Advances in Integrating Genomics and Bioinformatics in the Plant Breeding Pipeline



Abstract
1. Introduction
2. Third Generation Sequencing to Improve Crop Genome Assemblies
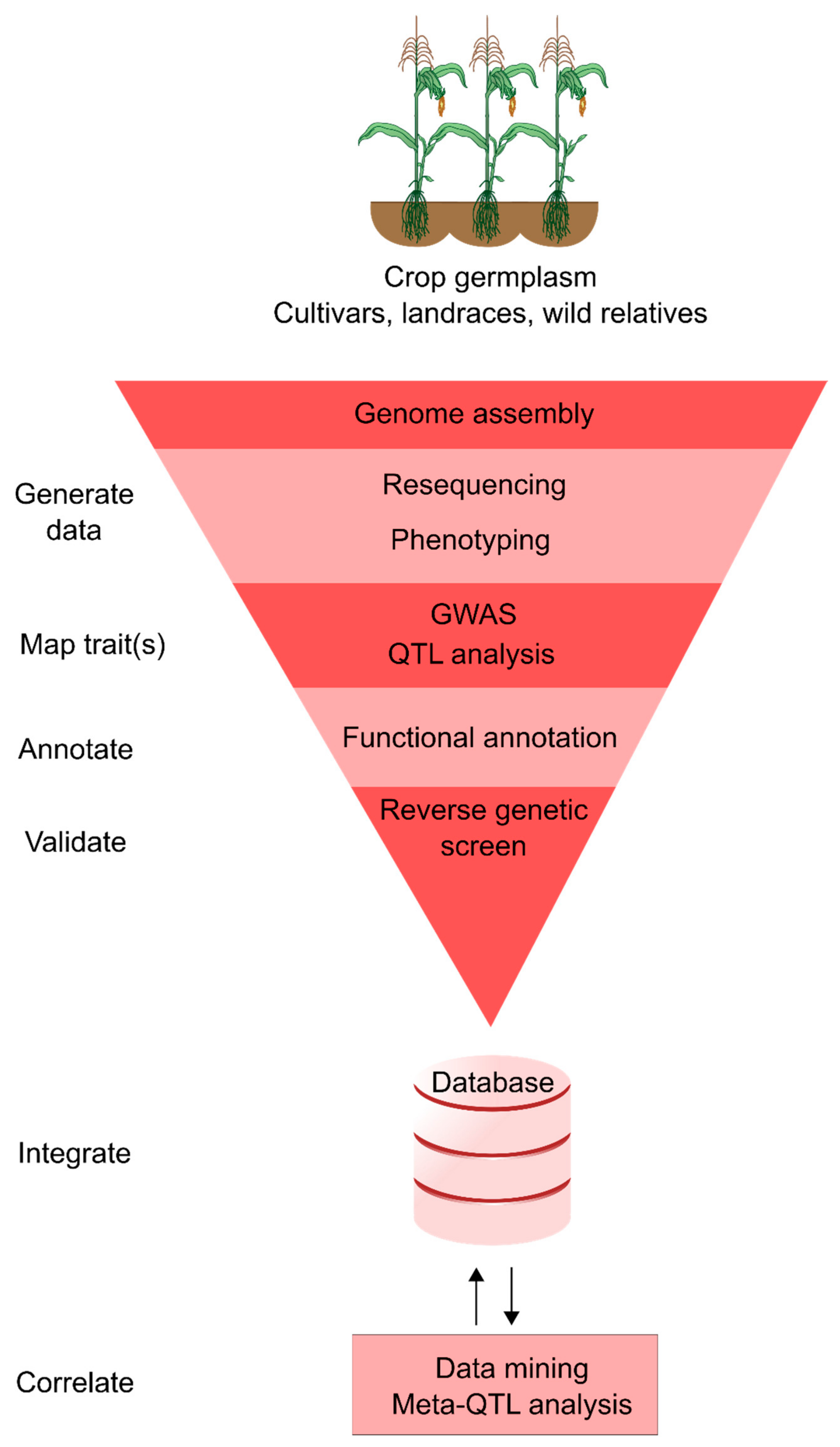
3. Integrated Crop Databases
4. Applying Integrative Genomics to Trait Discovery and Crop Improvement
4.1. Mining Quantitative Trait Loci Studies
4.2. Genome-Wide Association Studies for Identifying Breeding Targets
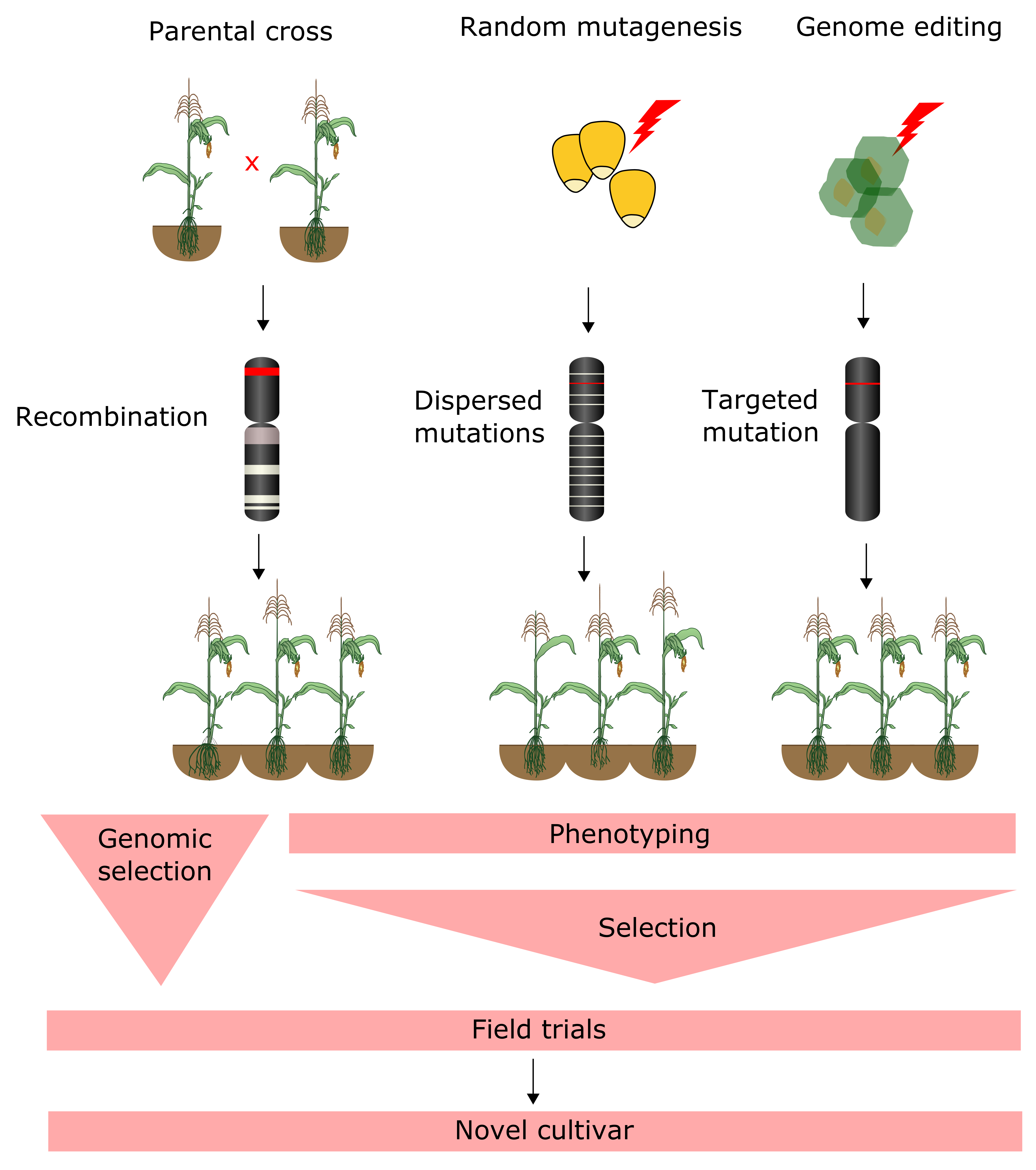
4.3. Forward and Reverse Genetic Screening
4.4. Genomic Selection
4.5. Beyond the Gene: Targeting Cis-Regulatory Elements for Crop Breeding
5. Applying Machine Learning to Crop Breeding
5.1. High Throughput Crop Phenotyping
5.2. Machine Learning in Crop Genomics Research
6. Genome Editing of Crops and Bioinformatics Challenges in Guide RNA Design
7. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Smit, E.; Nieto, F.J.; Crespo, C.J.; Mitchell, P. Estimates of animal and plant protein intake in US adults: Results from the Third National Health and Nutrition Examination Survey, 1988–1991. J. Acad. Nutr. Diet. 1999, 99, 813–820. [Google Scholar] [CrossRef]
- Ulijaszek, S.J. Human dietary change. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1991, 334, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Tilman, D.; Balzer, C.; Hill, J.; Befort, B.L. Global food demand and the sustainable intensification of agriculture. Proc. Natl. Acad. Sci. USA 2011, 108, 20260–20264. [Google Scholar] [CrossRef] [PubMed]
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Perez-de-Castro, A.M.; Vilanova, S.; Cañizares, J.; Pascual, L.; M Blanca, J.; J Diez, M.; Prohens, J.; Picó, B. Application of genomic tools in plant breeding. Curr. Genom. 2012, 13, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Abberton, M.; Batley, J.; Bentley, A.; Bryant, J.; Cai, H.; Cockram, J.; de Oliveira, A.C.; Cseke, L.J.; Dempewolf, H.; De Pace, C.; et al. Global agricultural intensification during climate change: A role for genomics. Plant Biotechnol. J. 2016, 14, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.; Batley, J.; Snowdon, R.J. Accessing complex crop genomes with next-generation sequencing. Theor. Appl. Genet. 2013, 126, 1–11. [Google Scholar] [CrossRef] [PubMed]
- IRGSP. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Troggio, M.; Cartwright, D.A.; Cestaro, A.; Pruss, D.; Pindo, M.; Fitzgerald, L.M.; Vezzulli, S.; Reid, J.; et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE 2007, 2, e1326. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; Jackson, S. The First 50 Plant Genomes. Plant Genome 2013, 6, 1–7. [Google Scholar] [CrossRef]
- Mousavi-Derazmahalleh, M.; Bayer, P.E.; Hane, J.K.; Babu, V.; Nguyen, H.T.; Nelson, M.N.; Erskine, W.; Varshney, R.K.; Papa, R.; Edwards, D. Adapting legume crops to climate change using genomic approaches. Plant Cell Environ 2018. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.L.; Scheben, A.; Edwards, D.; Spillane, C.; Ortiz, R. Assessing and exploiting functional diversity in germplasm pools to enhance abiotic stress adaptation and yield in cereals and food legumes. Front. Plant Sci. 2017, 8, 1461. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.H.; Asselbergs, F.W.; Williams, S.M. Bioinformatics challenges for genome-wide association studies. Bioinformatics 2010, 26, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Batley, J.; Edwards, D. The application of genomics and bioinformatics to accelerate crop improvement in a changing climate. Curr. Opin. Plant Biol. 2016, 30, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Pabinger, S.; Dander, A.; Fischer, M.; Snajder, R.; Sperk, M.; Efremova, M.; Krabichler, B.; Speicher, M.R.; Zschocke, J.; Trajanoski, Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief. Bioinform. 2014, 15, 256–278. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, E.; Lee, I.; Marcotte, E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 2015, 5, 17875. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Lee, H.; Darby, C.A.; Schatz, M.C. Piercing the dark matter: Bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet. 2018, 6, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Ong, Q.; Nguyen, P.; Phuong Thao, N.; Le, L. Bioinformatics approach in plant genomic research. Curr. Genom. 2016, 17, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Grierson, C.S.; Barnes, S.R.; Chase, M.W.; Clarke, M.; Grierson, D.; Edwards, K.J.; Jellis, G.J.; Jones, J.D.; Knapp, S.; Oldroyd, G.; et al. One hundred important questions facing plant science research. New Phytol. 2011, 192, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.E.; Carollo, V.L.; Lazo, G.R.; Anderson, O.D. GrainGenes, the genome database for small-grain crops. Nucleic Acids Res. 2003, 31, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Tello-Ruiz, M.K.; Naithani, S.; Stein, J.C.; Gupta, P.; Campbell, M.; Olson, A.; Wei, S.R.; Preece, J.; Geniza, M.J.; Jiao, Y.P.; et al. Gramene 2018: Unifying comparative genomics and pathway resources for plant research. Nucleic Acids Res. 2018, 46, D1181–D1189. [Google Scholar] [CrossRef] [PubMed]
- Scheben, A.; Batley, J.; Edwards, D. Revolution in genotyping platforms for crop improvement. In Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 1–16. [Google Scholar]
- Moose, S.P.; Mumm, R.H. Molecular plant breeding as the foundation for 21st century crop improvement. Plant Physiol. 2008, 147, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Ghosh, S.; Williams, M.J.; Cuddy, W.S.; Simmonds, J.; Rey, M.D.; Hatta, M.A.M.; Hinchliffe, A.; Steed, A.; Reynolds, D.; et al. Speed breeding is a powerful tool to accelerate crop research and breeding. Nat. Plants 2018, 4, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Algar, A.; Field, R.; Mayes, S. Integrating GIScience and Crop Science datasets: A study involving genetic, geographic and environmental data. PeerJ Preprints 2017, 5, e2248v2244. [Google Scholar] [CrossRef]
- Evans, K.; Jung, S.; Lee, T.; Brutcher, L.; Cho, I.; Peace, C.; Main, D. Addition of a breeding database in the Genome Database for Rosaceae. Database 2013, 2013, bat078. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lin, F.; An, D.; Wang, W.; Huang, R. Genome Sequencing and Assembly by Long Reads in Plants. Genes 2017, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Vlk, D.; Repkova, J. Application of next-generation sequencing in plant breeding. Czech J. Genet. Plant 2017, 53, 89–96. [Google Scholar] [CrossRef]
- Chen, K.; Ji, F.; Yuan, S.J.; Hao, W.T.; Wang, W.; Hu, Z.H. The performance of activated sludge exposed to arsanilic acid and amprolium hydrochloride in sequencing batch reactors. Int. Biodeterior. Biodegrad. 2017, 116, 260–265. [Google Scholar] [CrossRef]
- Jiao, W.-B.; Schneeberger, K. The impact of third generation genomic technologies on plant genome assembly. Curr. Opin. Plant Biol. 2017, 36, 64–70. [Google Scholar] [CrossRef] [PubMed]
- VanBuren, R.; Bryant, D.; Edger, P.P.; Tang, H.; Burgess, D.; Challabathula, D.; Spittle, K.; Hall, R.; Gu, J.; Lyons, E.; et al. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 2015, 527, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Stankova, H.; Hastie, A.R.; Chan, S.; Vrana, J.; Tulpova, Z.; Kubalakova, M.; Visendi, P.; Hayashi, S.; Luo, M.; Batley, J.; et al. BioNano genome mapping of individual chromosomes supports physical mapping and sequence assembly in complex plant genomes. Plant Biotechnol. J. 2016, 14, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A method to study the three-dimensional architecture of genomes. J. Vis. Exp. 2010, 39, e1869. [Google Scholar] [CrossRef] [PubMed]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; Jupe, F.; Bemm, F.; Motley, S.T.; Sandoval, J.P.; Lanz, C.; Loudet, O.; Weigel, D.; Ecker, J.R. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 2018, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; Gurtowski, J.; Ethe-Sayers, S.; Deshpande, P.; Schatz, M.C.; McCombie, W.R. Oxford Nanopore sequencing, hybrid error correction and de novo assembly of a eukaryotic genome. Genome Res. 2015, 25, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Med.stanford.edu. Stanford Medicine Sequencing Service Rates. Available online: http://med.stanford.edu/gssc/rates.html (accessed on 27 May 2018).
- Cgrb.oregonstate.edu. Illumina HiSeq 3000 Service Fees. Available online: http://cgrb.oregonstate.edu/core/illumina-hiseq-3000/illumina-hiseq-3000-service-fees (accessed on 27 May 2018).
- Allseq.com. General overview of Illumina Sequencing. Available online: http://allseq.com/knowledge-bank/sequencing-platforms/illumina/ (accessed on 27 May 2018).
- Gordon, D.; Huddleston, J.; Chaisson, M.J.P.; Hill, C.M.; Kronenberg, Z.N.; Munson, K.M.; Malig, M.; Raja, A.; Fiddes, I.; Hillier, L.W.; et al. Long-read sequence assembly of the gorilla genome. Science 2016, 352, aae0344. [Google Scholar] [CrossRef] [PubMed]
- Washington.edu. University of Washington PacBio Sequencing Services. Available online: https://pacbio.gs.washington.edu/ (accessed on 27 May 2018).
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Pankhurst, L.; Hubbard, A.; Votintseva, A.; Stoesser, N.; Sheppard, A.E.; Mathers, A.; Norris, R.; Navickaite, I.; Eaton, C.; et al. Resolving plasmid structures in Enterobacteriaceae using the MinION nanopore sequencer: Assessment of MinION and MinION/Illumina hybrid data assembly approaches. Microb. Genom. 2017, 3, e000118. [Google Scholar] [CrossRef] [PubMed]
- Nanoporetech.com. Available online: https://nanoporetech.com (accessed on 27 May 2018).
- Schmidt, M.H.W.; Vogel, A.; Denton, A.K.; Istace, B.; Wormit, A.; van de Geest, H.; Bolger, M.E.; Alseekh, S.; Mass, J.; Pfaff, C.; et al. De novo assembly of a new Solanum pennellii accession using nanopore sequencing. Plant Cell 2017, 29, 2336–2348. [Google Scholar] [CrossRef] [PubMed]
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. GenBank. Nucleic Acids Res. 2008, 36, D25. [Google Scholar] [CrossRef] [PubMed]
- Kanz, C.; Aldebert, P.; Althorpe, N.; Baker, W.; Baldwin, A.; Bates, K.; Browne, P.; van den Broek, A.; Castro, M.; Cochrane, G.; et al. The EMBL Nucleotide Sequence Database. Nucleic Acids Res. 2005, 33, D29–D33. [Google Scholar] [CrossRef] [PubMed]
- Duvick, J.; Fu, A.; Muppirala, U.; Sabharwal, M.; Wilkerson, M.D.; Lawrence, C.J.; Lushbough, C.; Brendel, V. PlantGDB: A resource for comparative plant genomics. Nucleic Acids Res. 2007, 36, D959–D965. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.; Lorenc, M.T.; Edwards, D. Genomic databases for crop improvement. Agronomy 2012, 2, 62. [Google Scholar] [CrossRef]
- Hassani-Pak, K.; Rawlings, C. Knowledge Discovery in Biological Databases for Revealing Candidate Genes Linked to Complex Phenotypes. J. Integr. Bioinform. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Hassani-Pak, K.; Castellote, M.; Esch, M.; Hindle, M.; Lysenko, A.; Taubert, J.; Rawlings, C. Developing integrated crop knowledge networks to advance candidate gene discovery. Appl. Transl. Genom. 2016, 11, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Scheben, A.; Chan, C.K.; Edwards, D. Databases for wheat genomics and crop improvement. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2017; Volume 1679, pp. 277–291. [Google Scholar]
- Furbank, R.T.; Tester, M. Phenomics–technologies to relieve the phenotyping bottleneck. Trends Plant Sci. 2011, 16, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F.; Stone, E.A.; Ayroles, J.F. The genetics of quantitative traits: Challenges and prospects. Nat. Rev. Genet. 2009, 10, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Mace, E.; Jordan, D. Integrating sorghum whole genome sequence information with a compendium of sorghum QTL studies reveals uneven distribution of QTL and of gene-rich regions with significant implications for crop improvement. Theor. Appl. Genet. 2011, 123, 169–191. [Google Scholar] [CrossRef] [PubMed]
- Veyrieras, J.B.; Goffinet, B.; Charcosset, A. MetaQTL: A package of new computational methods for the meta-analysis of QTL mapping experiments. BMC Bioinform. 2007, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Kumasaka, N.; Knights, A.J.; Gaffney, D.J. Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat. Genet. 2016, 48, 206. [Google Scholar] [CrossRef] [PubMed]
- Tecle, I.Y.; Menda, N.; Buels, R.M.; van der Knaap, E.; Mueller, L.A. solQTL: A tool for QTL analysis, visualization and linking to genomes at SGN database. BMC Bioinform. 2010, 11, 525. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.X.; Zhang, H.; Zhao, P.; Zhang, Z.X.; Liang, W.K.; Tian, Z.G.; Zheng, Y.L. Mining of candidate maize genes for nitrogen use efficiency by integrating gene expression and QTL data. Plant Mol. Biol. Report. 2012, 30, 297–308. [Google Scholar] [CrossRef]
- Said, J.I.; Lin, Z.; Zhang, X.; Song, M.; Zhang, J. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance and disease resistance in tetraploid cotton. BMC Genom. 2013, 14, 776. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.-M.; Wu, Q.; Han, X.; Sun, Y.-N.; Du, X.-Y.; Liu, C.-Y.; Jiang, H.-W.; Hu, G.-H.; Chen, Q.-S. Soybean oil content QTL mapping and integrating with meta-analysis method for mining genes. Euphytica 2011, 179, 499–514. [Google Scholar] [CrossRef]
- Hanocq, E.; Laperche, A.; Jaminon, O.; Laine, A.-L.; Le Gouis, J. Most significant genome regions involved in the control of earliness traits in bread wheat, as revealed by QTL meta-analysis. Theor. Appl. Genet. 2007, 114, 569–584. [Google Scholar] [CrossRef] [PubMed]
- Borevitz, J.O.; Nordborg, M. The impact of genomics on the study of natural variation in Arabidopsis. Plant Physiol. 2003, 132, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.; Roux, F. Towards identifying genes underlying ecologically relevant traits in Arabidopsis thaliana. Nat. Rev. Genet. 2010, 11, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Han, B. Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant Biol. 2014, 65, 531–551. [Google Scholar] [CrossRef] [PubMed]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Sonah, H.; O’Donoughue, L.; Cober, E.; Rajcan, I.; Belzile, F. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol. J. 2015, 13, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Gacek, K.; Bayer, P.E.; Bartkowiak-Broda, I.; Szala, L.; Bocianowski, J.; Edwards, D.; Batley, J. Genome-wide association study of genetic control of seed fatty acid biosynthesis in Brassica napus. Front. Plant Sci. 2017, 7, 2062. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Blackwell, T.; Boehnke, M.; Scott, L.J.; GoT2D Investigators. Recommended joint and meta-analysis strategies for case-control association testing of single low-count variants. Genet. Epidemiol. 2013, 37, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [PubMed]
- Jankowicz-Cieslak, J.; Till, B.J. Forward and reverse genetics in crop breeding. In Advances in Plant Breeding Strategies: Breeding, Biotechnology and Molecular Tools; Al-Khayri, J.M., Jain, S.M., Johnson, D.V., Eds.; Springer: Heidelberg, Germany, 2015; Volume 1, pp. 215–240. [Google Scholar]
- Sessions, A.; Burke, E.; Presting, G.; Aux, G.; McElver, J.; Patton, D.; Dietrich, B.; Ho, P.; Bacwaden, J.; Ko, C.; et al. A high-throughput Arabidopsis reverse genetics system. Plant Cell 2002, 14, 2985–2994. [Google Scholar] [CrossRef] [PubMed]
- Henry, I.M.; Nagalakshmi, U.; Lieberman, M.C.; Ngo, K.J.; Krasileva, K.V.; Vasquez-Gross, H.; Akhunova, A.; Akhunov, E.; Dubcovsky, J.; Tai, T.H.; et al. Efficient genome-wide detection and cataloging of EMS-induced mutations using exome capture and next-generation sequencing. Plant Cell 2014, 26, 1382–1397. [Google Scholar] [CrossRef] [PubMed]
- Slade, A.J.; Fuerstenberg, S.I.; Loeffler, D.; Steine, M.N.; Facciotti, D. A reverse genetic, nontransgenic approach to wheat crop improvement by TILLING. Nat. Biotechnol. 2005, 23, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Wang, T.L.; Welham, T.J.; Gardner, S.; Pike, J.M.; Yoshida, S.; Parniske, M. A TILLING reverse genetics tool and a web-accessible collection of mutants of the legume Lotus japonicus. Plant Physiol. 2003, 131, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Jannink, J.L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Bhat, J.A.; Ali, S.; Salgotra, R.K.; Mir, Z.A.; Dutta, S.; Jadon, V.; Tyagi, A.; Mushtaq, M.; Jain, N.; Singh, P.K.; et al. Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Front. Genet. 2016, 7, 221. [Google Scholar] [CrossRef] [PubMed]
- Heffner, E.L.; Sorrells, M.E.; Jannink, J.-L. Genomic selection for crop improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Lin, Z.; Cogan, N.O.; Pembleton, L.W.; Spangenberg, G.C.; Forster, J.W.; Hayes, B.J.; Daetwyler, H.D. Genetic gain and inbreeding from genomic selection in a simulated commercial breeding program for perennial ryegrass. Plant Genome 2016, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, E.J.; de Resende, M.D.V.; da Silva Santos, V.; Ferreira, C.F.; Oliveira, G.A.F.; da Silva, M.S.; de Oliveira, L.A.; Aguilar-Vildoso, C.I. Genome-wide selection in cassava. Euphytica 2012, 187, 263–276. [Google Scholar] [CrossRef]
- Scheben, A.; Batley, J.; Edwards, D. Genotyping-by-sequencing approaches to characterize crop genomes: Choosing the right tool for the right application. Plant Biotechnol. J. 2017, 15, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Voss-Fels, K.; Snowdon, R.J. Understanding and utilizing crop genome diversity via high-resolution genotyping. Plant Biotechnol. J. 2016, 14, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.; Endelman, J.; Dawson, J.; Rutkoski, J.; Wu, S.Y.; Manes, Y.; Dreisigacker, S.; Crossa, J.; Sanchez-Villeda, H.; Sorrells, M.; et al. Genomic Selection in Wheat Breeding using Genotyping-by-Sequencing. Plant Genome 2012, 5, 103–113. [Google Scholar] [CrossRef]
- Rutkoski, J.E.; Poland, J.A.; Singh, R.P.; Huerta-Espino, J.; Bhavani, S.; Barbier, H.; Rouse, M.N.; Jannink, J.L.; Sorrells, M.E. Genomic Selection for Quantitative Adult Plant Stem Rust Resistance in Wheat. Plant Genome 2014, 7, 1–10. [Google Scholar] [CrossRef]
- Crossa, J.; Beyene, Y.; Kassa, S.; Perez, P.; Hickey, J.M.; Chen, C.; de los Campos, G.; Burgueno, J.; Windhausen, V.S.; Buckler, E.; et al. Genomic prediction in maize breeding populations with genotyping-by-sequencing. G3 Genes Genomes Genet. 2013, 3, 1903–1926. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.S.; Purugganan, M.D. Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet. 2013, 14, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, G.; Goossens, A.; Pauwels, L. Lessons from domestication: Targeting cis-regulatory elements for crop improvement. Trends Plant Sci. 2016, 21, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, S.; Liu, Q.; Wu, K.; Zhang, J.; Wang, S.; Wang, Y.; Chen, X.; Zhang, Y.; Gao, C.; et al. The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat. Genet. 2015, 47, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Pauler, F.M.; Stricker, S.H.; Warczok, K.E.; Barlow, D.P. Long-range DNase I hypersensitivity mapping reveals the imprinted Igf2r and Air promoters share cis-regulatory elements. Genome Res. 2005, 15, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, G.; Lopes, R.; Ugalde, A.P.; Nevedomskaya, E.; Han, R.Q.; Myacheva, K.; Zwart, W.; Elkon, R.; Agami, R. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Rombauts, S.; Florquin, K.; Lescot, M.; Marchal, K.; Rouzé, P.; Van de Peer, Y. Computational approaches to identify promoters and cis-regulatory elements in plant genomes. Plant Physiol. 2003, 132, 1162–1176. [Google Scholar] [CrossRef] [PubMed]
- Van de Velde, J.; Heyndrickx, K.S.; Vandepoele, K. Inference of transcriptional networks in Arabidopsis through conserved noncoding sequence analysis. Plant Cell 2014, 26, 2729–2745. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.B.; Yu, H.; Zhang, Y.F.; Zhuang, F.; Song, X.G.; Gao, S.S.; Gao, C.X.; Li, J.Y. Construction of a genome-wide mutant library in rice using CRISPR/Cas9. Mol. Plant 2017, 10, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.M.; Ye, X.; Guo, R.M.; Huang, J.; Wang, W.; Tang, J.Y.; Tan, L.T.; Zhu, J.K.; Chu, C.C.; Qian, Y.W. Genome-wide targeted mutagenesis in rice using the CRISPR/Cas9 system. Mol. Plant 2017, 10, 1242–1245. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Leal, D.; Lemmon, Z.H.; Man, J.; Bartlett, M.E.; Lippman, Z.B. Engineering quantitative trait variation for crop improvement by genome editing. Cell 2017, 171, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.; Liebisch, F.; Hund, A. Plant phenotyping: From bean weighing to image analysis. Plant Methods 2015, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Koh, Z.X.; Goh, J.; Lin, Z.; Haaland, B.; Ting, B.P.; Ong, M.E.H. Prediction of adverse cardiac events in emergency department patients with chest pain using machine learning for variable selection. BMC Med. Inform. Decis. Mak. 2014, 14, 75. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Ganapathysubramanian, B.; Singh, A.K.; Sarkar, S. Machine learning for high-throughput stress phenotyping in plants. Trends Plant Sci. 2016, 21, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Naik, H.S.; Zhang, J.P.; Lofquist, A.; Assefa, T.; Sarkar, S.; Ackerman, D.; Singh, A.; Singh, A.K.; Ganapathysubramanian, B. A real-time phenotyping framework using machine learning for plant stress severity rating in soybean. Plant Methods 2017, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Blystone, D.; Singh, A.K.; Ganapathysubramanian, B.; Singh, A.; Sarkar, S. An explainable deep machine vision framework for plant stress phenotyping. Proc. Natl. Acad. Sci. USA 2018, 115, 201716999. [Google Scholar] [CrossRef] [PubMed]
- Ubbens, J.R.; Stavness, I. Deep Plant Phenomics: A deep learning platform for complex plant phenotyping tasks. Front. Plant Sci. 2017, 8, 1190. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, D.; Schluter, U.; Weber, A.P.M. Machine learning techniques for predicting crop photosynthetic capacity from leaf reflectance spectra. Mol. Plant 2017, 10, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, H.H.; Wang, X. Machine learning for Big Data analytics in plants. Trends Plant Sci. 2014, 19, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.; D’Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D.; et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Mapleson, D.; Venturini, L.; Kaithakottil, G.; Swarbreck, D. Efficient and accurate detection of splice junctions from RNAseq with Portcullis. bioRxiv 2017, 217620. [Google Scholar] [CrossRef]
- Hecker, M.; Lambeck, S.; Toepfer, S.; Van Someren, E.; Guthke, R. Gene regulatory network inference: Data integration in dynamic models—A review. Biosystems 2009, 96, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Roy, S.; Ay, F.; Meyer, P.E.; Candeias, R.; Kahveci, T.; Bristow, C.A.; Kellis, M. Predictive regulatory models in Drosophila melanogaster by integrative inference of transcriptional networks. Genome Res. 2012, 22, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Rafalski, J.A. Novel genetic mapping tools in plants: SNPs and LD-based approaches. Plant Sci. 2002, 162, 329–333. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Buggs, R.J.A.; Renny-Byfield, S.; Chester, M.; Jordon-Thaden, I.E.; Viccini, L.F.; Chamala, S.; Leitch, A.R.; Schnable, P.S.; Barbazuk, W.B.; Soltis, P.S.; et al. Next-generation sequencing and genome evolution in allopolyploids. Am. J. Bot. 2012, 99, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Clevenger, J.P.; Korani, W.; Ozias-Akins, P.; Jackson, S. Haplotype-based genotyping in polyploids. Front. Plant Sci. 2018, 9, 564. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Sedlazeck, F.J.; Lam, T.-W.; Schatz, M. Clairvoyante: A multi-task convolutional deep neural network for variant calling in Single Molecule Sequencing. bioRxiv 2018, 310458. [Google Scholar] [CrossRef]
- Gottschalk, W.; Wolff, G. Induced Mutations in Plant Breeding; Springer: Berlin, Germany, 1983. [Google Scholar]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Scheben, A.; Wolter, F.; Batley, J.; Puchta, H.; Edwards, D. Towards CRISPR/Cas crops—Bringing together genomics and genome editing. New Phytol. 2017, 216, 682–698. [Google Scholar] [CrossRef] [PubMed]
- Scheben, A.; Edwards, D. Genome editors take on crops. Science 2017, 355, 1122–1123. [Google Scholar] [CrossRef] [PubMed]
- Gao, C. The future of CRISPR technologies in agriculture. Nat. Rev. Mol. Cell Biol. 2018, 5, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Eeckhaut, T.; Lakshmanan, P.S.; Deryckere, D.; Van Bockstaele, E.; Van Huylenbroeck, J. Progress in plant protoplast research. Planta 2013, 238, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Wolter, F.; Klemm, J.; Puchta, H. Efficient in planta gene targeting in Arabidopsis using egg-cell specific expression of the Cas9 nuclease of Staphylococcus aureus. Plant J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Xiao, T.F.; Chen, C.H.; Li, W.; Meyer, C.A.; Wu, Q.; Wu, D.; Cong, L.; Zhang, F.; Liu, J.S.; et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015, 25, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.; Mali, P.; Moosburner, M.; Church, G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Meth. 2015, 12, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhang, H.M.; Lou, D.J.; Yu, D.Q. Selection of highly efficient sgRNAs for CRISPR/Cas9-based plant genome editing. Sci. Rep. 2016, 6, 21451. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Cradick, T.J.; Fine, E.J.; Bao, G. Nuclease target site selection for maximizing on-target activity and minimizing off-target effects in genome editing. Mol. Ther. 2016, 24, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Henry, V.J.; Bandrowski, A.E.; Pepin, A.S.; Gonzalez, B.J.; Desfeux, A. OMICtools: An informative directory for multi-omic data analysis. Database 2014, 2014, bau069. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ding, Y.; Zhou, Y.; Jin, W.; Xie, K.; Chen, L.L. CRISPR-P 2.0: An improved CRISPR-Cas9 tool for genome editing in plants. Mol. Plant 2017, 10, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Zhang, J.; Yang, Y. Genome-wide prediction of highly specific guide RNA spacers for CRISPR-Cas9-mediated genome editing in model plants and major crops. Mol. Plant 2014, 7, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wei, Z.; Dominguez, A.; Li, Y.; Wang, X.; Qi, L.S. CRISPR-ERA: A comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics 2015, 31, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Sattar, M.N.; Iqbal, Z.; Tahir, M.N.; Shahid, M.S.; Khurshid, M.; Al-Khateeb, A.A.; Al-Khateeb, S.A. CRISPR/Cas9: A practical approach in date palm genome editing. Front. Plant Sci. 2017, 8, 1469. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Illumina Short Reads (HiSeq) | PacBio (RSII/Sequel) | Oxford Nanopore Technologies (MinION) | |
|---|---|---|---|
| Sequencing method | Sequencing by synthesis | Single-molecule | Single-molecule |
| Average read length | 125 bp–300 bp | 10–15 kb (up to 100 kb) | 10–15 kb (up to 1Mb) |
| Raw error rate | ~0.1% | 10–15% | 10–38% |
| Corrected error rate | - | ~1% | 1–9% |
| Corrected error rate with short read polish | - | >0.1% | 0.1–2.8% |
| Cost/Gb with library prep * | $30–$45 | $240–$900 | $160–$250 |
| Applications for crop breeding | Resequencing studies; error correction of long reads | De novo assembly; scaffolding; gap filling | De novo assembly of genomes with long repetitive regions and high heterozygosity, scaffolding; gap filling |
| References | [11,40,41,42] | [34,40,43,44] | [45,46,47,48,49] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, H.; Scheben, A.; Edwards, D. Advances in Integrating Genomics and Bioinformatics in the Plant Breeding Pipeline. Agriculture 2018, 8, 75. https://doi.org/10.3390/agriculture8060075
Hu H, Scheben A, Edwards D. Advances in Integrating Genomics and Bioinformatics in the Plant Breeding Pipeline. Agriculture. 2018; 8(6):75. https://doi.org/10.3390/agriculture8060075
Chicago/Turabian StyleHu, Haifei, Armin Scheben, and David Edwards. 2018. "Advances in Integrating Genomics and Bioinformatics in the Plant Breeding Pipeline" Agriculture 8, no. 6: 75. https://doi.org/10.3390/agriculture8060075
APA StyleHu, H., Scheben, A., & Edwards, D. (2018). Advances in Integrating Genomics and Bioinformatics in the Plant Breeding Pipeline. Agriculture, 8(6), 75. https://doi.org/10.3390/agriculture8060075