlenmeyer flask, each containing 150 mL of Potato Dextrose Broth (PDB, Condalab S. L., Madrid, Spain). B. cinerea UCA992 was grown in 1 L Roux bottles containing 150 mL of Czapek-Dox modified medium (50 g/L glucose, 1 g/L yeast extract, 5 g/L K2HPO4, 2 g/L NaNO3, 0.5 g/L MgSO4⋅7H2O, and 0.01 g/L FeSO4⋅7H2O). Each flask was inoculated with fresh conidial suspensions to a final concentration of 1 × 106 conidia/mL and incubated at 25 °C under white light (daylight lamp).
After 2 to 6 days, 200 µL of an ethanol solution containing 22.5 mg of the xenobiotic substrates 1, 2, 2a, 3, or 3a (112.5 mg/mL) was added to each flask, achieving a final concentration of 150 ppm. Cultures were then incubated under the conditions described above for a further 3 to 12 days. The negative control consisted of the sterile medium with the substrate at the same concentration.
Once the biotransformation of each substrate was completed, the broth was filtered and extracted three times with ethyl acetate. The crude extract was dried over anhydrous Na2SO4, and the solvent was evaporated. The residue was chromatographed on a silica gel column using a solvent gradient from 100% hexane to 100% ethyl acetate, with 10% incremental increases in ethyl acetate content.
2.6. Biotransformation of Indene (1) by Purpureocillium lilacinum BC17-2
In accordance with the above procedure, thirty Erlenmeyer flasks, subcultured with the strain BC17-2 under shaking conditions, were fed with an EtOH solution of indene (1) on day 4 and then incubated for an additional 8 days. Extraction and purification of the culture broth, as outlined in the general method, yielded (R)-(−)-2-hydroxyindanone ((R)-5) (1.0 mg), (R)-(−)-3-hydroxyindanone ((R)-6) (1.3 mg), and syn-(1S,2R)-(−)-indane-1,2-diol (syn-(1S,2R)-7) (5.7 mg).
(R)-(−)-2-Hydroxyindanone ((R)-5). Colorless oil. − 1.84 (c 0.1, CHCl3), 43.0% ee. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA of 99:1, flow rate of 0.8 mL/min, tR of 46.3 min (R) and 48.8 min (S) (Figure S1).
(R)-(−)-3-Hydroxyindanone ((R)-6). Colorless oil. − 88.50 (c 0.05, CHCl3), 71.9% ee. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA 93:7, flow rate of 0.8 mL/min, tR of 17.2 min (S) and 19.9 min (R) (Figure S2).
syn-(1S,2R)-(−)-indane-1,2-diol (syn-(1S,2R)-7). Colorless oil. − 55.57 (c 0.15, CHCl3), 98.2% ee, 100% de. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA 95:5, flow rate of 0.8 mL/min, tR of 26.7 min (1R,2S), and 29.1 min (1S,2R) (Figure S3).
2.7. Biotransformation of Indene (1) by Emericellopsis maritima BC17
Thirty Erlenmeyer flasks, subcultured with E. maritima BC17 under shaking conditions, were fed with an EtOH solution of indene (1) on the sixth day. Extraction and purification of the broth 10 days post-feeding, as described in the general method, afforded (R)-(−)-2-hydroxyindanone ((R)-5) (0.5 mg) { − 0.56 (c 0.03, CHCl3), 4.3% ee} (Figure S1), (S)-(+)-3-hydroxyindanone ((S)-6) (1.2 mg) { + 60.71 (c 0.08, CHCl3), 47.1% ee} (Figure S2), syn-(1S,2R)-(−)-indane-1,2-diol (syn-(1S,2R)-7) (2.9 mg) { − 21.95 (c 0.23, CHCl3), 59.4% ee, 45.0% de} (Figure S3), and anti-(1R,2R)-(−)-indane-1,2-diol (anti-(1R,2R)-7) (1.1 mg).
anti-(1R,2R)-(−)-Indane-1,2-diol (anti-(1R,2R)-7). Colorless oil. − 7.45 (c 0.04, CHCl3), 68.4% ee. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA 96.5:3.5, flow rate of 0.8 mL/min, tR of 49.4 min (1R,2R), and 55.1 min (1S,2S) (Figure S4).
2.8. Biotransformation of Indanone (2) by Purpureocillium lilacinum BC17-2
Fourteen Roux bottles, subcultured with P. lilacinum BC12-2 under static conditions, were fed with an EtOH solution of indanone (2) on day 4 to a final concentration of 150 ppm. Extraction and purification of the broth 3 days post-feeding, as described in the general method, gave (S)-(+)-3-hydroxyindanone ((S)-6) (80.8 mg) { + 1.12 (c 8.1, CHCl3), 3.8% ee} (Figure S5) and indanol (8) (0.2 mg). In addition, 136.53 mg of unconsumed indanone (2) was recovered.
2.9. Biotransformation of Indanone (2) by Emericellopsis maritima BC17
Twenty Roux bottles, subcultured with the strain BC17 under static conditions, were fed with an EtOH solution of indanone (2) on day 3 and then incubated for an additional 12 days. Extraction and purification of the culture broth, as outlined in the general method, afforded (S)-(+)-3-hydroxyindanone ((S)-6) (4.3 mg) { + 36.07 (c 0.03, CHCl3), 43.6% ee} (Figure S5) and (R)-(−)-indanol ((R)-8) (0.8 mg). Additionally, 334.3 mg of unconsumed indanone (2) was recovered.
(R)-(−)-Indanol ((R)-8). Colorless oil. − 1.11 (c 0.05, CHCl3), 2.1% ee. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA 95:5, flow rate of 0.8 mL/min, tR of 13.3 min (S) and 14.6 min (R) (Figure S6).
2.10. Biotransformation of 5-Chloroindanone (2a) by Purpureocillium lilacinum BC17-2
Fourteen Roux bottles, subcultured with P. lilacinum BC12-2 under static conditions, were fed with an EtOH solution of 5-chloroindanone (2a) on day 4 to a final concentration of 150 ppm. Extraction and purification of the broth 3 days post-feeding, as described in the general method, gave (R)-(−)-5-chloro-3-hydroxyindanone ((R)-6a) (15.8 mg), (1R,2R)-anti-5-chloroindane-1,2-diol (anti-(1R,2R)-7a) (0.7 mg), and 5-chloroindanol (8a) (0.4 mg). In addition, 16.2 mg of unconsumed 5-chloroindanone (2a) was recovered.
(R)-(−)-5-Chloro-3-hydroxyindanone ((R)-6a). White solid. − 0.3 (c 0.11, CHCl3), 4.1% ee. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA 96:4, flow rate of 0.8 mL/min, tR of 29.6 min (S) and 31.1 min (R) (Figure S7).
(1R,2R)-(−)-anti-5-Chloroindane-1,2-diol (anti-(1R,2R)-7a). White solid. − 21.8 (c 0.10, CHCl3), 85.1% ee. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA 97:3, flow rate of 0.8 mL/min, tR of 38.1 min (minor) and 40.3 min (major) (Figure S8).
2.11. Biotransformation of 5-Chloroindanone (2a) by Emericellopsis maritima BC17
Twenty Roux bottles, subcultured with the strain BC17 under static conditions, were fed with an EtOH solution of 5-chloroindanone (2a) on day 4 and then incubated for an additional 3 days. Extraction and purification of the culture broth, as outlined in the general method, afforded (S)-(+)-5-chloro-3-hydroxyindanone ((S)-6a) (2.1 mg) { + 11.9 (c 0.12, CHCl3), 9.8% ee} (Figure S7) and (R)-(−)-5-chloroindanol ((R)-8a) (2.2 mg). In addition, 386.3 mg of unconsumed 2a was recovered.
(R)-(−)-5-Chloroindanol ((R)-8a). White solid. − 1.39 (c 0.07, CHCl3), 2.1% ee. HPLC conditions: Chiralcel OD, Daicel, Japan, hexane/IPA 95:5, flow rate of 0.8 mL/min, tR of 11.7 min (S) and 12.9 min (R) (Figure S9).
2.12. Biotransformation of (±)-1-Phenylpropyl Acetate (3) by Purpureocillium lilacinum BC17-2
Twenty Roux bottles, subcultured with P. lilacinum BC12-2 under static conditions, were fed with an EtOH solution of (±)-1-phenylpropyl acetate (3) on day 3 to a final concentration of 150 ppm. Extraction and purification of the broth after 7 days post-feeding, as described in the general method, gave (S)-(−)-1-phenylpropan-1-ol ((S)-9) (67.1 mg), (R)-(+)-1-phenylpropane-1,3-diol ((R)-10) (3.2 mg), (1R,2R)-(−)-threo-1-phenylpropane-1,2-diol (threo-(1R,2R)-11) (0.8 mg), and (1R,2S)-(−)-erythro-1-phenylpropane-1,2-diol (erythro-(1R,2S)-11) (0.5 mg).
(S)-(−)-1-Phenylpropan-1-ol ((S)-9). Colorless oil. − 1.35 (c 3.24, CHCl3), 1.2% ee. HPLC conditions: Chiralcel OD, Daicel, Japan, hexane/IPA of 99:1, flow rate of 0.8 mL/min, tR of 32.9 min (R) and 42.8 min (S) (Figure S10).
(R)-(+)-1-Phenylpropane-1,3-diol ((R)-10). Colorless oil. + 22.13 (c 0.22, CHCl3), 25.3% ee. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA 95:5, flow rate of 0.8 mL/min, tR of 32.0min (R) and 34.4 min (S) (Figure S11).
(1R,2R)-(−)-threo-1-Phenylpropane-1,2-diol (threo-(1R,2R)-11). Colorless oil. − 23.34 (c 0.02, CHCl3), 52.4% ee. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA 93:7, flow rate of 0.8 mL/min, tR of 14.8 min (1S,2S), and 15.5 min (1R,2R) (Figure S12).
(1R,2S)-(−)-erythro-1-Phenylpropane-1,2-diol (erythro-(1R,2S)-11). Colorless oil. − 6.34 (c 0.09, CHCl3), 28.8% ee, 23.1% de. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA 93:7, flow rate of 0.8 mL/min, tR of 20.2 min (1R,2R) and 21.6 min (1S,2S) (Figure S13).
2.13. Biotransformation of (±)-1-Phenylpropyl Acetate (3) by Emericellopsis maritima BC17
Twenty Roux bottles, subcultured with the strain BC17 under static conditions, were fed with an EtOH solution of (±)-1-phenylpropyl acetate (3) on day 5 and then incubated for an additional 10 days. Extraction and purification of the culture broth, as outlined in the general method, afforded (R)-(+)-phenylpropan-1-ol ((R)-9) (15.2 mg) { + 25.41 (c 1.52, CHCl3), 38.3% ee} (Figure S10). In addition, 98.7 mg of unconsumed 3 was recovered.
2.14. Biotransformation of (±)-1-Phenylpropyl Acetate (3) by Botrytis cinerea UCA992
Twenty Roux bottles, subcultured with B. cinerea UCA992 under static conditions, were fed with an EtOH solution of (±)-1-phenylpropyl acetate (3) on day 2 to a final concentration of 150 ppm. Extraction and purification of the broth after 6 days post-feeding, as described in the general method, gave (R)-(+)-1-phenylpropan-1-ol ((R)-9) (70.5 mg) { + 19.07 (c 7.05, CHCl3), 17.4% ee} (Figure S10), (R)-(+)-1-phenylpropane-1,3-diol ((R)-10) (2.4 mg) { + 12.6 (c 0.10, CHCl3), 5.4% ee} (Figure S11), (1R,2R)-(−)-threo-1-phenylpropane-1,2-diol (threo-(1R,2R)-11) (1.2 mg) { − 24.31 (c 0.04, CHCl3) (Figure S12), 49.5% ee}, and (±)-erythro-1-phenylpropane-1,2-diol (erythro-(1S,2R)-11) (2.8 mg) { + 0.4 (c 0.09, CHCl3), 0.8% ee, 40% de} (Figure S13).
2.15. Biotransformation of (±)-1-(4′-Chlorophenyl)propyl Acetate (3a) by Purpureocillium lilacinum BC17-2
Twenty Roux bottles, subcultured with the strain BC17-2 under static conditions, were fed with an EtOH solution of (±)-1-(4′-chlorophenyl)propyl acetate (3a) on day 3 and then incubated for an additional 7 days. Extraction and purification of the culture broth, as outlined in the general method, afforded (R)-(+)-1-(4′-chlorophenyl)propan-1-ol ((R)-9a) (80.7 mg), (R)-(+)-1-(4′-chlorophenyl)propan-1,3-diol ((R)-10a) (3.5 mg), (1S,2S)-(+)-threo-1-(4′-chlorophenyl)propan-1,2-diol (threo-(1S,2S)-11a) (13.0 mg), and (1S,2R)-(+)-erythro-1-(4′-chlorophenyl)propan-1,2-diol (erythro-(1S,2R)-11a) (26.6 mg).
(R)-(+)-1-(4′-Chlorophenyl)propan-1-ol ((R)-9a). Colorless oil. + 2.46 (c 6.94, CHCl3), 7.9% ee. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA of 99:1, flow rate of 0.8 mL/min, tR of 41.04 min (S), and 43.23 min (R) (Figure S14).
(R)-(+)-1-(4′-Chlorophenyl)propan-1,3-diol ((R)-10a). Colorless oil. + 2.65 (c 0.30, CHCl3), 6.6% ee. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, Hex/EtOAc 92:8, flow rate of 0.8 mL/min, tR of 51.5 min (R) and 58.5 min (S) (Figure S15).
(1S,2S)-(+)-threo-1-(4′-Chlorophenyl)propan-1,2-diol (threo-(1S,2S)-11a). Colorless oil. + 13.00 (c 0.11, CHCl3), 88.5% ee (HPLC: Chiralcel IB N-5, Daicel, Japan, hexane/IPA 96:4, flow rate of 0.6 mL/min, tR of 29.7 min (1S,2S) and 32.1 min (1R,1R) (Figure S16).
(1S,2R)-(+)-erythro-1-(4′-Chlorophenyl)propan-1,2-diol (erythro-(1S,2R)-11a). Colorless oil. + 27.43 (c 0.21, CHCl3), 91.2% ee, 34.2% de. HPLC conditions: Chiralcel IB N-5, Daicel, Japan, hexane/IPA 96:4, flow rate of 0.6 mL/min, tR of 31.1 min (1S,2R) and 34.0 min (1R,2S) (Figure S17).
2.16. Biotransformation of (±)-1-(4′-Chlorophenyl)propyl Acetate (3a) by Emericellopsis maritima BC17
Twenty Roux bottles, subcultured with E. maritima BC17 under static conditions, were fed on day 5 with an EtOH solution of (±)-1-(4′-chlorophenyl)propyl acetate (3a) to a final concentration of 150 ppm. Extraction and purification of the broth after 10 days post-feeding, as described in the general method, gave (R)-(+)-1-(4′-chlorophenyl)propan-1-ol ((R)-9a) (11.5 mg) {+5.53 (c 0.30, CHCl3), 9.5% ee}. In addition, 126.8 mg of unconsumed 3a was recovered.
2.17. Biotransformation of (±)-1-(4′-Chlorophenyl)propyl Acetate (3a) by Botrytis cinerea UCA992
Twenty Roux bottles, subcultured with B. cinerea UCA992 under static conditions, were fed on day 2 with an EtOH solution of 3a to a final concentration of 150 ppm. Extraction and purification of the broth after 6 days post-feeding, as described in the general method, gave (S)-(−)-1-(4′-chlorophenyl)propan-1-ol ((S)-9a) (71.5 mg) { − 1.00 (c 0.70, CHCl3), 0.7% ee} (Figure S14), (S)-(−)-1-(4′-chlorophenyl)propan-1,3-diol ((S)-10a) (20.9 mg) { − 26.41 (c 0.20, CHCl3), 50.5% ee} (Figure S15), (1R,2R)-(−)-threo-1-(4′-chlorophenyl)propan-1,2-diol (threo-(1R,2R)-11a) (2.8 mg) { − 5.04 (c 0.15, CHCl3), 11.9% ee} (Figure S16), (1R,2S)-(−)-eritro-1-(4′-chlorophenyl)propan-1,2-diol (erythro-(1R,2S)-11a) (8.1 mg) { − 9.22 (c 0.45, CHCl3), 21.1% ee, 48.7% de} (Figure S17). In addition, 12.5 mg of 3a was recovered.
2.18. Computational Details of ECD and ORD Calculations
All quantum mechanical calculations were performed using the Gaussian 16 package [31]. First, a conformational analysis of each enantiomer of compounds 6a and 11a was conducted using the Monte Carlo Minimum Method (MCMM) and the molecular mechanics OPLS_2005 force field with an energy cutoff of 5 kcal/mol in Schrodinger MacroModel (v 11.8). The resulting conformers were then geometrically optimized using density functional theory (DFT) within the framework of the B3LYP functional and the 6–311+G (2d, p) basis set, allowing the frequency check for compound 11a. The rotational strengths were subsequently calculated using TD-DFT at the same B3LYP framework and the 6-311+G (2d, p) basis set for 30 excited states for all conformers. For compound 6a, a second optimization and a frequency check were performed using the framework of the ωB97XD functional and the cc-pVTZ basis set. The rotational strength was then calculated for 6a using TD-DFT at the ωB97XD/cc-pVTZ level for 20 excited states on all conformers.
ECD spectra were obtained after Boltzmann averaging using SpecDis 1.71 [32]. To mimic the ECD spectrum of the conformers, a Gaussian function was used featuring a half-bandwidth of 0.33 eV.
The specific rotation for anti-7a and 11a enantiomers was studied following the previously indicated optimization and frequency check procedure. For anti-7a, the conformers were geometrically optimized through the ωB97XD functional and the cc-pVTZ basis set. The theoretical ORD was then calculated at multiple wavelengths ranging from 300 nm to 625 nm, including 589.3 nm (sodium-vapor lamp wavelength) at the CAM-B3LYP/aug-cc-pVTZ level. The ORD curve for each enantiomer was obtained finally after Boltzmann averaging using SpecDis 1.71.
The solvent effect was taken into account during calculations using the Polarizable Continuum Model (PCM) with the Implicit Solvation Energy (IEF) approach. Methanol and chloroform were used as solvents for ECD and ORD calculations, respectively.
2.19. In Vitro Antifungal Assay
The antifungal activity of biotransformation products (R)-5, (S)-6, syn-(1S,2R)-7, anti-(1R,2R)-7, (R)-8, (R)-9, threo-(1R,2R)-11, and erythro-(1R,2S)-11 was evaluated against the phytopathogenic fungus Botrytis cinerea UCA992 using the resazurin-based microdilution method. This method was adapted from the protocols of the Clinical and Laboratory Standards Institute (CLSI) and the European Committee on Antimicrobial Susceptibility Testing (EUCAST) [33].
B. cinerea UCA992 was cultured on tomato-agar plates and incubated for 7 days at 25 °C. Conidia were harvested in sterile distilled water, and their concentration was determined using a Neubauer chamber. The resulting spore suspension was adjusted to 1·107 conidia/mL in modified RPMI-1640 medium (containing 0.3 g/L glutamine) with 165 mmol MOPS buffer (pH 7.0).
For the assay, 20 µL/well of the diluted conidial suspension was mixed with 178 µL/well of RPMI-1640 modified medium and 2 µL/well of compound dissolved in DMSO, resulting in a final concentration of 1·106 conidia/mL per well. The final concentration of DMSO in each well was 1% (v/v). Control wells containing 1% (v/v) DMSO were included in each experiment to confirm that this concentration had no effect on fungal growth. RPMI-1640 was prepared by adding resazurin at a concentration of 0.002%. An amphotericin B dose-response curve was used as a positive control.
All compounds were tested in triplicate using a six-point curve by 1:2 serial dilutions starting at 256 µg/mL. Fungal growth was monitored by measuring absorbance at 570 and 600 nm with a Multiskan GO Microplate Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) at time zero and after 48 h of incubation at 25 °C.
Growth inhibition percentages were calculated using a normalization equation [34]. RZ’ factor values ranging from 0.80 to 0.85 confirmed the robustness and reliability of the assay [35].
3. Results and Discussion
Five substrates, indene (1), indanone (2), 5-chloroindanone (2a), (±)-1-phenylpropyl acetate (3), and (±)-1-(4′-chlorophenyl)propyl acetate (3a), were subjected to biotransformation by the marine-derived strains P. lilacinum BC17-2 and E. maritima BC17 to evaluate their biocatalytic potential.
Substrates 3 and 3a were synthesized from the commercially available ketones propiophenone (4) and 4-chloropropiophenone (4a), respectively, through a chemical reduction followed by acetylation, as described in the Material and Methods section. The structures of the resulting acetates were confirmed by comparison of their NMR data with those previously reported in the literature [11,29].
The structures of the biotransformation products were established through NMR spectroscopic analysis and by comparison of their spectroscopic data with those reported in the literature. Their absolute configurations were determined either by comparing their optical activities with those published in the literature or through computational methods, including electronic circular dichroism (ECD) and optical rotation dispersion (ORD) calculations. Enantiomeric and diastereomeric excesses were determined by HPLC analysis using Chiralcel IB N-5 and Chiralcel OD columns.
3.1. Biotransformation of Indene (1)
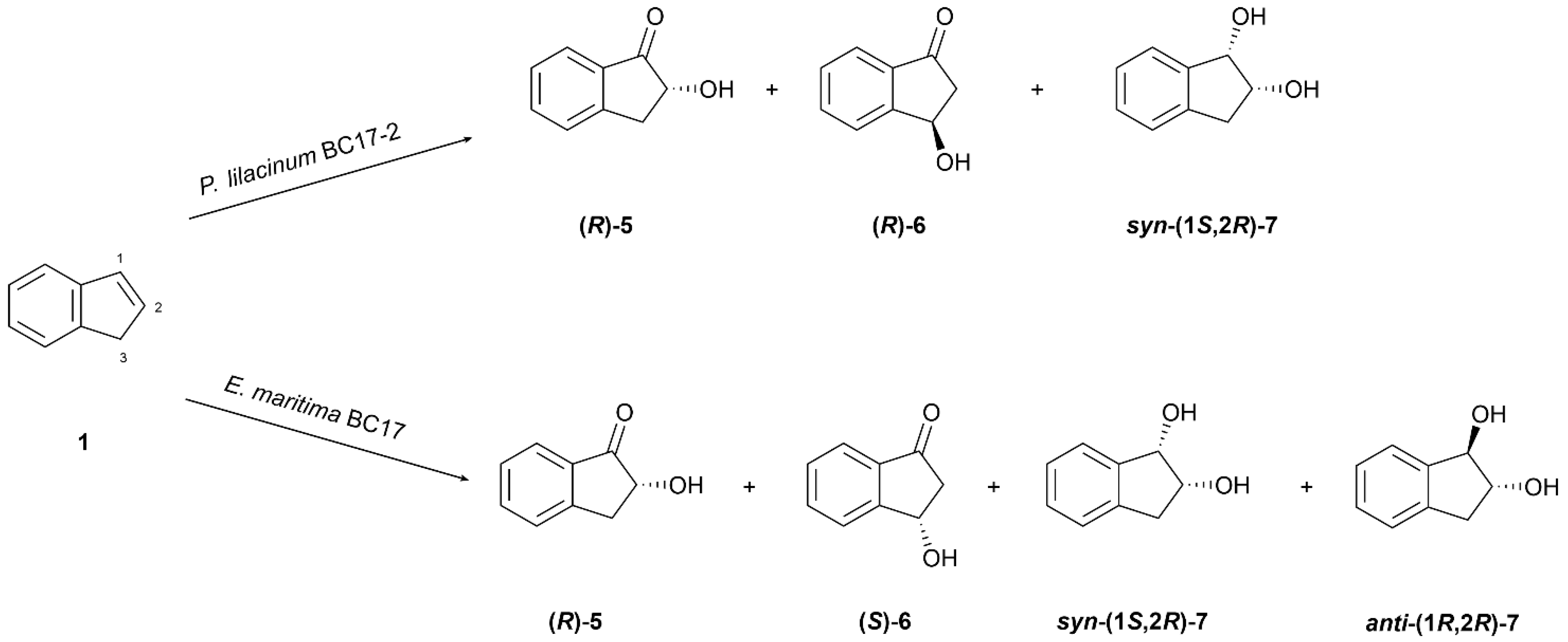
The biotransformation of indene (1) by P. lilacinum BC17-2 yielded three known compounds: (R)-(−)-2-hydroxyindanone ((R)-5) [36], (R)-(−)-3-hydroxyindanone ((R)-6) [37], and syn-(1S,2R)-(−)-indane-1,2-diol (syn-(1S,2R)-7) [38] (Figure 3).
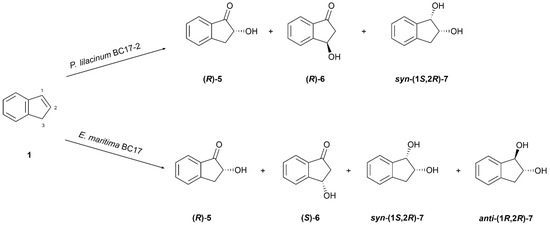
Figure 3.
Biotransformation of indene (1) by Purpureocillium lilacinum BC17-2 and Emericellopsis maritima BC17.
Both hydroxyindanones 5 and 6 were assigned the R-configuration by comparing their specific rotation values with the reported literature data [39]. Compound (R)-5 was obtained with a moderate ee of 43.0%, whereas (R)-6 was produced with a significantly higher enantioselectivity of 71.9%.
The enantioselective formation of compound 6 is particularly noteworthy, as the 3-hydroxyindanone scaffold is a key structural motif in a variety of biologically active natural products with anticancer, antispasmodic, antifungal, and antiviral activities, including tripartin, pterosins, the fused-indanone ligustrone C, spiroindanone coleophomone D, and sibiricine [40].
Additionally, the biotransformation afforded the enantiomer syn-(1S,2R)-(−)-indane-1,2-diol (syn-(1S,2R)-7) as evidenced by its specific rotation and NMR data [38]. This compound exhibited the highest enantioselectivity among all the products obtained, with a 98.2% ee and 100% de. This is particularly noteworthy given the synthetic importance of syn-(1S,2R)-7 as a key intermediate in the production of Crixivan® (indinavir sulfate), a potent HIV protease inhibitor. It is widely acknowledged that the chiral resolution is considered to be one of the most difficult steps in the synthetic pathway. Consequently, the enantioselective biological production of this intermediate offers a promising alternative for future large-scale applications [41].
Incubation of E. maritima BC17 with 1 resulted in the formation of four known compounds: (R)-(-)-2-hydroxyindanone ((R)-5), (S)-(+)-3-hydroxyindanone ((S)-6), syn-(1S,2R)-(−)-indane-1,2-diol (syn-(1S,2R)-7) [38], and anti-(1R,2R)-(−)-indane-1,2-diol (anti-(1R,2R)-7) [42] (Figure 3).
Compound 5 was produced as a nearly racemic mixture, with a slight preference for the R-enantiomer (4.3% ee), whereas compound 6 exhibited a moderate enantioselectivity of 47.1% in favor of the S-enantiomer. In contrast to the results obtained with P. lilacinum BC17-2, both diastereoisomers syn-(1S,2R)-7 and anti-(1R,2R)-7 were isolated in this biotransformation, with a moderate diastereoselectivity of 45.0% and a moderate enantioselectivity of 59.4% and 68.4%, respectively. The absolute and relative configurations of all products were assigned by comparison with previously reported spectroscopic data [38,42,43].
3.2. Biotransformation of Indanone (2) and 5-Chloroindanone (2a)
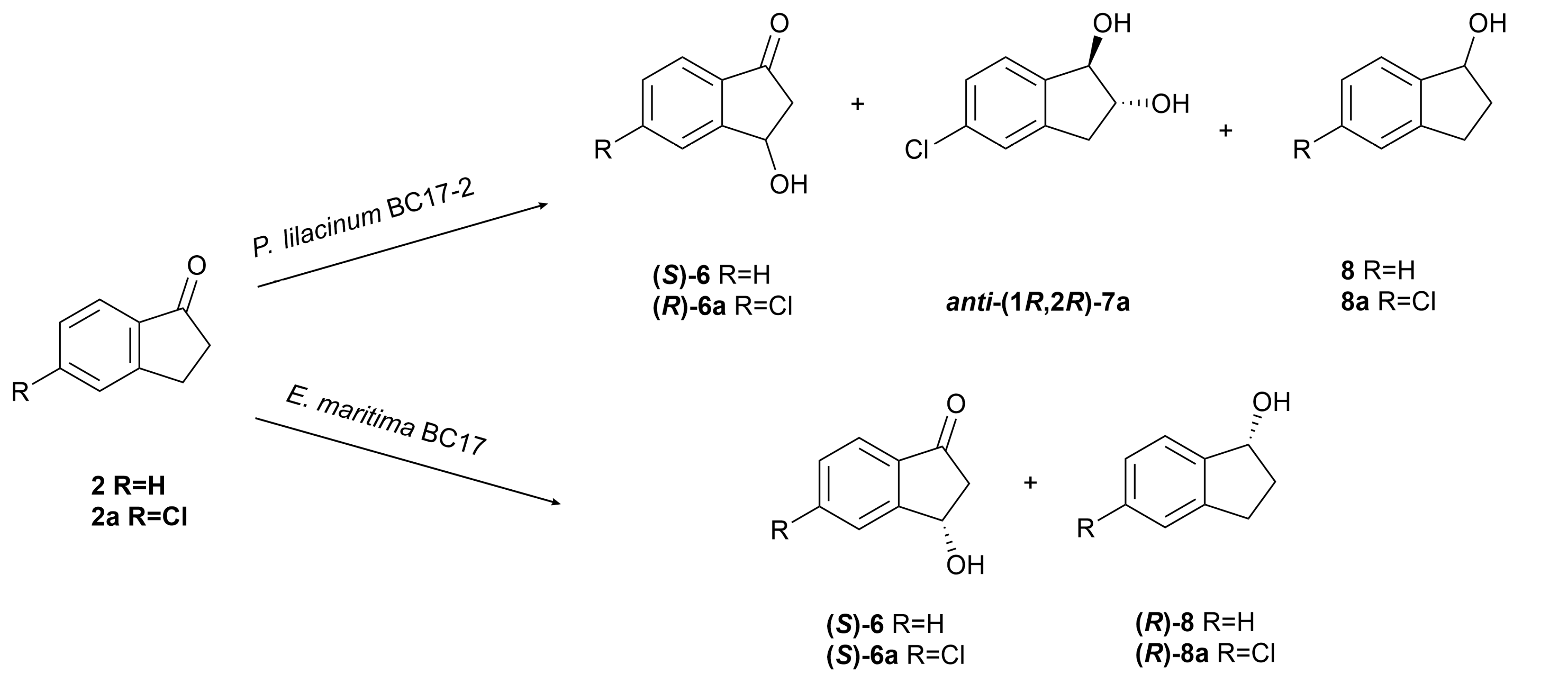
The biotransformation of indanone (2) by P. lilacinum BC17-2 afforded two known compounds: (S)-(+)-3-hydroxyindanone ((S)-6) [37] and indanol (8) [44], in addition to the unreacted starting material. In contrast, incubation of the chlorinated analogue 2a with strain BC17-2 resulted in the production of three known compounds: (R)-(+)-5-chloro-3-hydroxyindanone ((R)-6a) [40], anti-(1R,2R)-(−)-5-chloroindane-1,2-diol (anti-(1R,2R)-7a) [27], and 5-chloroindanol (8a) [45] (Figure 4).
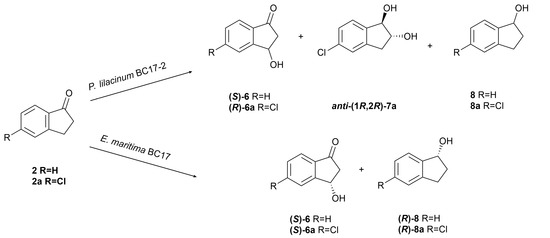
Figure 4.
Biotransformation of indanone (2) and 5-chloroindanone (2a) by Purpureocillium lilacinum BC17-2 and Emericellopsis maritima BC17.
Compound (S)-6 was isolated in a noteworthy yield (80.8 mg, 30% yield). Its absolute configuration was established by comparing its specific rotation value, + 1.12 (c 8.1, CHCl3), with literature data [39]. However, the low ee of 3.8% (Figure S5) indicated that the product was formed almost racemic. Similarly, compound 6a was also isolated with a low ee of 4.3% (Figure S7), consistent with the modest enantioselectivity observed for its non-chlorinated counterpart (6).
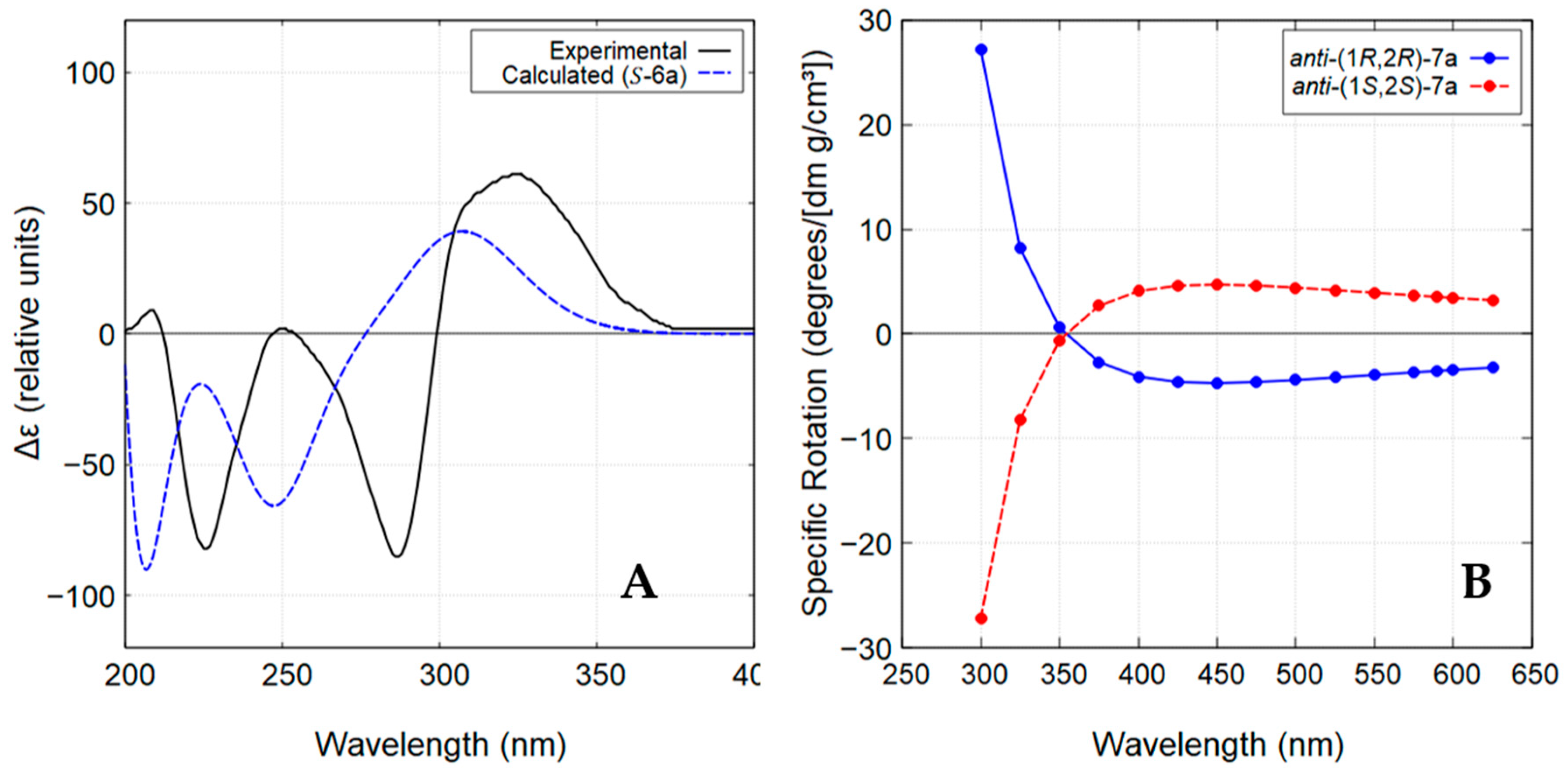
The stereochemistry of compound 6a was determined by comparison of the experimental ECD spectrum of the enantiomer with the lower retention time (minor enantiomer; Figure S7) with the theoretical spectrum calculated for the S-configuration (Figure 5A). As illustrated in Figure 5A, the experimental ECD spectrum exhibited positive and negative Cotton effects that closely matched the theoretical ECD curve obtained using the time-dependent density functional theory (TD-DFT), thereby confirming the assignment of its absolute configuration as S. Consequently, the major enantiomer was assigned the R-configuration.
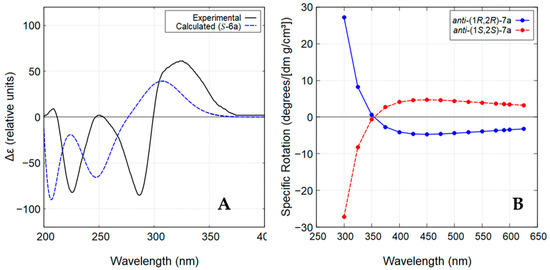
Figure 5.
(A) Experimental and calculated ECD spectra (methanol) for the lower retention time enantiomer of compound 6a. (B) ORD curve calculated for anti-(1R,2R)-7a and anti-(1S,2S)-7a.
In contrast to the chlorohydroxyindanone 6a, the chloroindanediol anti-7a was obtained with high diastereoselectivity (>99% de) and in an almost enantiomerically pure form (85.1% ee) (Figure S8). The structure of anti-7a was established by comparison of its NMR data with those published in the literature [27]. Its absolute configuration was determined through theoretical calculations of optical rotatory dispersion (ORD) in the wavelength range of 300-625 nm. Based on the obtained ORD curves (Figure 5B), the experimentally observed negative specific optical rotation at 589.3 nm (sodium D-line) for compound anti-7a, − 21.8 (c 0.10, CHCl3), supports the assignment of the (1R,2R) configuration.
Indanol (8) and 5-chloroindanol (8a) were identified by comparison of their NMR data with literature values. However, due to insufficient sample availability, stereochemical characterization of these compounds could not be performed.
The biotransformation of indanone (2) by E. maritima BC17 afforded two known compounds: (S)-(+)-3-hydroxyindanone ((S)-6) [37] and (R)-(−)-indanol ((R)-8) [44], along with the unreacted starting material. Similarly, the bioconversion of 5-chloroindanone (2a) yielded the two known compounds (S)-(+)-5-chloro-3-hydroxyindanone ((S)-6a) [40] and (R)-5-chloroindanol ((R)-8a) [45] (Figure 4).
Compound (S)-(+)-3-hydroxyindanone ((S)-6) was obtained through the regioselective bio-oxidation at C-3, with a moderate ee of 43.6% (Figure S5). This result suggests that E. maritima displays higher stereoselectivity in the bioconversion of indanone (2) compared to P. lilacinum. In contrast, its chlorinated homologue (S)-(+)-5-chloro-3-hydroxyindanone ((S)-6a) was isolated with a significantly lower ee of 9.8% (Figure S7), indicating a reduced stereoselectivity. Indanol (8) and 5-chloroindanol (8a) were obtained almost racemic (Figures S6 and S9, respectively). However, their negative optical rotations, − 1.11 (c 0.05, CHCl3) for 8 and − 1.39 (c 0.07, CHCl3) for 8a, suggested a slight enrichment in the R-enantiomer [46,47].
3.3. Biotransformation of (±)-1-Phenylpropyl Acetate (3) and (±)-1-(4′-Chlorophenyl)propyl Acetate (3a)
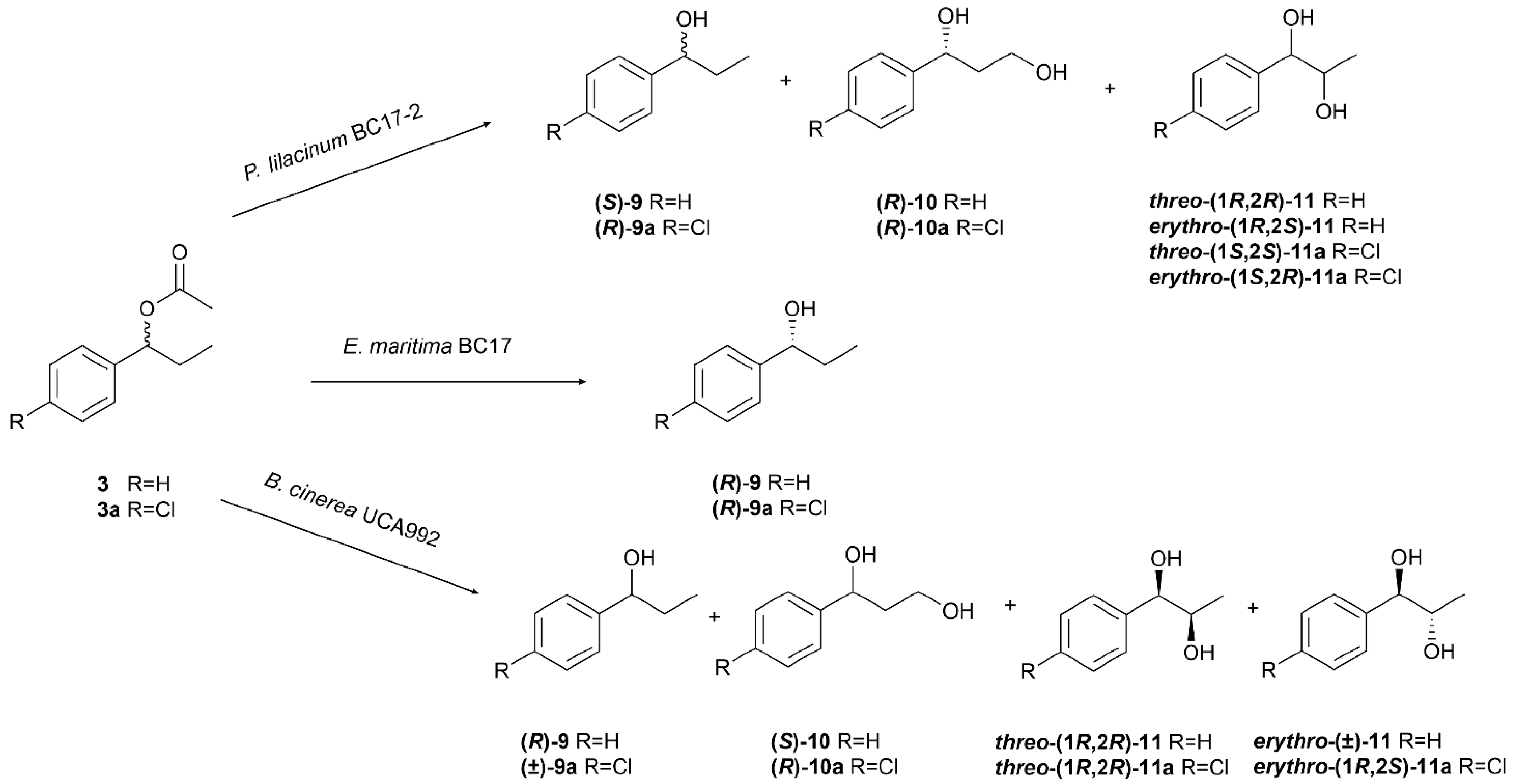
The biotransformation of (±)-1-phenylpropyl acetate (3) by P. lilacinum BC17-2 led to the formation of four known compounds: (S)-(−)-1-phenylpropan-1-ol ((S)-9) [48], (R)-(+)-1-phenylpropane-1,3-diol ((R)-10) [49], threo-(1R,2R)-(−)-1-phenylpropane-1,2-diol (threo-(1R,2R)-11) [50], and erythro-(1R,2S)-(−)-1-phenylpropane-1,2-diol (erythro-(1R,2S)-11) [50]. Similarly, biotransformation of its chlorinated derivative 3a yielded the corresponding known chlorinated analogues: (R)-(+)-1-(4′-chlorophenyl)propan-1-ol ((R)-9a) [48], (R)-(+)-1-(4′-chlorophenyl)propane-1,3-diol ((R)-10a) [26], threo-(1S,2S)-(+)-1-(4′-chlorophenyl)propane-1,2-diol (threo-(1S,2S)-11a) [51], and erythro-(1S,2R)-(+)-1-(4′-chlorophenyl)propane-1,2-diol (erythro-(1S,2R)-11a) [51] (Figure 6).
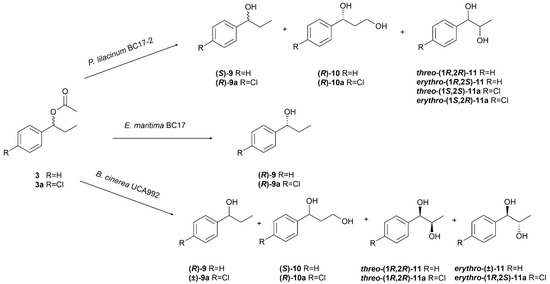
Figure 6.
Biotransformation of 1-phenylpropyl acetate (3) and 1-(4′-chlorophenyl)propyl acetate (3a) by Purpureocillium lilacinum BC17-2, Emericellopsis maritima BC17, and Botrytis cinerea UCA992.
Phenylpropanols 9 and 9a, obtained through hydrolysis of the acetyl group at C-1, were isolated almost racemic, exhibiting 1.35 (c 3.24, CHCl3) (1.2% ee) (Figure S10) and + 2.56 (c 6.94, CHCl3) (7.9% ee) (Figure S14) in favor of configurations S and R, respectively, in accordance with data reported in the literature [48].
Compounds 10 and 10a showed the absence of signals corresponding to the methyl group and the appearance of resonances attributable to a hydroxymethyl moiety in their NMR spectra, suggesting hydroxylation at the C-3 position. Comparative analysis of their spectroscopic data with previously reported values enabled their identification as 1-phenylpropane-1,3-diol (10) [49] and 1-(4′-chlorophenyl)propane-1,3-diol (10a) [26], respectively. Both compounds displayed positive optical rotation, + 22.13 for 10 and + 2.65 for 10a, consistent with the assignment of the R absolute configuration based on comparison with literature data. Compound 10 was obtained with a low ee of 25.3% (Figure S11), whereas the chlorinated homologue 10a was isolated as an almost racemic mixture, with 6.6% ee (Figure S15).
Diols 11 and 11a were obtained as a mixture of diastereoisomers, exhibiting diastereomeric excesses of 23.1% and 34.1% in favor of the threo and erythro configurations, respectively. Diastereomeric excesses were determined from the isolated quantities of each enantiomer. The non-chlorinated diols threo-11 and erythro-11 were produced with a moderate to low enantioselectivity of 52.4% (Figure S12) and 28.8% (Figure S13) in favor of the (1R,2R) ( − 23.34) and (1R,2S) ( − 6.34) configurations, respectively, in accordance with the specific rotation reported in the literature [50].
In contrast, the chlorinated diols threo-11a and erythro-11a were isolated with remarkably high enantioselectivity of 88.5% (Figure S16) and 91.2% (Figure S17) in favor of the (1S,2S) and (1S,2R) configurations, respectively. These results indicate an opposite stereochemical preference of P. lillacinum compared to the non-chlorinated substrate.
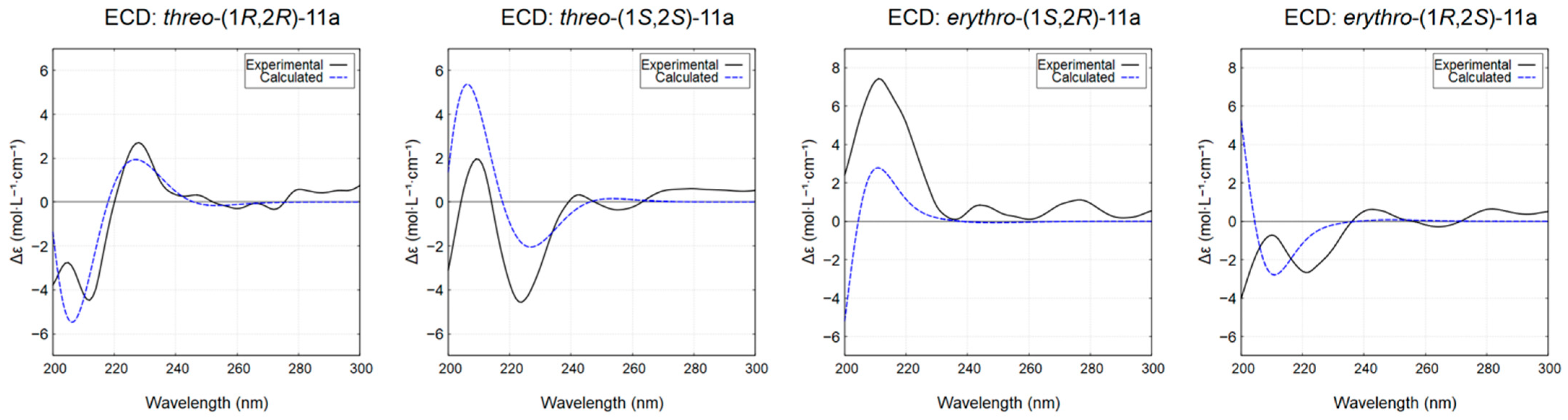
The relative configuration of the chlorinated diol 11a was established according to NMR literature data [51]. Furthermore, stereochemical characterization of its diastereoisomers was studied, given the lack of information in the available literature. Hence, the absolute configurations of threo-11a and erythro-11a were determined by comparison of their experimental ECD spectra with calculated theoretical spectra using the TD-DFT method. The experimental ECD spectra of the major diastereisomers, with tR of 32.1 min for threo-11a (Figure S16) and 31.1 min for erythro-11a (Figure S17), were in good agreement with the calculated ECD spectrum for the threo-(1S,2S) and erythro-(1S,2R) configurations, respectively (Figure 7).
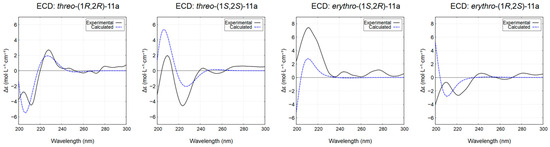
Figure 7.
Experimental and calculated ECD spectra (methanol) for the diastereoisomers of compound 11a. The calculated spectra for threo-(1R,2R)-11a and threo-(1S,2S)-11a are compared with the experimental spectra of the enantiomers with tR of 29.7 and 32.1 min, respectively (see Figure S16). Similarly, the calculated spectra for erythro-(1S,2R)-11a and erythro-(1R,2S)-11a are compared with the experimental spectra of the enantiomers with tR of 31.1 and 34.0 min, respectively.
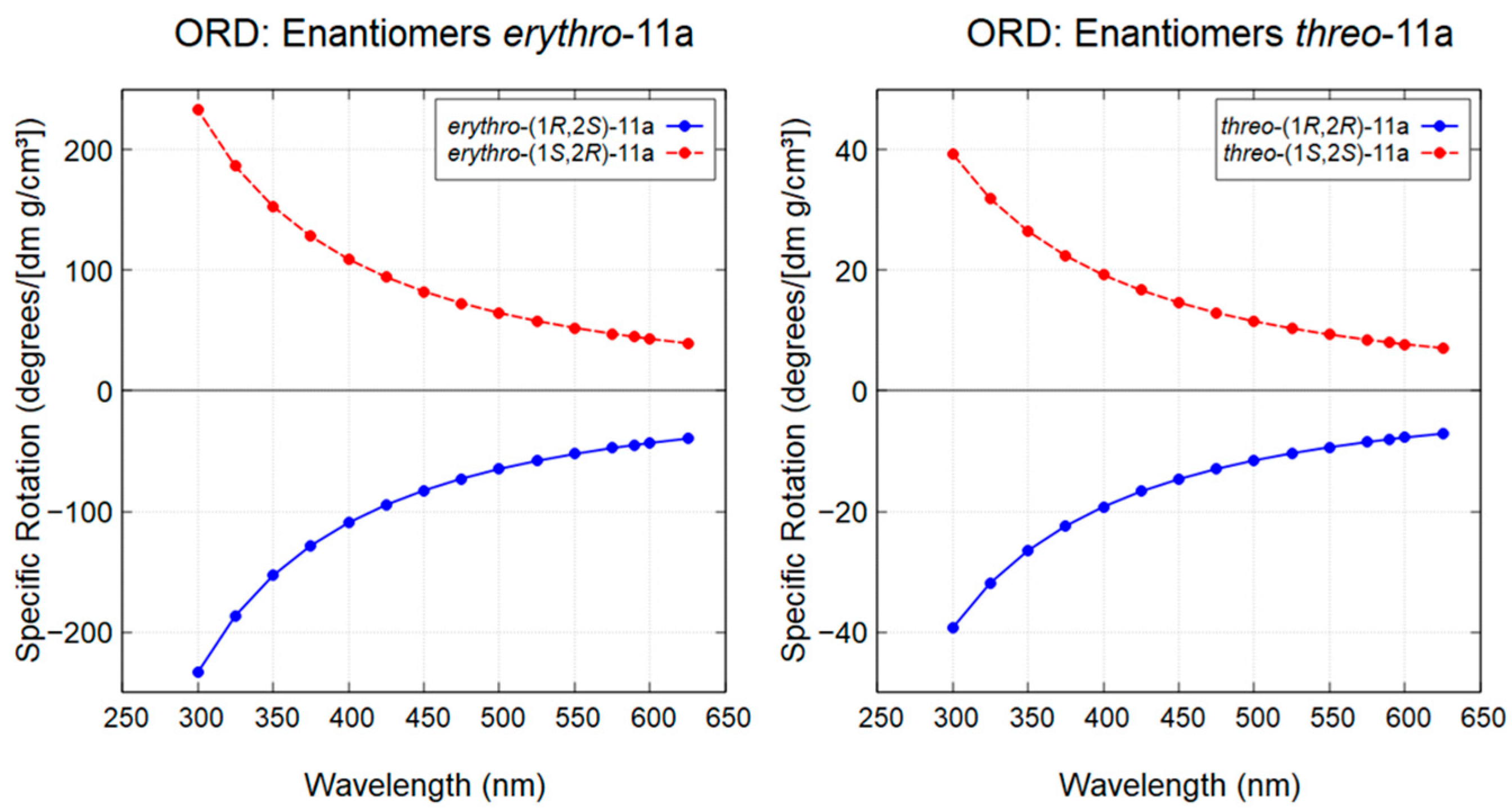
To further support these assignments, theoretical ORD curves were calculated for all diastereoisomers over the 300-625 nm range (Figure 8). The experimentally observed positive specific optical rotations at 589.3 nm (sodium D-line) for both threo-(1S,2S)-11a and erythro-(1S,2R)-11a were consistent with the predicted ORD curves, thereby confirming the assignment of their absolute configurations.
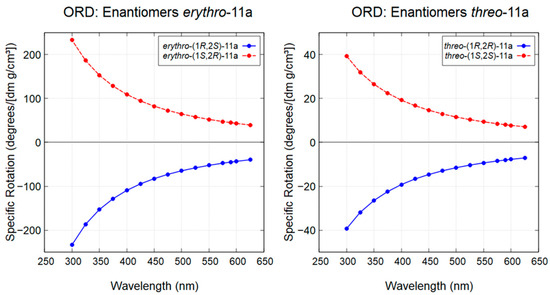
Figure 8.
Theoretical ORD curves for diastereoisomers threo-(1R,2R)-11a, threo-(1S,2S)-11a, erythro-(1S,2R)-11a, and erythro-(1R,2S)-11a.
While various asymmetric chemical approaches have been developed for the synthesis of optically pure diols—including olefin dihydroxylation using cinchona alkaloid-derived ligands, rhodium-catalyzed hydrogenation, and Corey–Bakshi–Shibata (CBS) reduction—these methods often require complex ligand synthesis and involve multiple steps such as protection and deprotection of functional groups [52,53,54].
Therefore, the isolation and characterization of optically enriched 1-phenylethane-1,2-diols—threo-11, erythro-11, threo-11a, and erythro-11a—by biocatalytic means is particularly noteworthy, as these compounds represent valuable intermediates in the synthesis of pharmaceuticals, fatty acid amide hydrolase inhibitors [55], antivirals [56], and anticancer agents [57]. Moreover, they are precursors to bioactive molecules such as (R)-selegiline—used in the treatment of Parkinson’s and Alzheimer’s diseases—and scyphostatin, a potent inhibitor of neutral sphingomyelinase [58,59].
The biotransformation of (±)-1-phenylpropyl acetate (3) and (±)-1-(4′-chlorophenyl)propyl acetate (3a) by E. maritima BC17 afforded (R)-(+)-1-phenylpropan-1-ol ((R)-9) [48] and (R)-(+)-1-(4′-chlorophenyl)propan-1-ol ((R)-9a) [48], respectively (Figure 6). Both biotransformation products exhibited moderate to low enantioselectivity, with ee of 38.3% ( + 25.41) for (R)-9 (Figure S10) and 9.5% ( + 5.53) for (R)-9a (Figure S14). Despite this relatively low selectivity, this fungal strain demonstrated higher enantioselectivity than P. lilacinum BC17-2 to hydrolyze 3.
In light of the reported biocatalytic potential of the phytopathogenic fungus B. cinerea to modify the structures of certain antifungal phenylpropyl derivatives [26], compounds 3 and 3a were subjected to biotransformation by the strain UCA992 of B. cinerea in order to explore novel structural modifications. Previous research had demonstrated the capacity of this fungus to biotransform antifungal secondary alcohols through hydroxylation at various positions [26].
The separate biotransformation of compounds 3 and 3a by B. cinerea UCA992 resulted in the formation of the same products previously obtained by P. lilacinum BC17-2. Specifically, the biotransformation of 3 produced (R)-(+)-1-phenylpropan-1-ol ((R)-9) [48], (R)-(+)-1-phenylpropane-1,3-diol ((R)-10) [49], threo-(1R,2R)-(−)-1-phenylpropane-1,2-diol (threo-(1R,2R)-11) [50], and erythro-(±)-1-phenylpropane-1,2-diol (erythro-11) [50]. Similarly, the bioconversion of 3a yielded (±)-1-(4′-chlorophenyl)propan-1-ol (9a) [48], (S)-(−)-1-(4′-chlorophenyl)propane-1,3-diol ((S)-10a) [26], threo-(1R,2R)-(−)-1-(4′-chlorophenyl)propane-1,2-diol (threo-(1R,2R)-11a) [51], and erythro-(1S,2R)-(+)-1-(4′-chlorophenyl)propane-1,2-diol (erythro-(1R,2S)-11a) [51] (Figure 6).
Compounds 9, 9a, and 10 were obtained with low enantioselectivity or as racemic mixtures, as observed with E. maritima and P. lilacinum. In contrast, B. cinerea catalyzed the hydrolysis and C-3 oxidation of the chlorinated derivative 3a to give (S)-10a with moderate enantioselectivity (50.5% ee).
Compounds 11 and 11a were obtained with moderate diastereoselectivity favoring the erythro configuration (40.0% and 48.7% de, respectively), as previously observed with P. lilacinum. Although erythro-11 diol was obtained as a racemic mixture, threo-(1R,2R)-11 was formed with the same absolute configuration and a moderate enantioselectivity of 49.5% ee (Figures S12 and S13), comparable to that observed with the BC17-2 strain. In contrast, the chlorinated diols threo-(1R,2R)-11a and erythro-(1R,2S)-11a, although also obtained with the same absolute configurations, displayed significantly lower enantioselectivities than those achieved with BC17-2, with ee of 11.9% and 21.1%, respectively.
In summary, most biotransformation products have been formed by hydroxylations on aliphatic chains, oxidation of alcohols, reduction of ketone groups to alcohol, or by hydrolysis of an acetate group. Therefore, the enzymes that could be involved in such transformations would probably be cytochrome P450 monooxygenases, dehydrogenases, and hydrolytic enzymes, respectively.
Despite the evident ability of these fungal strains to obtain products with remarkable stereoselectivity in some cases, the enzymatic basis for this selectivity has not yet been elucidated.
3.4. Antifungal Assays Against Botrytis cinerea UCA992
The antifungal activity of the biotransformation products (R)-5, (S)-6, syn-(1S,2R)-7, anti-(1R,2R)-7, (R)-8, (R)-9, threo-(1R,2R)-11, and erythro-(1R,2S)-11 was evaluated against the phytopathogenic strain B. cinerea UCA992, but no significant values of inhibition were detected for these compounds (Table 1).

Table 1.
Average inhibition percentages obtained from in vitro antimicrobial assays against B. cinerea UCA992. Positive values indicate growth inhibition, while negative values reflect fungal overgrowth compared to the control.
In this regard, previous studies have demonstrated the antifungal potential of structurally related compounds bearing a chlorine substituent. Specifically, Bustillo et al. reported significant antifungal activity against B. cinerea UCA992 for the compounds 1-(4′-chlorophenyl)propan-1-ol (9a) and 1-(4′-chlorophenyl)propane-1,3-diol (10a) [11]. Similarly, Pinedo-Rivilla et al. demonstrated the antifungal properties of 5-chloroindanone (2a) and 5-chloroindanol (8a) [27]. In contrast, their non-chlorinated homologues did not show significant inhibitory activity, suggesting that antifungal effects are crucially influenced by the incorporation of the halogen atom.
4. Conclusions
In light of the mounting significance of biocatalysis as an alternative approach to conventional chemistry in addressing pivotal stages in the synthesis of bioactive compounds characterized by high enantiomeric purity, this study undertakes an evaluation of the biocatalytic capacity of the marine-derived fungal strains E. maritima BC17 and P. lilacinum BC17-2, along with the phytopathogenic fungus B. cinerea UCA992. This evaluation is conducted through the examination of microbiological transformations, thereby showcasing the enzyme versatility exhibited by the employed fungal strains.
Biotransformation of indene (1), indanone (2), 5-chloroindanone (2a), (±)-1-phenylpropyl acetate (3), and (±)-1-(4′-chlorophenyl)propyl acetate (3a) by E. maritima BC17, P. lilacinum BC17-2, and B. cinerea UCA992 yielded sixteen known derivatives. Stereochemical characterization of the optically active biotransformation products 6a, 7a, and 11a was performed using computational calculations of ECD and ORD.
These marine fungal strains demonstrated a remarkable ability to biotransform indane and phenylpropyl derivatives, suggesting an interesting potential as biocatalysts for modifying such structures, particularly through oxidation reactions.
This study demonstrated that the fungal strain P. lilacinum BC17-2 exhibits a higher capacity for enantioselective biotransformation of the tested substrates compared to E. maritima BC17 and B. cinerea UCA992. This ability is particularly valuable for the production of the enantiomerically enriched diols syn-(1S,2R)-(−)-indane-1,2-diol (syn-(1S,2R)-7), anti-(1R,2R)-(−)-5-chloroindane-1,2-diol (anti-(1R,2R)-7a), threo-(1S,2S)-(+)-1-phenylpropane-1,2-diol (threo-(1S,2S)-11a), and erythro-(1S,2R)-(+)-1-phenylpropane-1,2-diol (erythro-(1S,2R)-11a), whose scaffolds are considered valuable building blocks in drug design and organic synthesis. Of particular significance is the isolation of enantiomerically pure syn-(1S,2R)-7, a key intermediate in the synthesis of Crixivan®, an HIV protease inhibitor [41]. Nevertheless, the relatively low yields obtained for some of the biotransformation products limited their full characterization.
Thus, the results obtained in this study represent a promising starting point for assessing the biocatalytic potential of E. maritima BC17 and P. lilacinum BC17-2, paving the way for future investigations using substrates of greater synthetic or pharmacological relevance, particularly where hydroxylation of aliphatic chains, ketone reduction, or acetate hydrolysis could enhance the bioactivity or functional value of the resulting compounds.
Unfortunately, the specific enzymatic mechanisms involved remain largely unexplored, particularly considering that E. maritima BC17 and P. lilacinum BC17-2 have scarcely been studied for their biocatalytic potential. Although these strains demonstrated the ability to generate products with remarkable stereoselectivity, the enzymatic basis for such selectivity remains unknown. Therefore, future studies could aim to deepen our understanding of the metabolic pathways involved, through enzymatic or omics-based approaches, in order to unlock and optimize the biocatalytic capabilities of these underexplored marine-derived fungi.
Antifungal activity of the compounds (R)-5, (S)-6, syn-(1S,2R)-7, anti-(1R,2R)-7, (R)-8, (R)-9, threo-(1R,2R)-11, and erythro-(1R,2S)-11 was evaluated against B. cinerea UCA992; however, no significant inhibitory activity was observed under the tested conditions, suggesting that the chlorine atom plays a key role in this effect. Therefore, future biotransformation studies involving similar scaffolds should prioritize chlorinated homologues to further investigate their antifungal potential.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jmse13081386/s1, Figure S1: Indene (1) biotransformation. ee Determination of 2-hydroxyindanone (5) from P. lilacinum BC17-2 (Chromatogram I) and E. maritima BC17 (Chromatogram II); Figure S2: Indene (1) biotransformation. ee Determination of 3-hydroxyindanone (6) from P. lilacinum BC17-2 (Chromatogram I) and E. maritima BC17 (Chromatogram II); Figure S3: Indene (1) biotransformation. ee Determination of syn-indane-1,2-diol (syn-7) from P. lilacinum BC17-2 (Chromatogram I) and E. maritima BC17 (Chromatogram II); Figure S4: Indene (1) biotransformation. ee Determination of anti-indane-1,2-diol (anti-7) from E. maritima BC17 (Chromatogram I); Figure S5: Indanone (2) biotransformation. ee Determination of 3-hydroxyindanone (6) from P. lilacinum BC17-2 (Chromatogram I) and E. maritima BC17 (Chromatogram II); Figure S6: Indanone (2) biotransformation. ee Determination of indanol (8) from E. maritima BC17 (Chromatogram I); Figure S7: 5-Chloroindanone (2a) biotransformation. ee Determination of 5-chloro-3-hydroxyindanone (6a) from P. lilacinum BC17-2 (Chromatogram I) and E. maritima BC17 (Chromatogram II); Figure S8: 5-Chloroindanone (2a) biotransformation. ee Determination of anti-5-chloroindane-1,2-diol (anti-7a) from P. lilacinum BC17-2 (Chromatogram I); Figure S9: 5-Chloroindanone (2a) biotransformation. ee Determination of 5-chloroindanol (8a) from E. maritima BC17 (Chromatogram I); Figure S10: (±)-1-Phenylpropyl acetate (3) biotransformation. ee Determination of 1-phenylpropan-1-ol (9) from P. lilacinum BC17-2 (Chromatogram I), E. maritima BC17 (Chromatogram II), and B. cinerea UCA992 (Chromatogram III); Figure S11: (±)-1-Phenylpropyl acetate (3) biotransformation. ee Determination of 1-phenylpropane-1,3-diol (10) from P. lilacinum BC17-2 (Chromatogram I) and B. cinerea UCA992 (Chromatogram II); Figure S12: (±)-1-Phenylpropyl acetate (3) biotransformation. ee Determination of threo-1-phenylpropane-1,2-diol (threo-11) from P. lilacinum BC17-2 (Chromatogram I) and B. cinerea UCA992 (Chromatogram II); Figure S13 (±)-1-Phenylpropyl acetate (3) biotransformation. ee Determination of erythro-phenylpropane-1,2-diol (erythro-11) from P. lilacinum BC17-2 (Chromatogram I) and B. cinerea UCA992 (Chromatogram II). Chromatogram II was deconvoluted in order to obtain an accurate result; Figure S14: (±)-1-(4′-chlorophenyl)propyl acetate (3a) biotransformation. ee Determination of 1-(4′-chlorophenyl)propan-1-ol (9a) from P. lilacinum BC17-2 (Chromatogram I), E. maritima BC17 (Chromatogram II), and B. cinerea UCA992 (Chromatogram III); Figure S15: (±)-1-(4′-chlorophenyl)propyl acetate (3a) biotransformation. ee Determination of 1-(4′-chlorophenyl)1-phenylpropane-1,3-diol (10a) from P. lilacinum BC17-2 (Chromatogram I) and B. cinerea UCA992 (Chromatogram II); Figure S16: (±)-1-(4′-chlorophenyl)propyl acetate (3a) biotransformation. ee Determination of threo-1-(4′-chlorophenyl)propane-1,2-diol (threo-11a) from P. lilacinum BC17-2 (Chromatogram I) and B. cinerea UCA992 (Chromatogram II); Figure S17: (±)-1-(4′-chlorophenyl)propyl acetate (3a) biotransformation. ee Determination of erythro-1-(4′-chlorophenyl)propane-1,2-diol (erythro-11a) from P. lilacinum BC17-2 (Chromatogram I) and B. cinerea UCA992 (Chromatogram II).
Author Contributions
Conceptualization, J.A. and R.D.-P.; funding acquisition, J.A. and R.D.-P.; investigation, J.R.V.-S., S.M.-M., N.C.-G., M.P. and M.G.-M.; computational calculations J.R.V.-S.; methodology, J.M. and J.A.; project administration, J.A.; supervision, J.M. and J.A.; writing—original draft, J.R.V.-S. and J.A.; writing—review and editing, J.R.V.-S., R.D.-P. and J.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the 2014–2020 ERDF Operational Programme and by the Department of Economy, Knowledge, Business and University of the Regional Government of Andalusia. Project reference: FEDER-UCA18-105749.
Data Availability Statement
Data are contained within this article and the Supplementary Materials.
Acknowledgments
All simulations were performed using computational facilities from the Information Systems Area (HPC). We also thank the Systems Unit of the HPC of the University of Cadiz for computer resources and technical support. J.R.V.-S thanks the Spanish Ministry of Universities for a grant from the FPU 2022 National Programme.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Yamanish, Y.; Iimura, Y.; Kawakami, Y. Donepezil hydrochloride (E2020) and other acetylcholinesterase inhibitors. Curr. Med. Chem. 2000, 7, 303–339. [Google Scholar] [CrossRef] [PubMed]
- Leoni, L.M.; Hamel, E.; Genini, D.; Shih, H.; Carrera, C.J.; Cottam, H.B.; Carson, D.A. Indanocine, a microtubule-binding indanone and a selective inducer of apoptosis in multidrug-resistant cancer cells. JNCI J. Natl. Cancer Inst. 2000, 92, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Vilums, M.; Heuberger, J.; Heitman, L.H.; IJzerman, A.P. Indanes—Properties, preparation, and presence in ligands for G protein coupled receptors. Med. Res. Rev. 2015, 35, 1097–1126. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, A.; Shah, S.; Kanwal; Hameed, S.; Khan, K.M.; Khan, S.U.; Zaib, S.; Iqbal, J.; Perveen, S. Indane-1,3-diones: As potential and selective α-glucosidase inhibitors, their synthesis, in vitro and in silico studies. Med. Chem. 2021, 17, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Bano, B.; Kanwal; Khan, K.M.; Begum, F.; Lodhi, M.A.; Salar, U.; Khalil, R.; Ul-Haq, Z.; Perveen, S. Benzylidine indane-1,3-diones: As novel urease inhibitors; synthesis, in vitro, and in silico studies. Bioorg. Chem. 2018, 81, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Ugliarolo, E.A.; Gagey, D.; Lantaño, B.; Moltrasio, G.Y.; Campos, R.H.; Cavallaro, L.V.; Moglioni, A.G. Synthesis and biological evaluation of novel homochiral carbocyclic nucleosides from 1-amino-2-indanols. Bioorg. Med. Chem. 2012, 20, 5986–5991. [Google Scholar] [CrossRef] [PubMed]
- Plosker, G.L.; Noble, S. Indinavir: A review of its use in the management of HIV infection. Drugs 1999, 58, 1165–1203. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.S.; Elghany, R.A.A.; Sharaky, M.; Diab, H.M.; Abdelhamid, I.A.; Elwahy, A.H.M. Synthesis, cytotoxicity, antiproliferative, antioxidant, anti-inflammatory, and anti-metastatic activities of novel 2-arylidene-indane-1,3-dione scaffolds incorporating 2-phenoxy-N-arylacetamide moiety: Induction of apoptosis in Hela cells. J. Mol. Struct. 2025, 1328, 141424. [Google Scholar] [CrossRef]
- Saiz-Urra, L.; Pérez, A.J.B.; Cruz-Monteagudo, M.; Pinedo-Rivilla, C.; Aleu, J.; Hernández-Galan, R.; Collado, I.G. Global antifungal profile optimization of chlorophenyl derivatives against Botrytis cinerea and Colletotrichum gloeosporioides. J. Agric. Food Chem. 2009, 57, 4838–4843. [Google Scholar] [CrossRef] [PubMed]
- Bustillo, A.J.; Aleu, J.; Hernández-Galán, R.; Collado, I.G. Biocatalytically assisted preparation of antifungal chlorophenylpropanols. Tetrahedron Asymmetry 2002, 13, 1681–1686. [Google Scholar] [CrossRef]
- McGinty, D.; Letizia, C.S.; Api, A.M. Fragrance material review on ethyl phenyl carbinyl acetate. Food Chem. Toxicol. 2012, 50, S512–S515. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef] [PubMed]
- Muñoz Solano, D.; Hoyos, P.; Hernáiz, M.J.; Alcántara, A.R.; Sánchez-Montero, J.M. Industrial biotransformations in the synthesis of building blocks leading to enantiopure drugs. Bioresour. Technol. 2012, 115, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Trincone, A. Marine biocatalysts: Enzymatic features and applications. Mar. Drugs 2011, 9, 478–499. [Google Scholar] [CrossRef] [PubMed]
- Virués-Segovia, J.R.; Muñoz-Mira, S.; Durán-Patrón, R.; Aleu, J. Marine-derived fungi as biocatalysts. Front. Microbiol. 2023, 14, 1125639. [Google Scholar] [CrossRef] [PubMed]
- Debashish, G.; Malay, S.; Barindra, S.; Joydeep, M. Marine enzymes. In Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2005; Volume 96, pp. 189–218. [Google Scholar] [CrossRef]
- Dowd, B.; Tuohy, M.G. Induction and characterisation of lignocellulolytic activities from novel deep-sea fungal secretomes. Fermentation 2023, 9, 780. [Google Scholar] [CrossRef]
- Batista-García, R.A.; Sutton, T.; Jackson, S.A.; Tovar-Herrera, O.E.; Balcázar-López, E.; Sánchez-Carbente, M.D.R.; Sánchez-Reyes, A.; Dobson, A.D.W.; Folch-Mallol, J.L. Characterization of lignocellulolytic activities from fungi isolated from the deep-sea sponge Stelletta normani. PLoS ONE 2017, 12, e0173750. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.; Qiu, H.; Li, S.; Zhang, T. Biological degradation of aflatoxin B1 by Emericellopsis sp. 1912 and Sarocladium sp. 10A. Lect. Notes Electr. Eng. 2018, 444, 525–532. [Google Scholar] [CrossRef]
- Pradeep, S.; Faseela, P.; Josh, M.K.S.; Balachandran, S.; Devi, R.S.; Benjamin, S. Fungal biodegradation of phthalate plasticizer in situ. Biodegradation 2013, 24, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.-S.; Lee, M.-J.; Wu, J.-A.; Kuo, S.-L.; Chang, S.-L.; Huang, S.-J.; Liu, C.-T. Poly(butylene adipate-co-terephthalate) biodegradation by Purpureocillium lilacinum strain BA1S. Appl. Microbiol. Biotechnol. 2023, 107, 6057–6070. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Ten, L.N.; Das, K.; You, Y.-H.; Jung, H.-Y. Biodegradative activities of fungal strains isolated from terrestrial environments in Korea. Mycobiology 2021, 49, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Cavello, I.A. Study of the production of alkaline keratinases in submerged cultures as an alternative for solid waste treatment generated in leather technology. J. Microbiol. Biotechnol. 2013, 23, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Virués-Segovia, J.R.; Pinedo-Rivilla, C.; Muñoz-Mira, S.; Ansino, M.; González-Rodríguez, V.E.; Ezzanad, A.; Galán-Sánchez, F.; Durán-Patrón, R.; Aleu, J. Biotransformation of thiochroman derivatives using marine-derived fungi: Isolation, characterization, and antimicrobial activity. Int. J. Mol. Sci. 2025, 26, 908. [Google Scholar] [CrossRef] [PubMed]
- Bustillo, A.J.; Aleu, J.; Hernández-Galán, R.; Collado, I.G. Biotransformation of the fungistatic compound (R)-(+)-1-(4′-chlorophenyl)propan-1-ol by Botrytis cinerea. J. Mol. Catal. B Enzym. 2003, 21, 267–271. [Google Scholar] [CrossRef]
- Pinedo-Rivilla, C.; Moraga, J.; Pérez-Sasián, G.; Peña-Hernández, A.; Collado, I.G.; Aleu, J. Biocatalytic preparation of chloroindanol derivatives antifungal activity and detoxification by the phytopathogenic fungus Botrytis cinerea. Plants 2020, 9, 1648. [Google Scholar] [CrossRef]
- Virués-Segovia, J.R.; Millán, C.; Pinedo, C.; González-Rodríguez, V.E.; Papaspyrou, S.; Zorrilla, D.; Mackenzie, T.A.; Ramos, M.C.; de la Cruz, M.; Aleu, J.; et al. New eremophilane-type sesquiterpenes from the marine sediment-derived fungus Emericellopsis maritima BC17 and their cytotoxic and antimicrobial activities. Mar. Drugs 2023, 21, 634. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.E.D.; Contreras Redondo, M.A.; Wills, M. Applications of N′-alkylated derivatives of TsDPEN in the asymmetric transfer hydrogenation of C=O and C=N bonds. Tetrahedron Asymmetry 2010, 21, 2258–2264. [Google Scholar] [CrossRef]
- Senaweera, S.; Cartwright, K.C.; Tunge, J.A. Decarboxylative acetoxylation of aliphatic carboxylic acids. J. Org. Chem. 2019, 84, 12553–12561. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental Electronic Circular Dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Serrano, R.; González-Menéndez, V.; Tormo, J.R.; Genilloud, O. Development and validation of a HTS platform for the discovery of new antifungal agents against four relevant fungal phytopathogens. J. Fungi 2023, 9, 883. [Google Scholar] [CrossRef] [PubMed]
- Algaffar, S.O.A.; Verbon, A.; van de Sande, W.W.J.; Khalid, S.A. Development and validation of an in vitro resazurin-based susceptibility assay against Madurella mycetomatis. Antimicrob. Agents Chemother. 2021, 65, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. SLAS Discov. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kroc, M.A.; Patil, A.; Carlos, A.; Ballantine, J.; Aguilar, S.; Mo, D.-L.; Wang, H.-Y.; Mueller, D.S.; Wink, D.J.; Anderson, L.L. Synthesis of α-oxygenated ketones and substituted catechols via the rearrangement of N-enoxy- and N-aryloxyphthalimides. Tetrahedron 2017, 73, 4125–4137. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Silva, A.M.S.; Tagliatesta, P.; Cavaleiro, J.A.S. Oxidation of bicyclic arenes with hydrogen peroxide catalysed by Mn(III) porphyrins. J. Mol. Catal. A Chem. 2005, 232, 135–142. [Google Scholar] [CrossRef]
- Fernández, R.; Ros, A.; Magriz, A.; Dietrich, H.; Lassaletta, J.M. Enantioselective synthesis of cis-α-substituted cycloalkanols and trans-cycloalkyl amines thereof. Tetrahedron 2007, 63, 6755–6763. [Google Scholar] [CrossRef]
- Murahashi, S.I.; Noji, S.; Hirabayashi, T.; Komiya, N. Manganese-catalyzed enantioselective oxidation of C-H bonds of alkanes and silyl ethers to optically active ketones. Tetrahedron Asymmetry 2005, 16, 3527–3535. [Google Scholar] [CrossRef]
- He, G.; Wu, C.; Zhou, J.; Yang, Q.; Zhang, C.; Zhou, Y.; Zhang, H.; Liu, H. A method for synthesis of 3-hydroxy-1-indanones via Cu-catalyzed intramolecular annulation reactions. J. Org. Chem. 2018, 83, 13356–13362. [Google Scholar] [CrossRef] [PubMed]
- Dafoe, J.T.S.; Daugulis, A.J. Bioproduction of cis-(1S,2R)-indandiol, a chiral pharmaceutical intermediate, using a solid–liquid two-phase partitioning bioreactor for enhanced removal of inhibitors. J. Chem. Technol. Biotechnol. 2011, 86, 1379–1385. [Google Scholar] [CrossRef]
- Stahl, S.; Ikemoto, N.; King, A.; Greasham, R.; Chartrain, M. Asymmetric direduction of 1,2-indanedione to cis (1s,2r) indanediol by Trichosporon cutaneum MY 1506. J. Biosci. Bioeng. 1999, 88, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Boyd, D.R.; Sharma, N.D.; Smith, A.E. Chemical synthesis and optical purity determination of optically active 1,2-epoxyindan and alcohol products which are also derived from mammalian or microbial metabolism of indene or indanones. J. Chem. Soc. Perkin Trans. 1982, 1, 2767. [Google Scholar] [CrossRef]
- Kišić, A.; Stephan, M.; Mohar, B. ansa-Ruthenium(II) complexes of R 2 NSO 2 DPEN-(CH 2) n (η 6-aryl) conjugate ligands for asymmetric transfer hydrogenation of aryl ketones. Adv. Synth. Catal. 2015, 357, 2540–2546. [Google Scholar] [CrossRef]
- Arp, F.O.; Fu, G.C. Catalytic enantioselective negishi reactions of racemic secondary benzylic halides. J. Am. Chem. Soc. 2005, 127, 10482–10483. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, T.; Utsumi, N.; Tsutsumi, K.; Watanabe, M.; Murata, K.; Arai, N.; Kurono, N. Asymmetric hydrogenation of aromatic heterocyclic ketones catalyzed by the MsDPEN–Cp*Ir(III) Complex. Heterocycles 2010, 80, 141. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, S.; Yamada, A.; Nakata, K. Silylative kinetic resolution of racemic 1-indanol derivatives catalyzed by chiral guanidine. J. Org. Chem. 2018, 83, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-X.; Liu, G.-J.; Wang, J.; Li, F.; Liu, L. Synthesis of MeO-PEG2000-supported chiral ferrocenyl oxazoline carbinol ligand and its application in asymmetric catalysis. Tetrahedron Asymmetry 2016, 27, 1139–1144. [Google Scholar] [CrossRef]
- Passays, J.; Ayad, T.; Ratovelomanana-Vidal, V.; Gaumont, A.-C.; Jubault, P.; Leclerc, E. Synthesis and evaluation of a broad range of new chiral phosphine–carbene ligands for asymmetric hydrogenation. Tetrahedron Asymmetry 2011, 22, 562–574. [Google Scholar] [CrossRef]
- Zang, C.; Liu, Y.; Xu, Z.; Tse, C.; Guan, X.; Wei, J.; Huang, J.; Che, C. Highly enantioselective iron-catalyzed cis-dihydroxylation of alkenes with hydrogen peroxide oxidant via an Fe III-OOH reactive intermediate. Angew. Chem. Int. Ed. 2016, 55, 10253–10257. [Google Scholar] [CrossRef] [PubMed]
- Pelter, A.; Peverall, S.; Pitchford, A. Hindered organoboron groups in organic chemistry. 30. The production of erythro-1,2-diols by the condensation of dimesitylboron stabilised carbanions with aromatic aldehydes. Tetrahedron 1996, 52, 1085–1094. [Google Scholar] [CrossRef]
- Kuang, Y.-Q.; Zhang, S.-Y.; Jiang, R.; Wei, L.-L. A free ligand for the asymmetric dihydroxylation of olefins utilizing one-phase catalysis and two-phase separation. Tetrahedron Lett. 2002, 43, 3669–3671. [Google Scholar] [CrossRef]
- Adams, H.; Gilmore, N.J.; Jones, S.; Muldowney, M.P.; von Reuss, S.H.; Vemula, R. Asymmetric synthesis of corsifuran a by an enantioselective oxazaborolidine reduction. Org. Lett. 2008, 10, 1457–1460. [Google Scholar] [CrossRef] [PubMed]
- Hannedouche, J.; Clarkson, G.J.; Wills, M. A new class of “Tethered” Ruthenium(II) catalyst for asymmetric transfer hydrogenation reactions. J. Am. Chem. Soc. 2004, 126, 986–987. [Google Scholar] [CrossRef] [PubMed]
- Behnke, M.L.; Castro, A.C.; Evans, C.A.; Grenier, L.; Grogan, M.J.; Liu, T.; Snyder, D.A.; Tibbitts, T.T. Inhibitors of fatty acid amide hydrolase. PCT WO2009003140A1, 15 October 2009. [Google Scholar]
- Schnute, M.E.; Anderson, D.J. 4-Oxo-4,7-dihydrothieno[2,3-b]pyridine-5-carboxamides as antiviral agents. PCT WO2005003140A1, 13 January 2005. [Google Scholar]
- Kang, S.H.; Kim, C.M.; Youn, J.-H. An enantiocontrolled synthesis of the masked taxol C-13 side chain, oxazoline carboxylic acid. Tetrahedron Lett. 1999, 40, 3581–3582. [Google Scholar] [CrossRef]
- Fujioka, H.; Sawama, Y.; Kotoku, N.; Ohnaka, T.; Okitsu, T.; Murata, N.; Kubo, O.; Li, R.; Kita, Y. Concise asymmetric total synthesis of scyphostatin, a potent inhibitor of neutral sphingomyelinase. Chem. A Eur. J. 2007, 13, 10225–10238. [Google Scholar] [CrossRef] [PubMed]
- Sayyed, I.A.; Sudalai, A. Asymmetric synthesis of L-DOPA and (R)-selegiline via OsO4-catalyzed asymmetric dihydroxylation. Tetrahedron Asymmetry 2004, 15, 3111–3116. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).