
Metagenomic Insights into the Structure of Microbial Communities Involved in Nitrogen Cycling in Two Integrated Multitrophic Aquaculture (IMTA) Ponds


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sampling
2.2. Measurement of Water and Sediment Physiochemical Indexes
2.3. Microbial DNA Extraction
2.4. Illumina 16S rRNA Gene Sequencing Analysis
2.5. Metagenome Sequencing, Data Processing, and Assembly
2.6. Statistical Analysis
3. Results
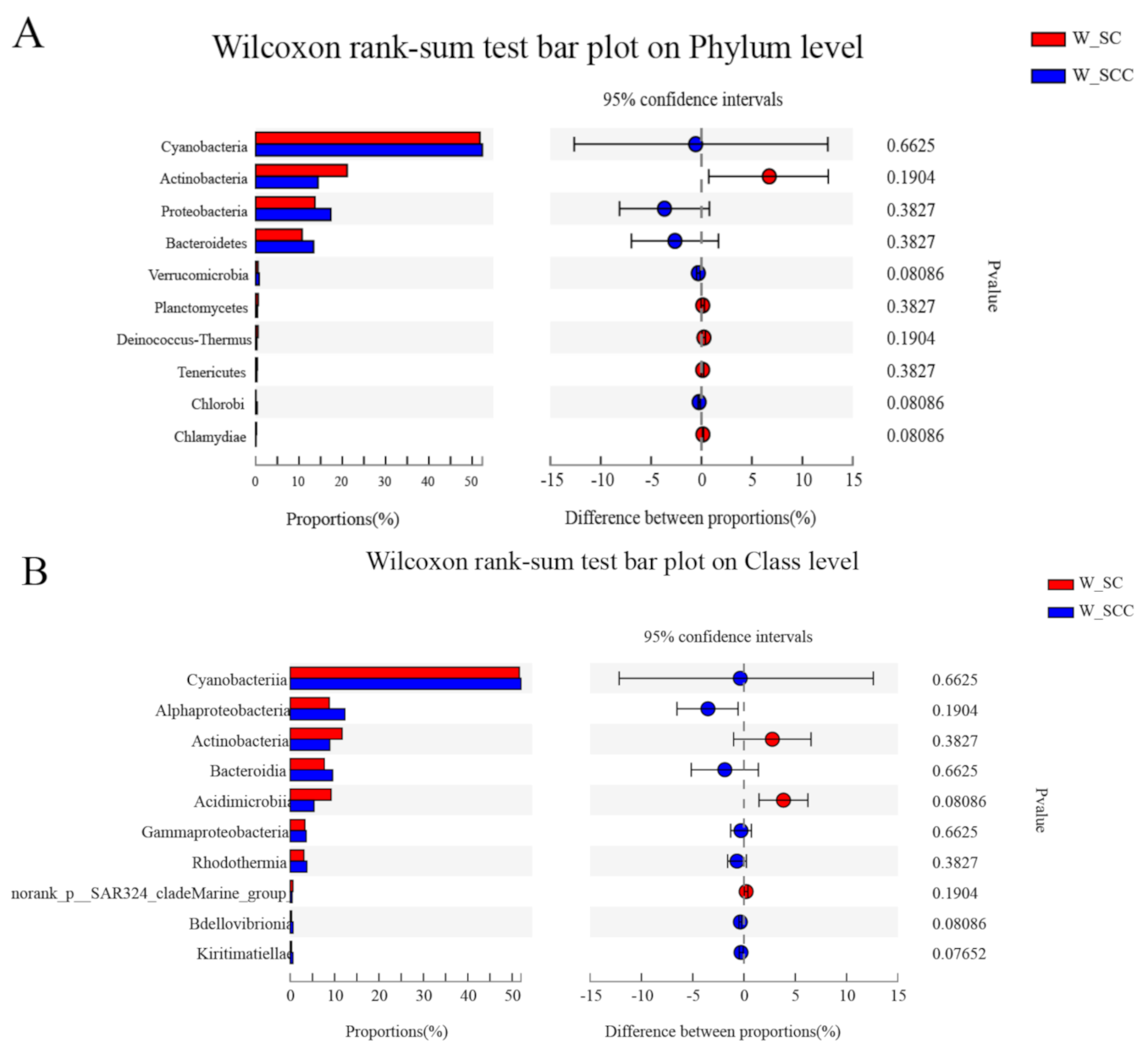
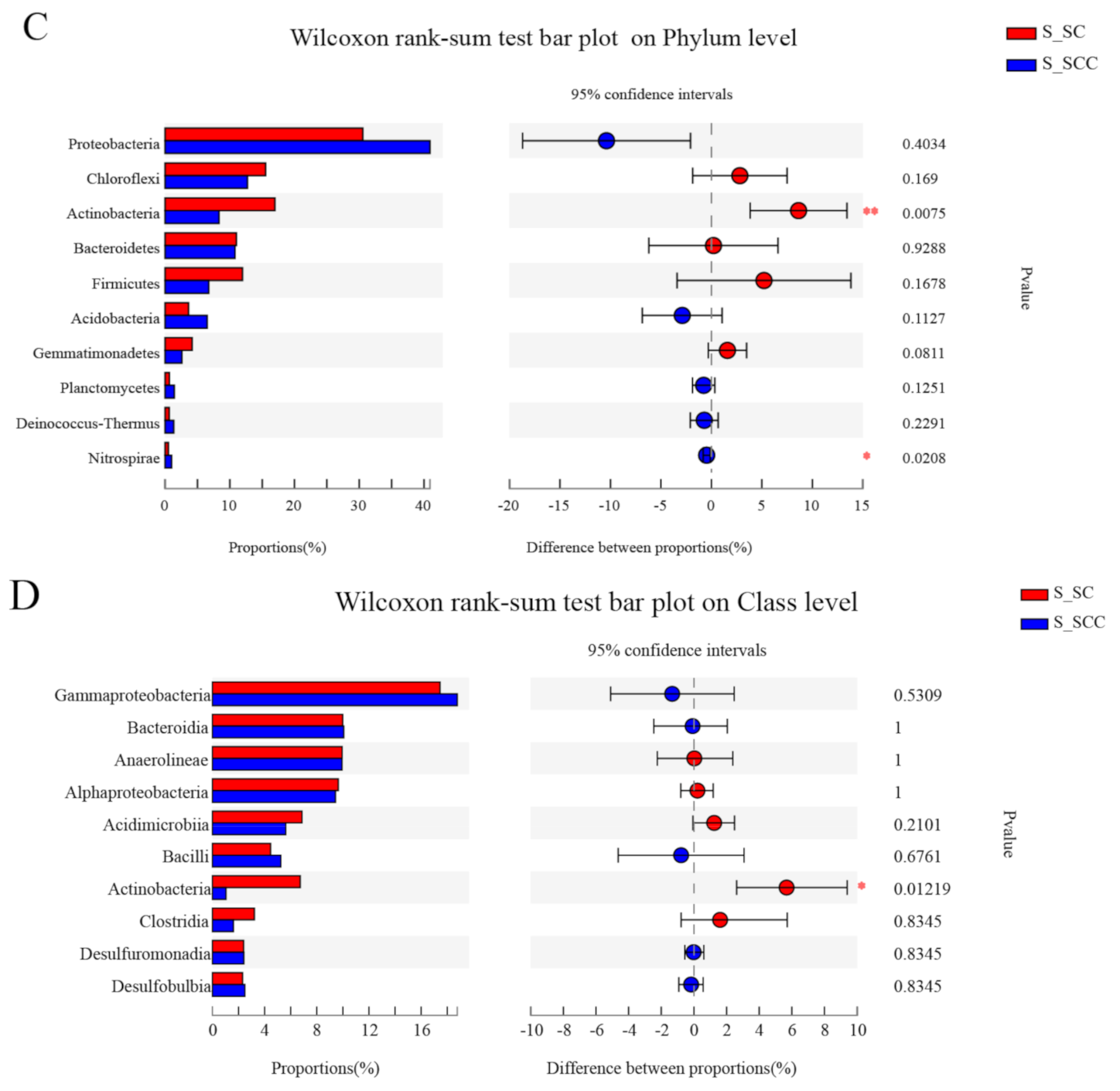
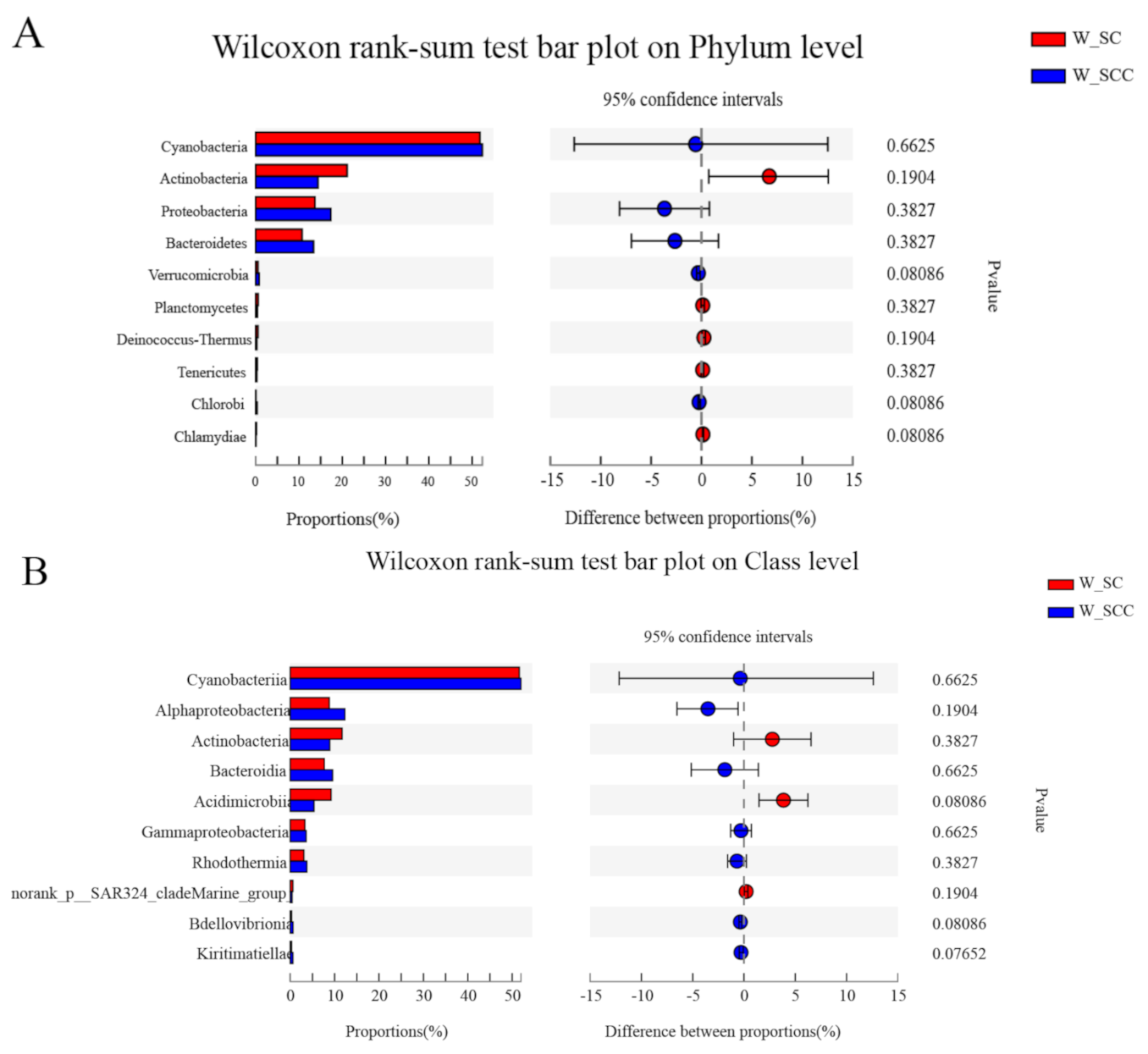
3.1. Microbial Community Composition in Water and Sediment
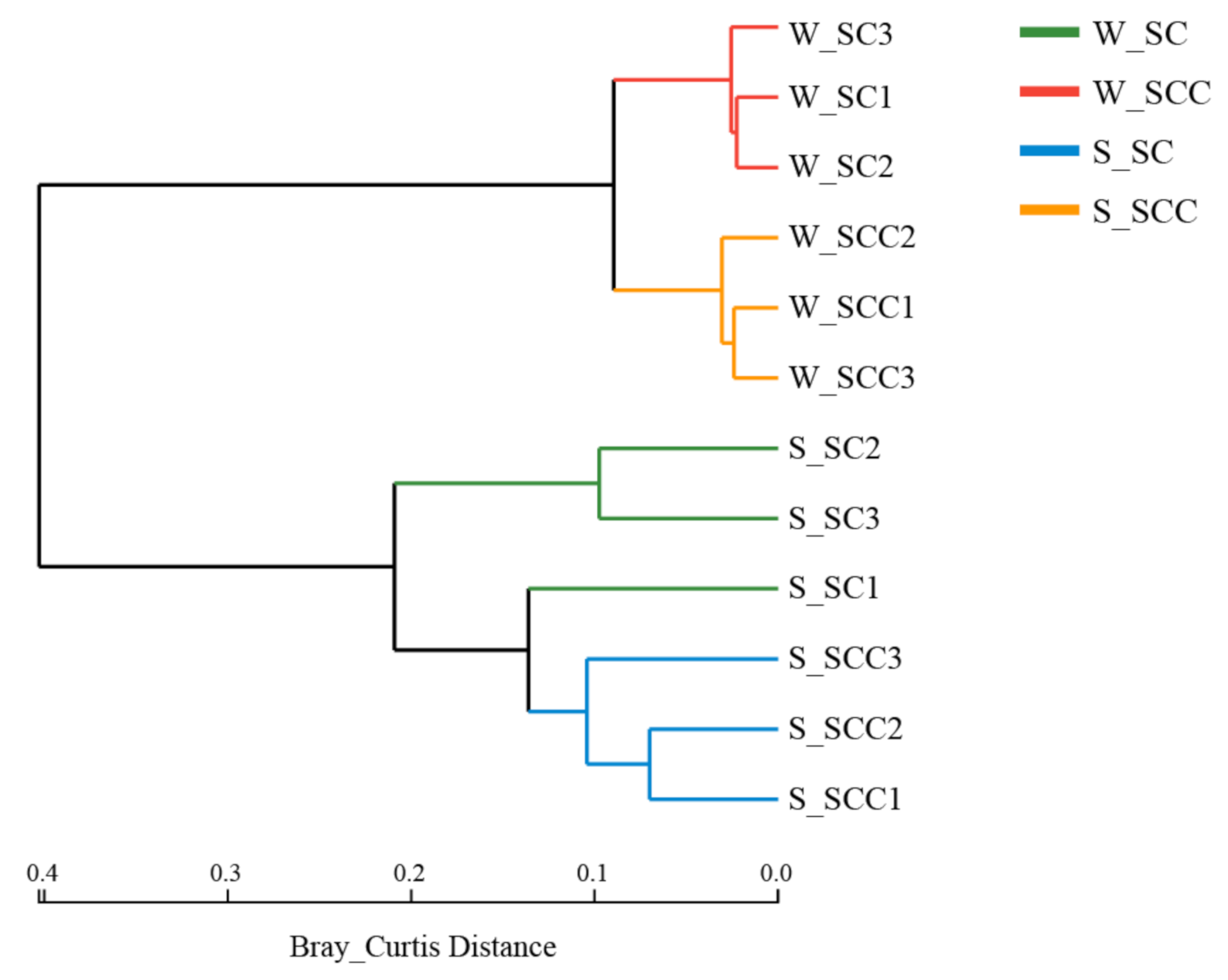
3.2. Microbial Community β Diversity in Water and Sediment
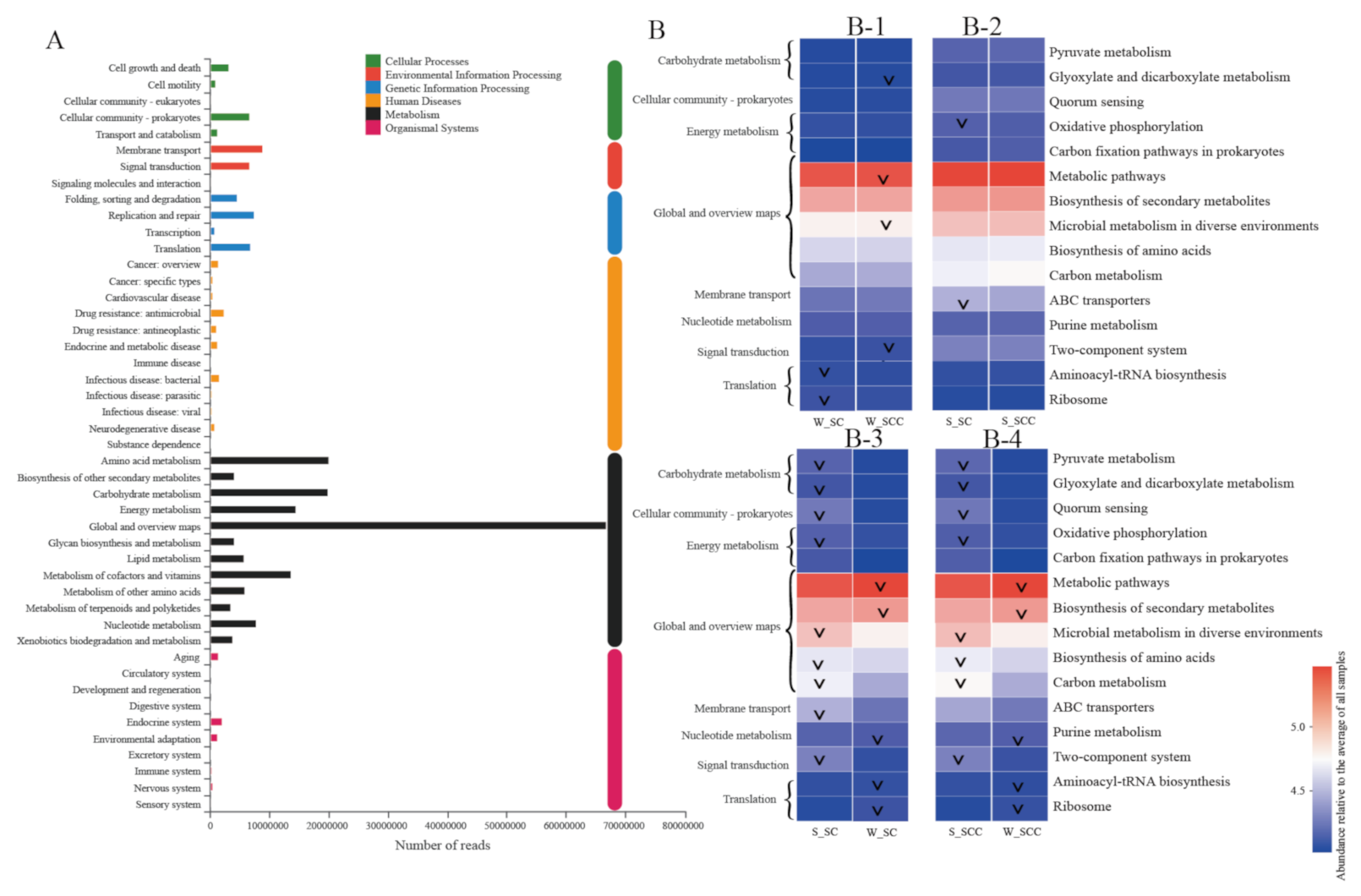
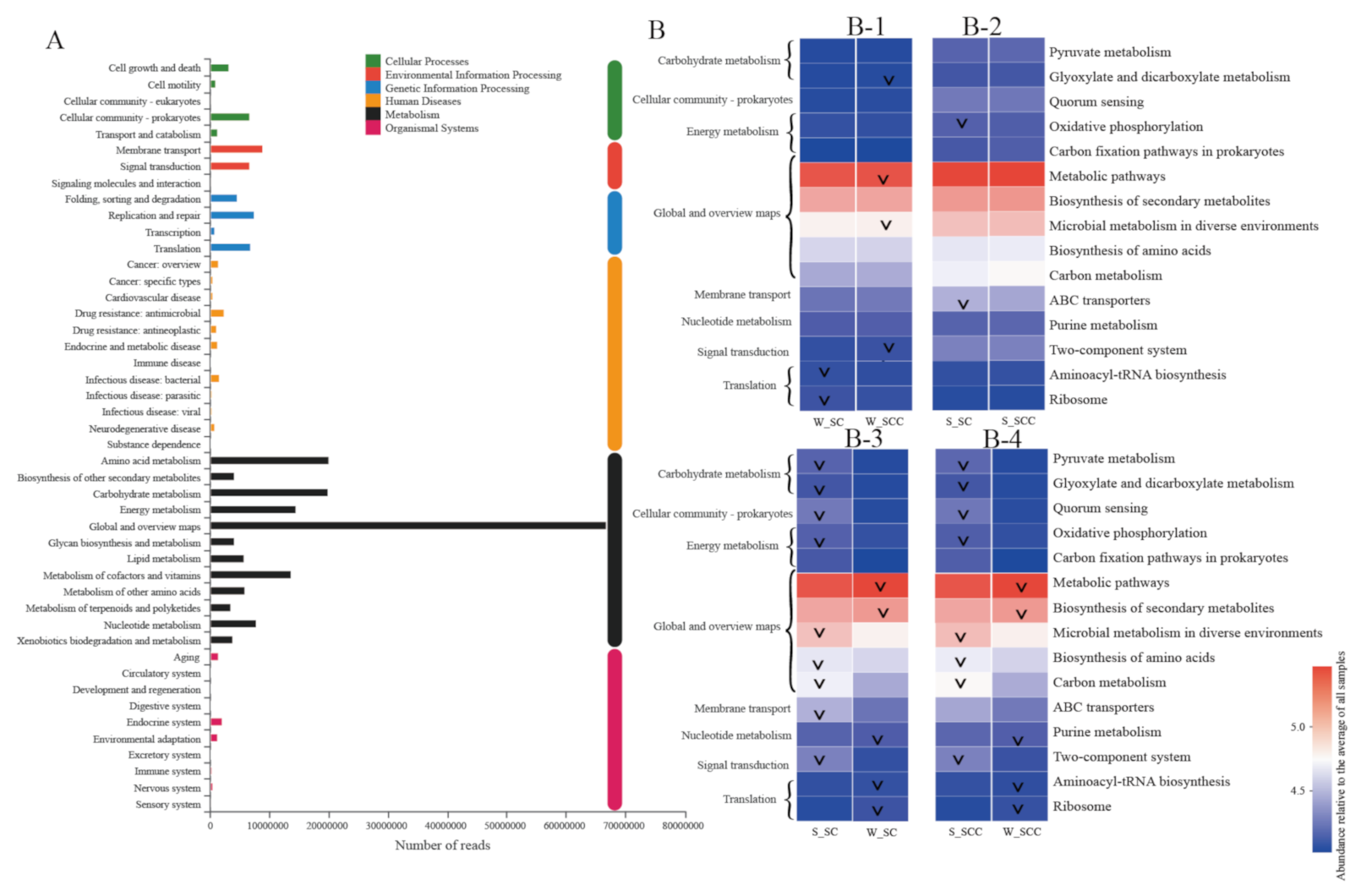
3.3. Microbial Functional Profile-Based Feedback Direction
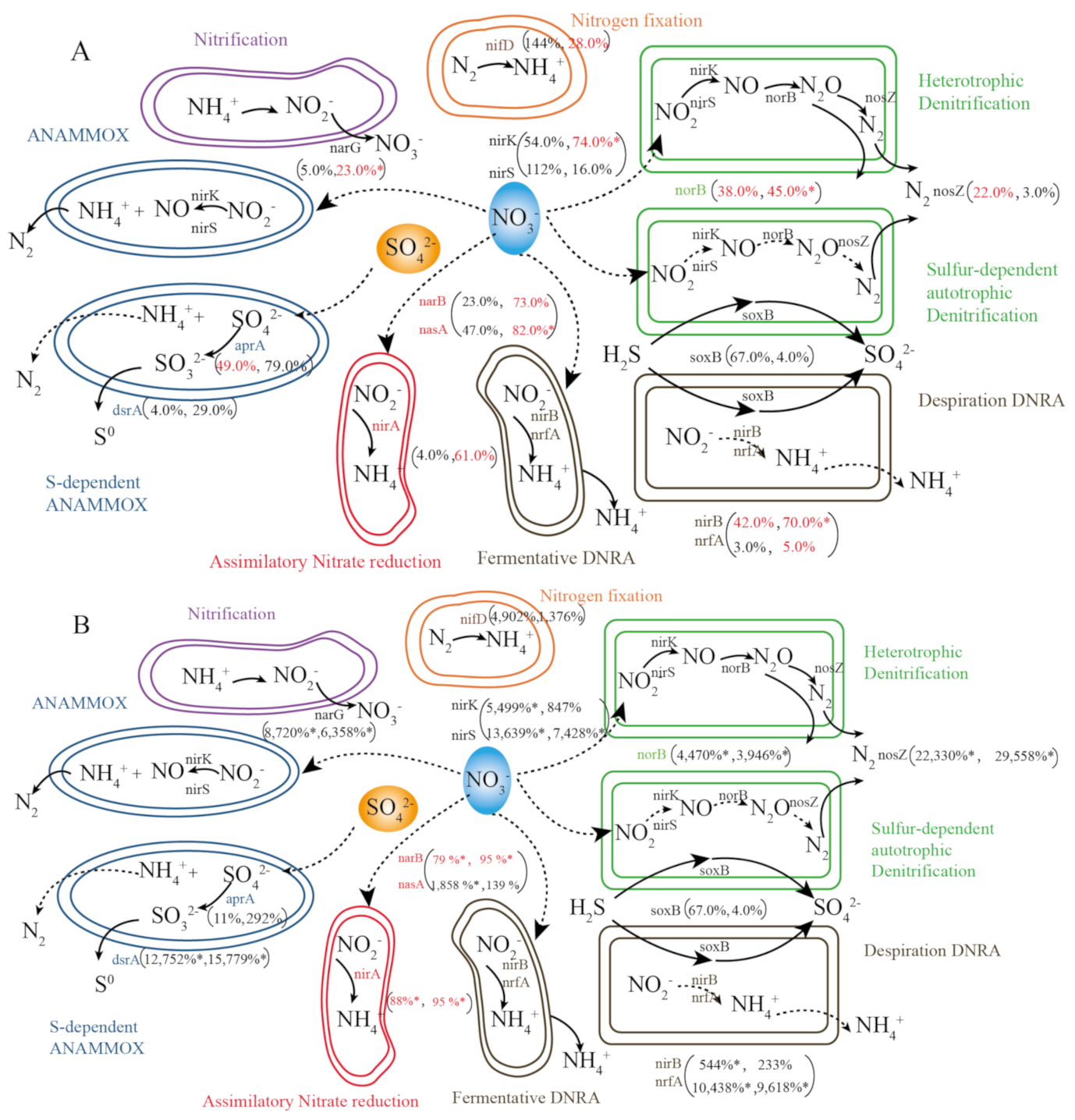
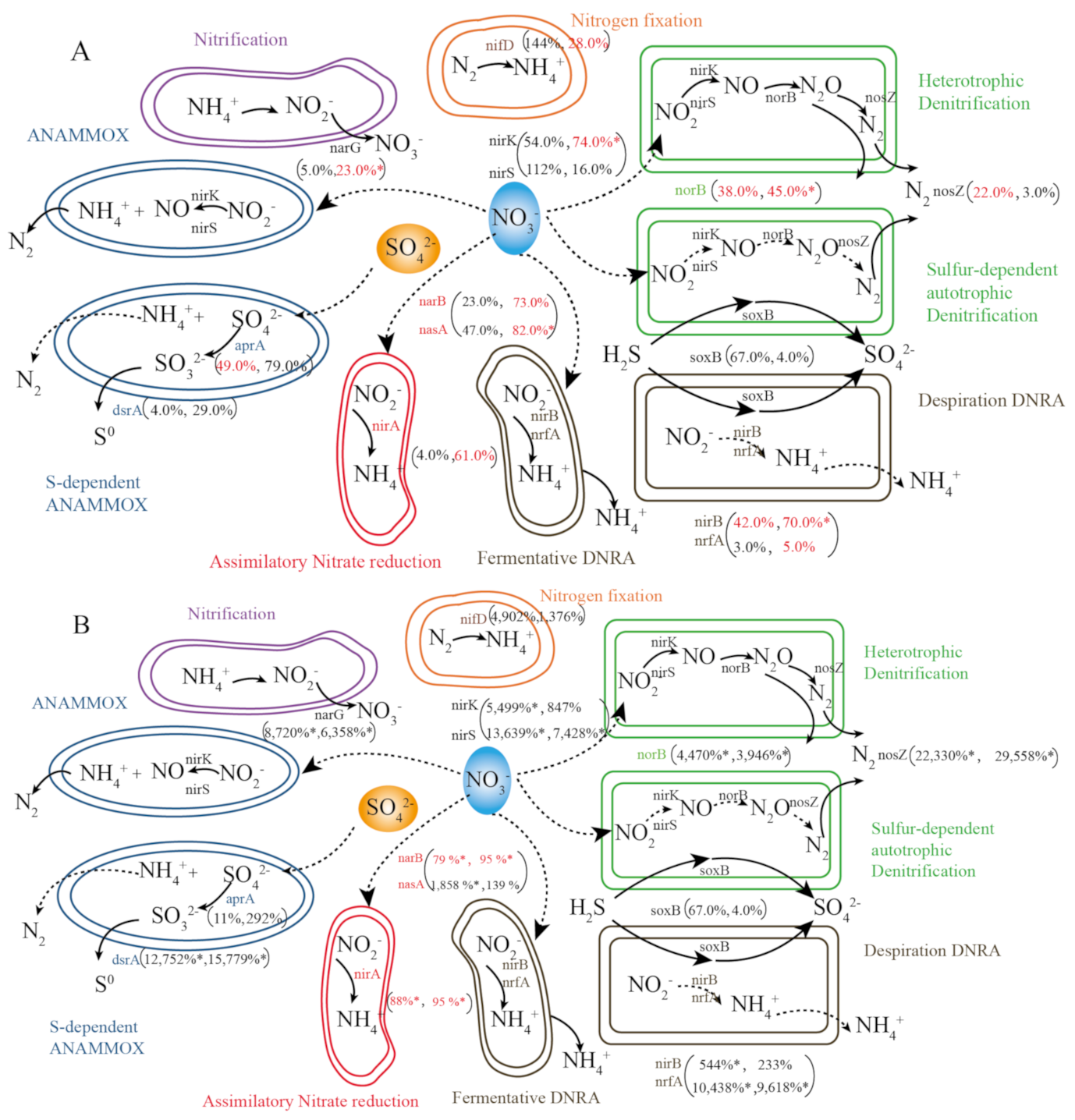
3.4. Expression of N Cycling Genes in Microbes
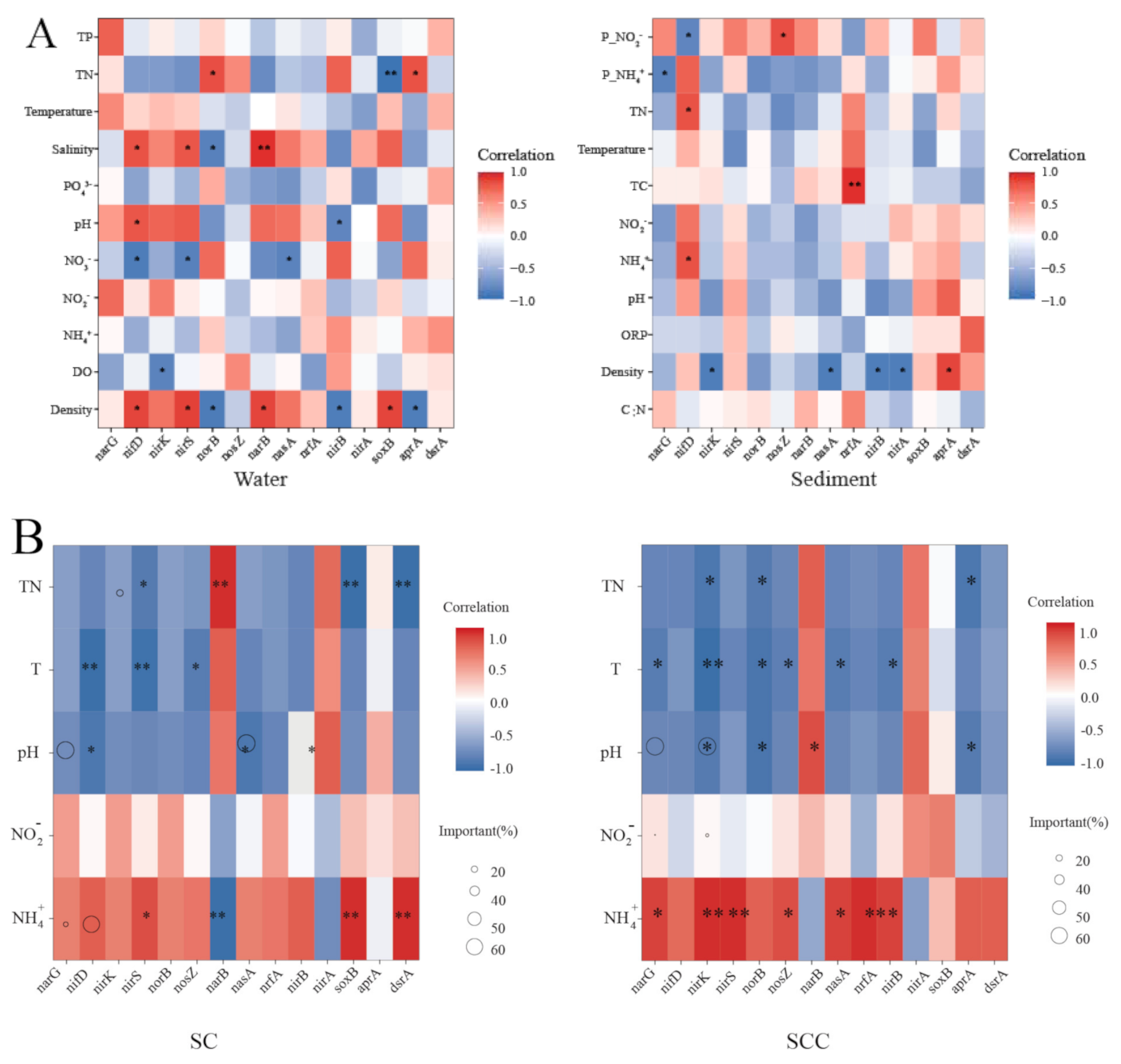
3.5. Drivers of the Differences in the Relative Abundance of N Cycling Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, M.; Pan, L.; Huang, F.; Gao, S.; Su, C.; Zhang, M.; He, Z. Metagenomic analysis of composition, function and cycling processes of microbial community in water, sediment and effluent of Litopenaeus vannamei farming environments under different culture modes. Aquaculture 2019, 506, 280–293. [Google Scholar] [CrossRef]
- Gonzalez, S.V.; Johnston, E.; Gribben, P.E.; Dafforn, K. The application of bioturbators for aquatic bioremediation: Review and meta-analysis. Environ. Pollut. 2019, 250, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Pan, L.; Zhang, M.; Huang, F.; Gao, S.; Tian, C. Changes of water, sediment, and intestinal bacterial communities in Penaeus japonicus cultivation and their impacts on shrimp physiological health. Aquacult. Int. 2020, 28, 1847–1865. [Google Scholar] [CrossRef]
- Wei, D.; Xing, C.; Hou, D.; Zeng, S.; Zhou, R.; Yu, L.; Wang, H.; Huang, Z.; Deng, Z.; Weng, H.; et al. Distinct bacterial communities in the environmental water, sediment and intestine between two crayfish-plant coculture ecosystems. Appl. Microbiol. Biot. 2021, 105, 5087–5101. [Google Scholar] [CrossRef]
- Lin, X.; Gao, D.; Lu, K.; Li, X. Bacterial Community Shifts Driven by Nitrogen Pollution in River Sediments of a Highly Urbanized City. Int. J. Environ. Res. Public Health 2019, 16, 3794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, K.; Huang, L.; Dong, P.; Wang, S.; Chen, H.; Lu, Z.; Hou, D.; Zhang, D. Fine-scale succession patterns and assembly mechanisms of bacterial community of Litopenaeus vannamei larvae across the developmental cycle. Microbiome 2020, 8, 106. [Google Scholar] [CrossRef]
- Hu, W.; Liang, J.; Ju, F.; Wang, Q.; Liu, R.; Bai, Y.; Liu, H.; Qu, J. Metagenomics unravels differential microbiome composition and metabolic potential in rapid sand filters purifying surface water versus groundwater. Environ. Sci. Technol. 2020, 54, 5197–5206. [Google Scholar] [CrossRef]
- Zeng, S.; Khoruamkid, S.; Kongpakdee, W.; Wei, D.; Yu, L.; Wang, H.; Deng, Z.; Weng, S.; Huang, Z.; He, J.; et al. Dissimilarity of microbial diversity of pond water, shrimp intestine and sediment in Aquamimicry system. AMB Express 2020, 10, 180. [Google Scholar] [CrossRef]
- Huang, F.; Pan, L.; Song, M.; Tian, C.; Gao, S. Microbiota assemblages of water, sediment, and intestine and their associations with environmental factors and shrimp physiological health. Appl. Microbiol. Biot. 2018, 102, 8585–8598. [Google Scholar] [CrossRef]
- Buck, B.H.; Troell, M.F.; Krause, G.; Angel, D.L.; Grote, B.; Chopin, T. State of the art and challenges for offshore Integrated Multi-Trophic Aquaculture (IMTA). Front. Mar. Sci. 2018, 5, 165. [Google Scholar] [CrossRef]
- Samocha, T.M.; Fricker, J.; Ali, A.M.; Shpigel, M.; Neori, A. Growth and nutrient uptake of the macroalga Gracilaria tikvahiae Cultured with the shrimp Litopenaeus vannamei in an Integrated Multi-Trophic Aquaculture (IMTA) system. Aquaculture 2015, 446, 263–371. [Google Scholar] [CrossRef]
- Nobre, A.M.; Robertson-Andersson, D.; Neori, A.; Sankar, K. Ecological-economic assessment of aquaculture options: Comparison between abalone monoculture and integrated multi-trophic aquaculture of abalone and seaweeds. Aquaculture 2010, 306, 116–126. [Google Scholar] [CrossRef]
- Santoro, A.E. Microbial nitrogen cycling at the saltwater-freshwater interface. Hydrogeol. J. 2010, 18, 187–202. [Google Scholar] [CrossRef]
- Huang, F.; Lin, X.; Hu, W.; Zeng, F.; He, L.; Yin, K. Nitrogen cycling processes in sediments of the Pearl River Estuary: Spatial variations, controlling factors, and environmental implications. Catena 2021, 206, 105545. [Google Scholar] [CrossRef]
- Lin, X.; Li, X.; Gao, D.; Liu, M.; Cheng, L. Ammonium Production and Removal in the Sediments of Shanghai River Networks: Spatiotemporal Variations, Controlling Factors, and Environmental Implications. J. Geophys. Res.-Biogeosci. 2017, 122, 2461–2478. [Google Scholar] [CrossRef]
- Zhang, Y.; Hua, Z.; Lu, H.; Oehmen, A.; Guo, J. Elucidating functional microorganisms and metabolic mechanisms in a novel engineered ecosystem integrating C, N, P and S biotransformation by metagenomics. Water Res. 2019, 148, 219–230. [Google Scholar] [CrossRef]
- Murphy, A.E.; Bulseco, A.N.; Ackerman, R.; Vineis, J.H.; Bowen, J.L. Sulphide addition favours respiratory ammonification (DNRA) over complete denitrification and alters the active microbial community in salt marsh sediments. Environ. Microbiol. 2020, 22, 2124–2139. [Google Scholar] [CrossRef]
- Slobodkina, G.B.; Reysenbach, A.L.; Kolganova, T.V.; Novikov, A.A.; Bonch-Osmolovskaya, E.A.; Slobodkin, A.I. Thermosulfuriphilus ammonigenes gen. nov., sp. nov., a thermophilic, chemolithoautotrophic bacterium capable of respiratory ammonification of nitrate with elemental sulfur. Int. J. Syst. Evol. Microbiol. 2017, 67, 3474–3479. [Google Scholar] [CrossRef]
- Lin, X.; Liu, M.; Hou, L.; Gao, D.; Li, X.; Lu, K.; Gao, J. Nitrogen losses in sediments of the east china sea: Spatiotemporal variations, controlling factors, and environmental implications. J. Geophys. Res.-Biogeosci. 2017, 122, 2699–2715. [Google Scholar] [CrossRef]
- Zhu, J.; He, Y.; Zhu, Y.; Huang, M.; Zhang, Y. Biogeochemical sulfur cycling coupling with dissimilatory nitrate reduction processes in freshwater sediments. Environ. Rev. 2018, 26, 121–132. [Google Scholar] [CrossRef]
- Song, B.; Lisa, J.A.; Tobias, C.R. Linking DNRA community structure and activity in a shallow lagoonal estuarine system. Front. Microbiol. 2014, 5, 460. [Google Scholar] [CrossRef] [Green Version]
- Rikmann, E.; Zekker, I.; Tomingas, M.; Tenno, T.; Menert, A.; Loorits, L.; Tenno, T. Sulfate-Reducing anaerobic ammonium oxidation as a potential treatment method for high nitrogen-content wastewater. Biodegradation 2012, 23, 509–524. [Google Scholar] [CrossRef]
- Levy-Booth, D.J.; Giesbrecht, I.J.W.; Kellogg, C.T.E.; Heger, T.J.; D’Amore, D.V.; Keeling, P.J.; Hallam, S.J.; Mohn, W.W. Seasonal and ecohydrological regulation of active microbial populations involved in DOC, CO2, and CH4 fluxes in temperate rainforest soil. ISME J. 2019, 13, 950–963. [Google Scholar] [CrossRef]
- Burgin, A.J.; Hamilton, S.K. NO3−-Driven SO42− Production in Freshwater Ecosystems: Implications for N and S Cycling. Ecosystems 2008, 11, 908–922. [Google Scholar] [CrossRef]
- Lin, X.; Hou, L.; Liu, M.; Li, X.; Zheng, Y.; Yin, G.; Gao, J.; Jiang, X. Nitrogen mineralization and immobilization in sediments of the East China Sea: Spatiotemporal variations and environmental implications. J. Geophys. Res.-Biogeosci. 2016, 121, 2842–2855. [Google Scholar] [CrossRef]
- Zhang, X.; Johnston, E.R.; Li, L.; Konstantinidis, K.T.; Han, X. Experimental warming reveals positive feedbacks to climate change in the Eurasian Steppe. ISME J. 2017, 11, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Gao, D.; Liu, Y.; Lin, X. Sediment nitrate reduction processes in response to environmental gradients along an urban river-estuary-sea continuum. Sci. Total Environ. 2020, 718, 137185. [Google Scholar] [CrossRef]
- Santoro, A.E.; Francis, C.A.; De Sieyes, N.R.; Boehm, A.B. Shifts in the relative abundance of ammonia-oxidizing bacteria and archaea across physicochemical gradients in a subterranean estuary. Environ. Microbiol. 2008, 10, 1068–1079. [Google Scholar] [CrossRef]
- Lin, X.; Hou, L.; Liu, M.; Li, X.; Yin, G.; Zheng, Y.; Deng, F. Gross nitrogen mineralization in surface sediments of the Yangtze Estuary. PLoS ONE 2016, 11, e0151930. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.; Huang, J.; Lu, J.; Su, M.; Hu, B.; Lin, X. Geochemical and microbial insights into vertical distributions of genetic potential of N-cycling processes in deep-sea sediments. Ecol. Indic. 2021, 125, 107461. [Google Scholar] [CrossRef]
- Zhang, D.; Tian, X.; Dong, S.; Chen, Y.; Feng, J.; He, R.P.; Zhang, K. Methane fluxes from typical marine polyculture ponds of swimming crab with kuruma shrimp and short-necked clam in eastern China. Aquac. Environ. Interact. 2019, 11, 63–73. [Google Scholar] [CrossRef]
- Hou, D.; Huang, Z.; Zeng, S.; Liu, J.; Wei, D.; Deng, X.; Weng, S.; He, Z.; He, J. Environmental Factors Shape Water Microbial Community Structure and Function in Shrimp Cultural Enclosure Ecosystems. Front. Microbiol. 2017, 8, 2359. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Xiao, B.; Xiong, J.; Chen, X. A simple miniaturised photometrical method for rapid determination of nitrate and nitrite in freshwater. Talanta 2010, 82, 976–983. [Google Scholar] [CrossRef]
- Laskov, C.; Herzog, C.; Lewandowski, J.; Hupfer, M. Miniaturized photometrical methods for the rapid analysis of phosphate, ammonium, ferrous iron, and sulfate in pore water of freshwater sediments. Limnol. Oceanogr. Methods 2007, 5, 63–71. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, H.; Park, J.; Takagi, T. MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006, 34, 5623–5630. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Li, Y.; Zhang, J.; Niu, L.; Zhang, W.; Cai, W.; Zhu, X. Sediment bacterial communities in a eutrophic lake influenced by multiple inflow-rivers. Environ. Sci. Pollut. Res. 2017, 24, 19795–19806. [Google Scholar] [CrossRef]
- Vargas, S.; Leiva, L.; Wörheide, G. Short-Term Exposure to High-Temperature Water Causes a Shift in the Microbiome of the Common Aquarium Sponge Lendenfeldia chondrodes. Microb. Ecol. 2021, 81, 213–222. [Google Scholar] [CrossRef]
- Hou, D.; Zhou, R.; Zeng, S.; Wei, D.; Deng, X.; Xing, C.; Weng, S.; He, J.; Huang, Z. Stochastic processes shape the bacterial community assembly in shrimp cultural pond sediments. Appl. Microbiol. Biot. 2021, 105, 5013–5022. [Google Scholar] [CrossRef]
- Fike, D.A.; Bradley, A.S.; Rose, C.V. Rethinking the ancient sulfur cycle. Annu. Rev. Earth Planet. Sci. 2015, 43, 593–622. [Google Scholar] [CrossRef] [Green Version]
- Zhi, L.; Li, X.; Bai, J.; Guan, Y. Integrating ecological and socioeconomic networks using nitrogen metabolism in the Yellow River Delta, China. Resour. Conserv. Recycl. 2020, 162, 105012. [Google Scholar] [CrossRef]
- Dai, Z.; Zang, H.; Chen, J.; Fu, Y.; Wang, X.; Liu, H.; Shen, C.; Xu, J.; Wang, J.; Chen, H.; et al. Metagenomic insights into soil microbial communities involved in carbon cycling along an elevation climosequences. Environ. Microbiol. 2021, 23, 4631–4645. [Google Scholar] [CrossRef]
- Adachi, K.; Oiwa, K.; Yoshida, M.; Nishizaka, T.; Kinosita, K. Controlled rotation of the F1-ATPase reveals differential and continuous binding changes for ATP synthesis. Nat. Commun. 2012, 3, 1022. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, Y.; Aoki, K.F.; Mamitsuka, H.; Kuma, K.I.; Kanehisa, M. The Evolutionary Repertoires of the Eukaryotic-Type ABC Transporters in Terms of the Phylogeny of ATP-Binding Domains in Eukaryotes and Prokaryotes. Mol. Biol. Evol. 2004, 21, 2149–2160. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, P.; Delgado-Baquerizo, M.; Trivedi, C.; Hu, H.; Anderson, I.C.; Jeffries, T.C.; Zhou, J.; Singh, B.K. Microbial regulation of the soil carbon cycle: Evidence from gene-enzyme relationships. ISME J. 2016, 10, 2593. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, T.; Doi, H.; Uramoto, G.; Wörmer, L.; Adhikari, R.R.; Xiao, N.; Morono, Y.; D’Hondt, S.; Hinrichs, K.U.; Inagaki, F. Global Diversity of Microbial Communities in Marine Sediment. Proc. Natl. Acad. Sci. USA 2020, 117, 27587–27597. [Google Scholar] [CrossRef]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Feng, G.; Sun, W.; Zhang, F.; Karthik, L.; Li, Z. Inhabitancy of active Nitrosopumilus-like ammonia-oxidizing archaea and Nitrospira nitrite-oxidizing bacteria in the sponge Theonella swinhoei. Sci. Rep. 2016, 6, 24966. [Google Scholar] [CrossRef] [Green Version]
- Székely, A.J.; Berga, M.; Langenheder, S. Mechanisms determining the fate of dispersed bacterial communities in new environments. ISME J. 2013, 7, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ren, H.; Shen, L.; Lou, L.; Tian, G.; Zheng, P.; Hu, B. pH levels drive bacterial community structure in the Qiantang River as determined by 454 pyrosequencing. Front. Microbiol. 2015, 6, 285. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Li, J.; Shan, H.; Xie, Y. Metagenomic Insights into the Structure of Microbial Communities Involved in Nitrogen Cycling in Two Integrated Multitrophic Aquaculture (IMTA) Ponds. J. Mar. Sci. Eng. 2022, 10, 171. https://doi.org/10.3390/jmse10020171
Liu Q, Li J, Shan H, Xie Y. Metagenomic Insights into the Structure of Microbial Communities Involved in Nitrogen Cycling in Two Integrated Multitrophic Aquaculture (IMTA) Ponds. Journal of Marine Science and Engineering. 2022; 10(2):171. https://doi.org/10.3390/jmse10020171
Chicago/Turabian StyleLiu, Qian, Junnan Li, Hongwei Shan, and Yicheng Xie. 2022. "Metagenomic Insights into the Structure of Microbial Communities Involved in Nitrogen Cycling in Two Integrated Multitrophic Aquaculture (IMTA) Ponds" Journal of Marine Science and Engineering 10, no. 2: 171. https://doi.org/10.3390/jmse10020171
APA StyleLiu, Q., Li, J., Shan, H., & Xie, Y. (2022). Metagenomic Insights into the Structure of Microbial Communities Involved in Nitrogen Cycling in Two Integrated Multitrophic Aquaculture (IMTA) Ponds. Journal of Marine Science and Engineering, 10(2), 171. https://doi.org/10.3390/jmse10020171