Effects of Auricularia heimuer Residue Amendment on Soil Quality, Microbial Communities, and Maize Growth in the Black Soil Region of Northeast China

Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sample Collection
2.2. Physicochemical Properties, Lignocellulose Content and Lignocellulase Activity
2.3. High-Throughput Sequencing 16S and ITS Microbial Diversity Analysis
2.4. Statistical Analysis
3. Results
3.1. Changes in Compost Fermentation Temperature and Water Content
3.2. pH Changes
3.3. Maize Growth Indicators
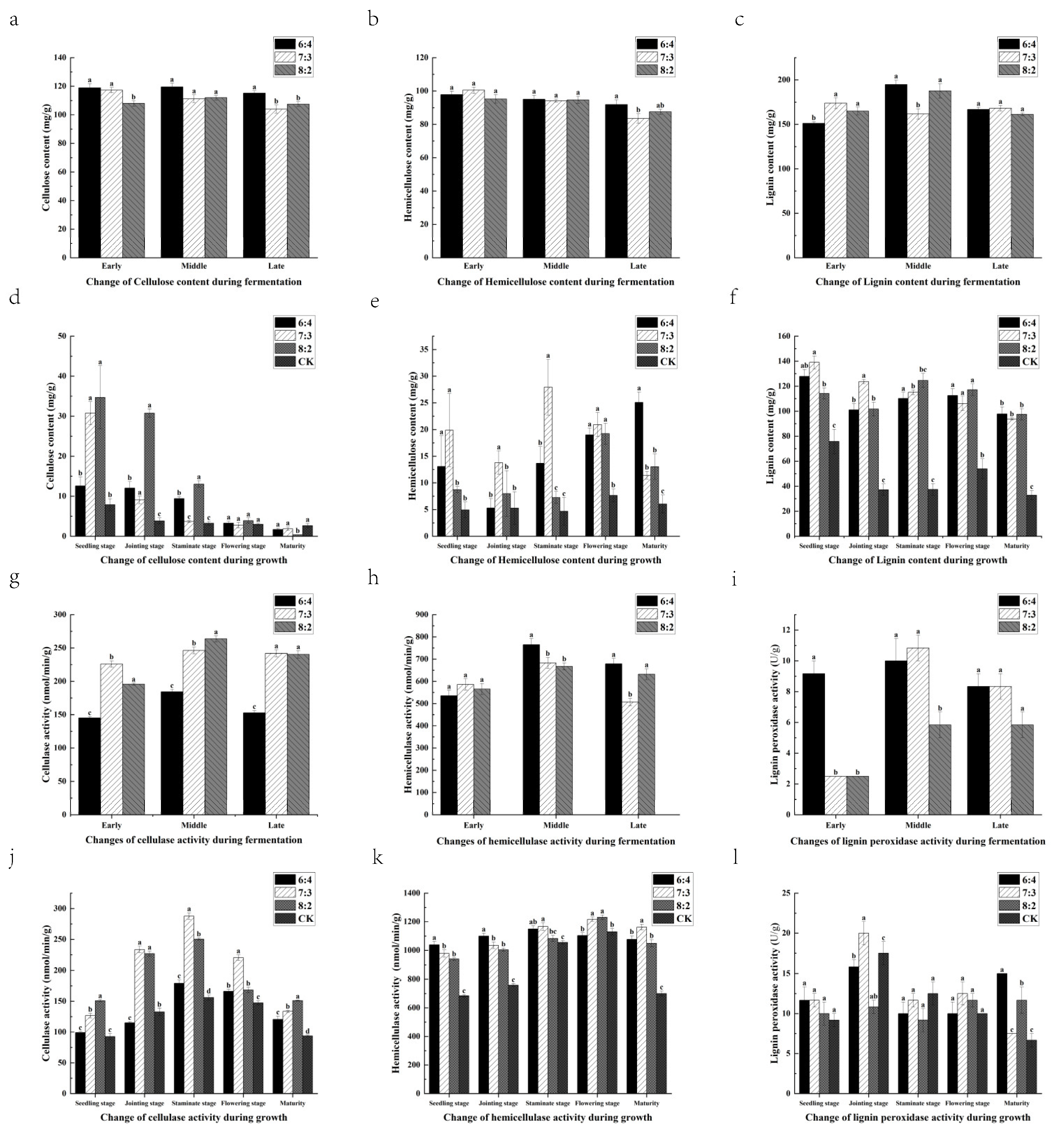
3.4. Changes in Lignocellulose Content
3.5. Changes in Lignocellulose Activity
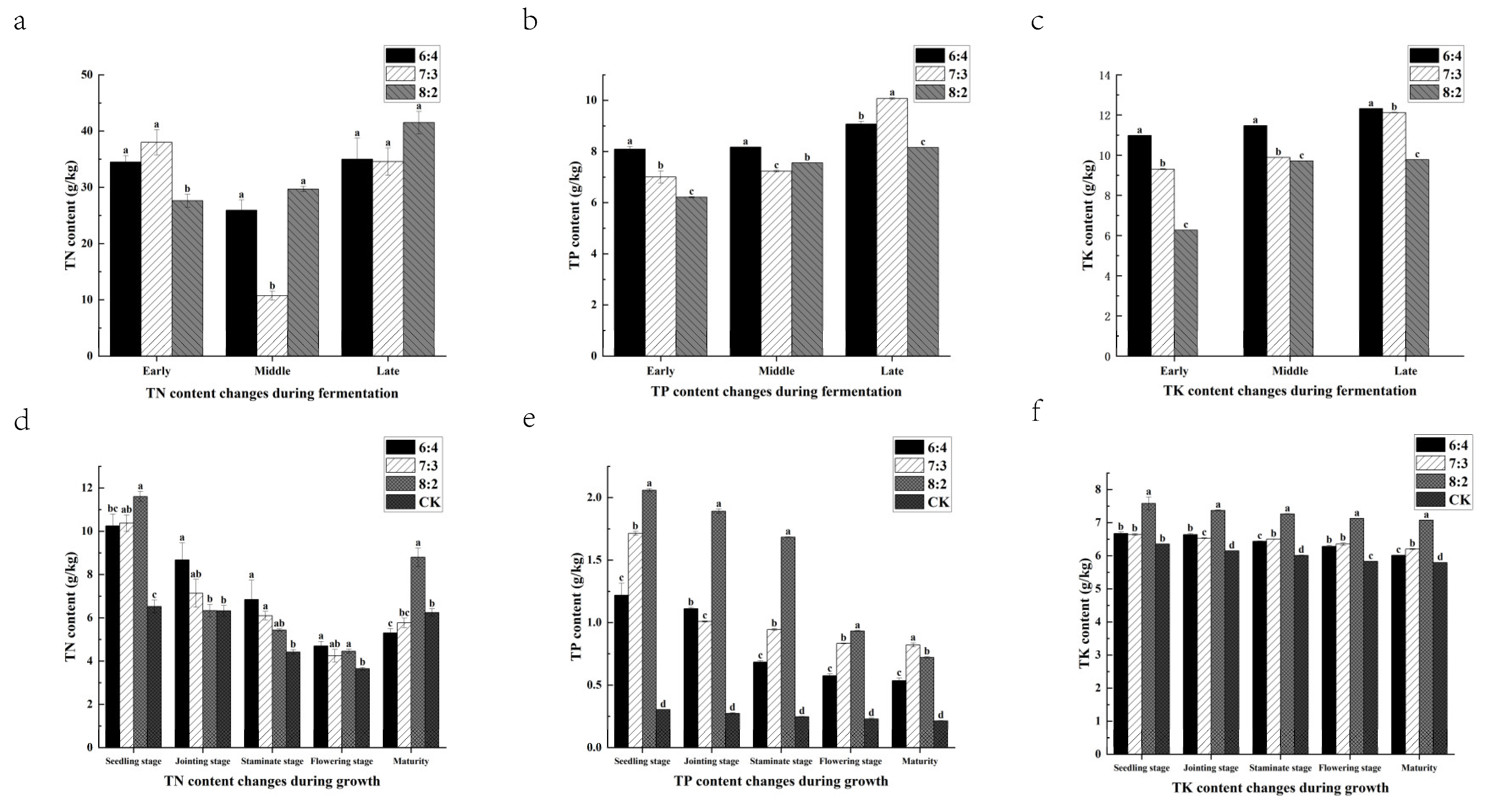
3.6. Changes in Total Nitrogen, Total Phosphorus, and Total Potassium Content
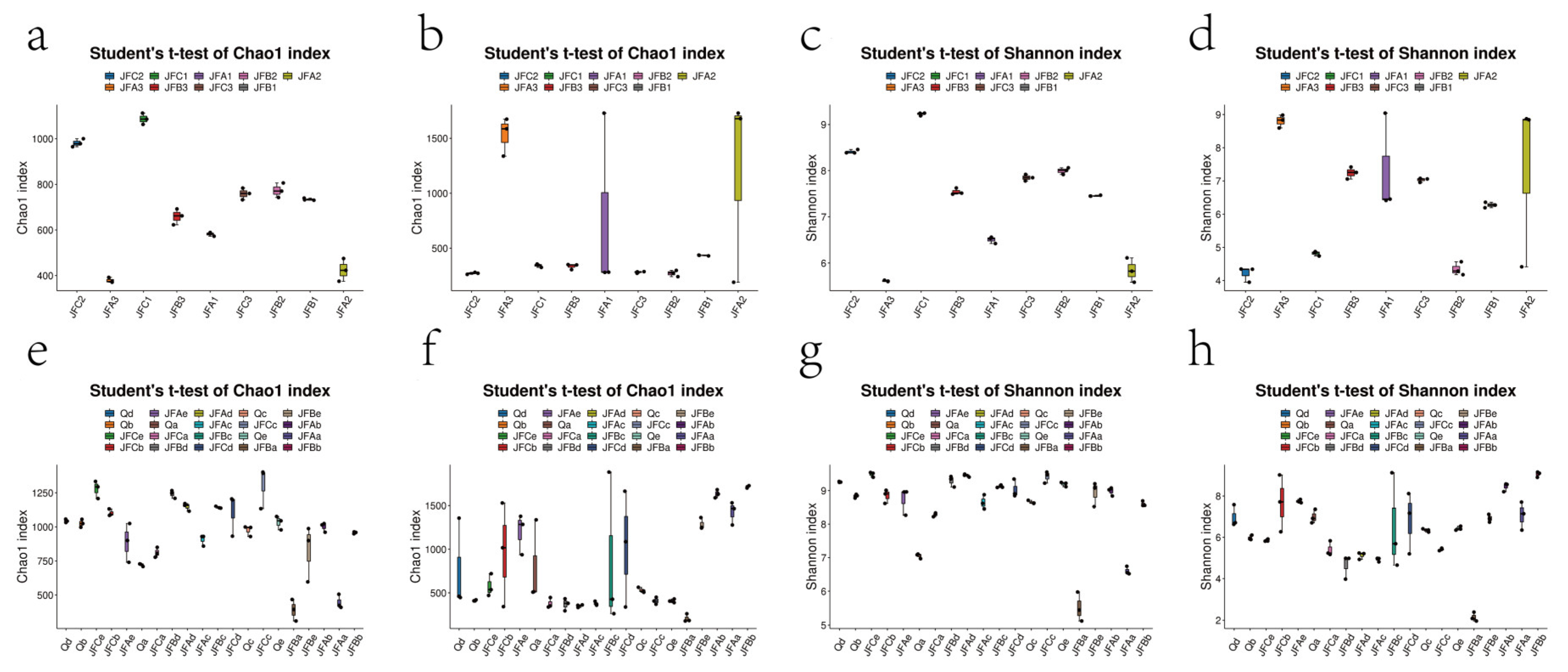
3.7. Changes in Microbial Community Diversity
3.7.1. OTU Analysis
3.7.2. Effects of Microbial Alpha Diversity on Bacterial and Fungal Communities
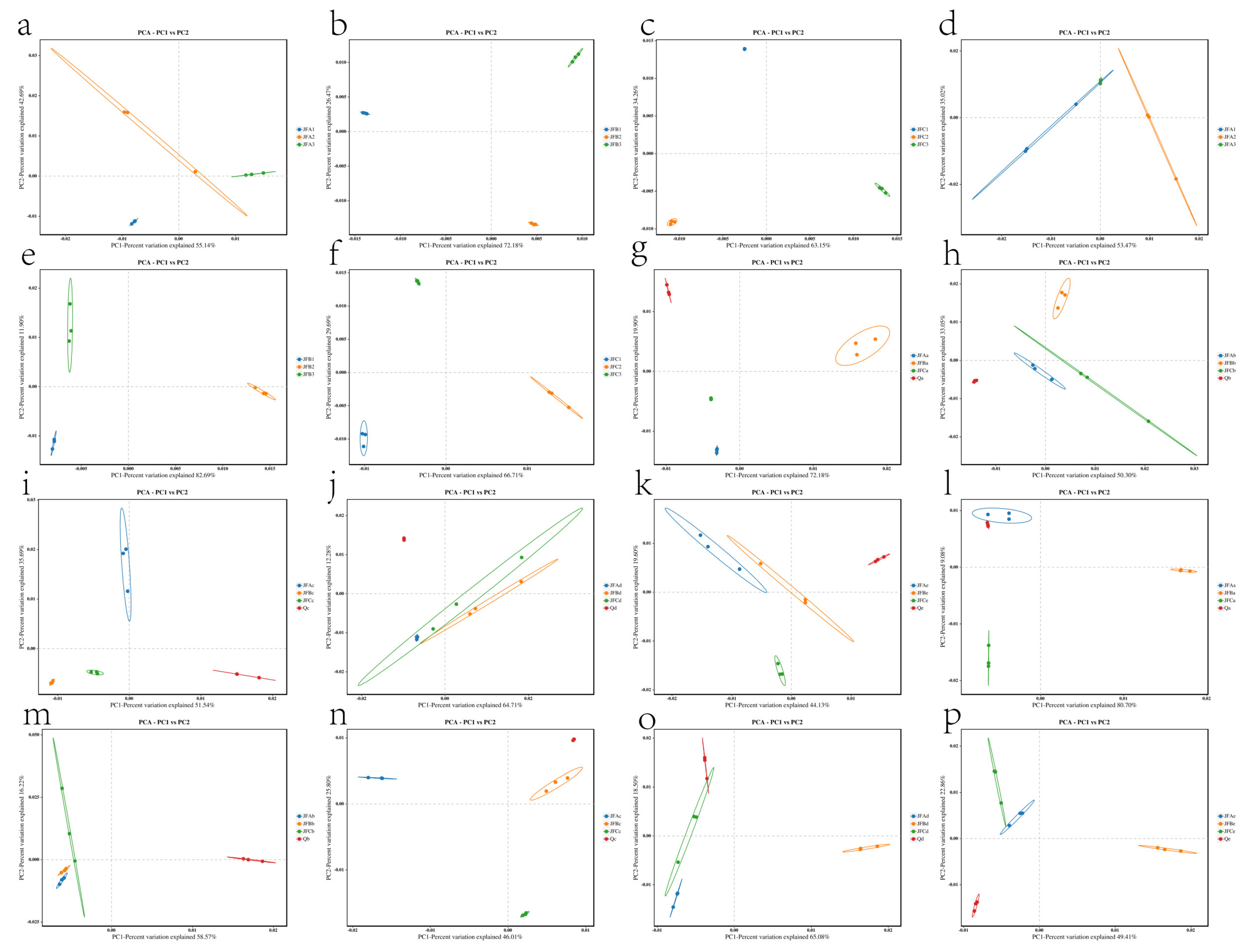
3.7.3. Effects of Beta Diversity of Microbial Bacterial and Fungal Communities
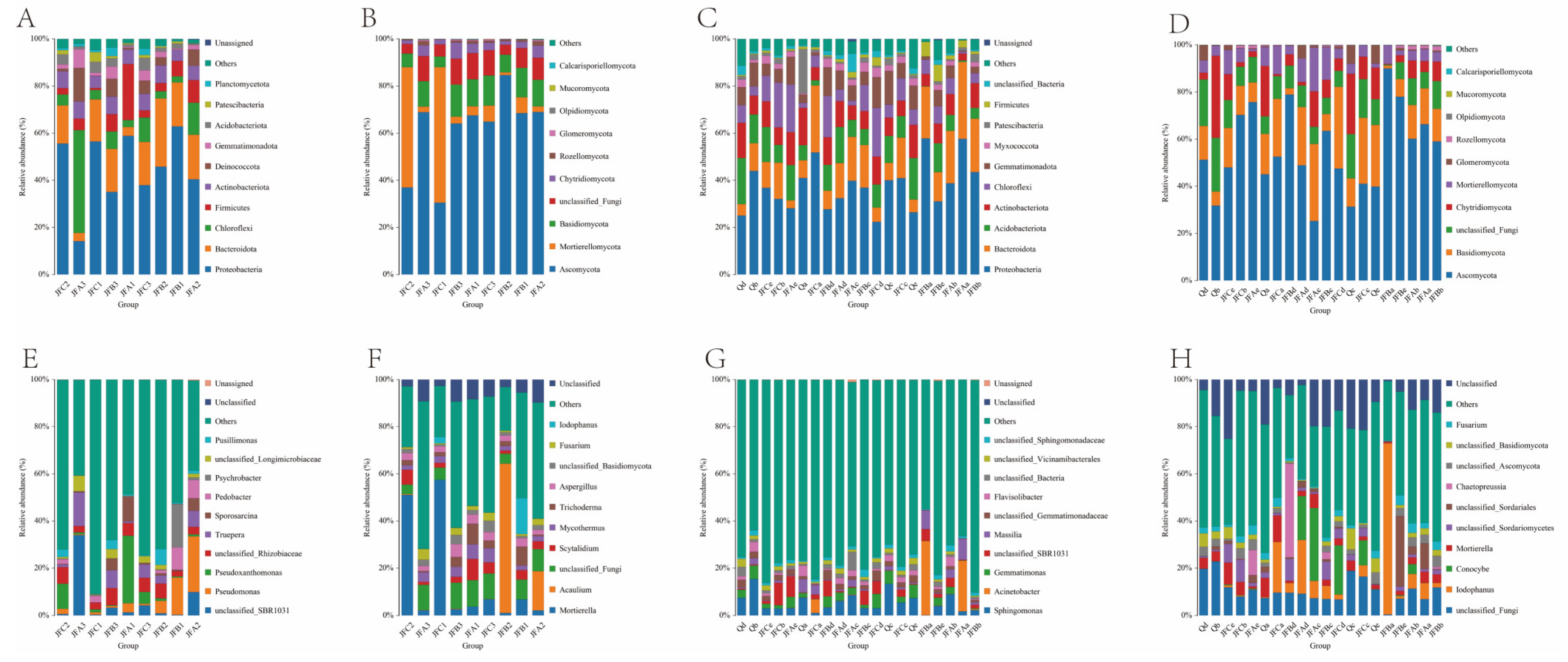
3.7.4. Composition and Relative Abundance at the Phylum Level of Microbial Bacteria and Fungi
3.7.5. Genus-Level Composition and Relative Abundance of Microbial Bacteria and Fungi
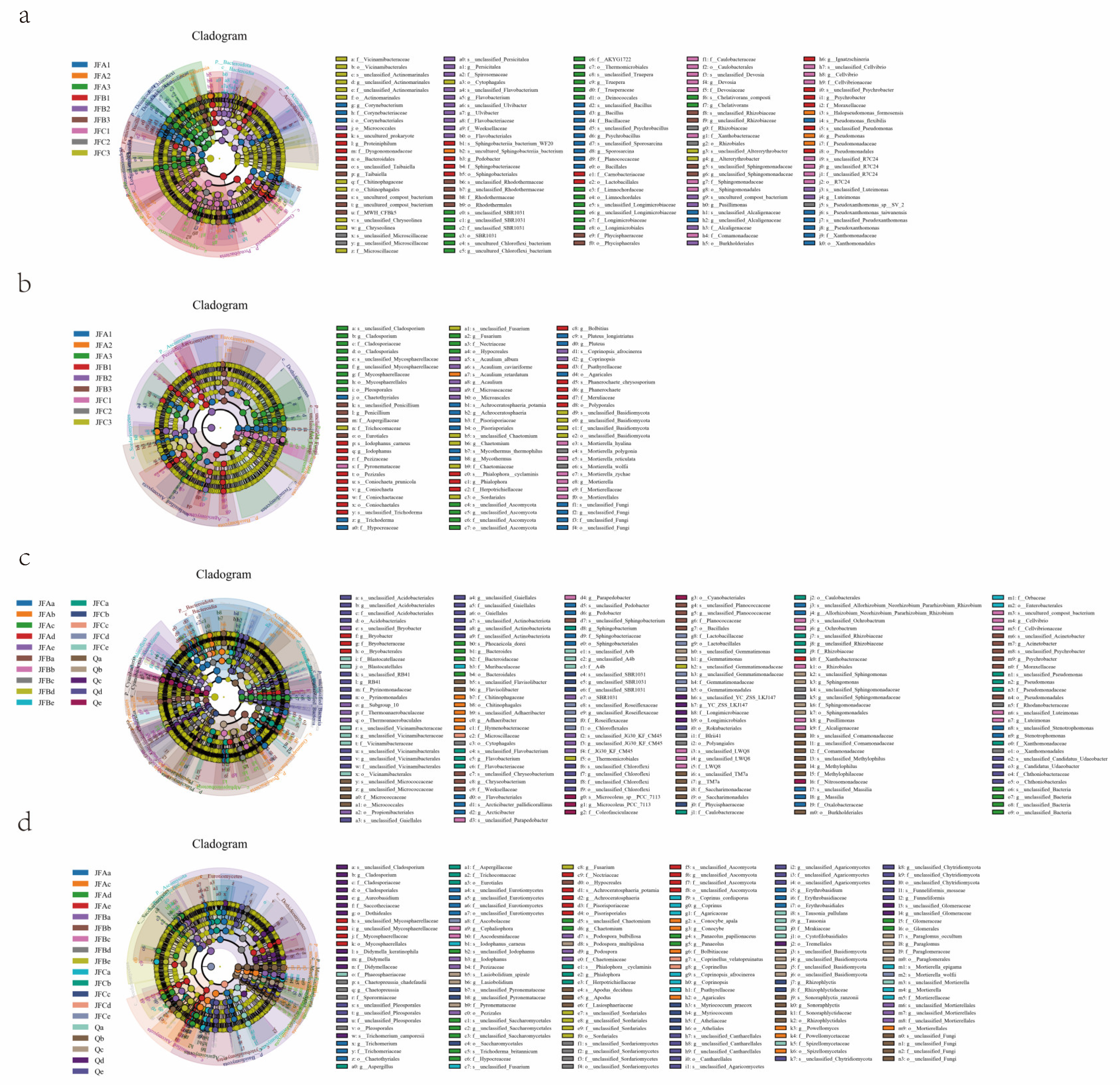
3.7.6. Differential Species Analysis of Soil Microbial Communities by Different Composting Treatments
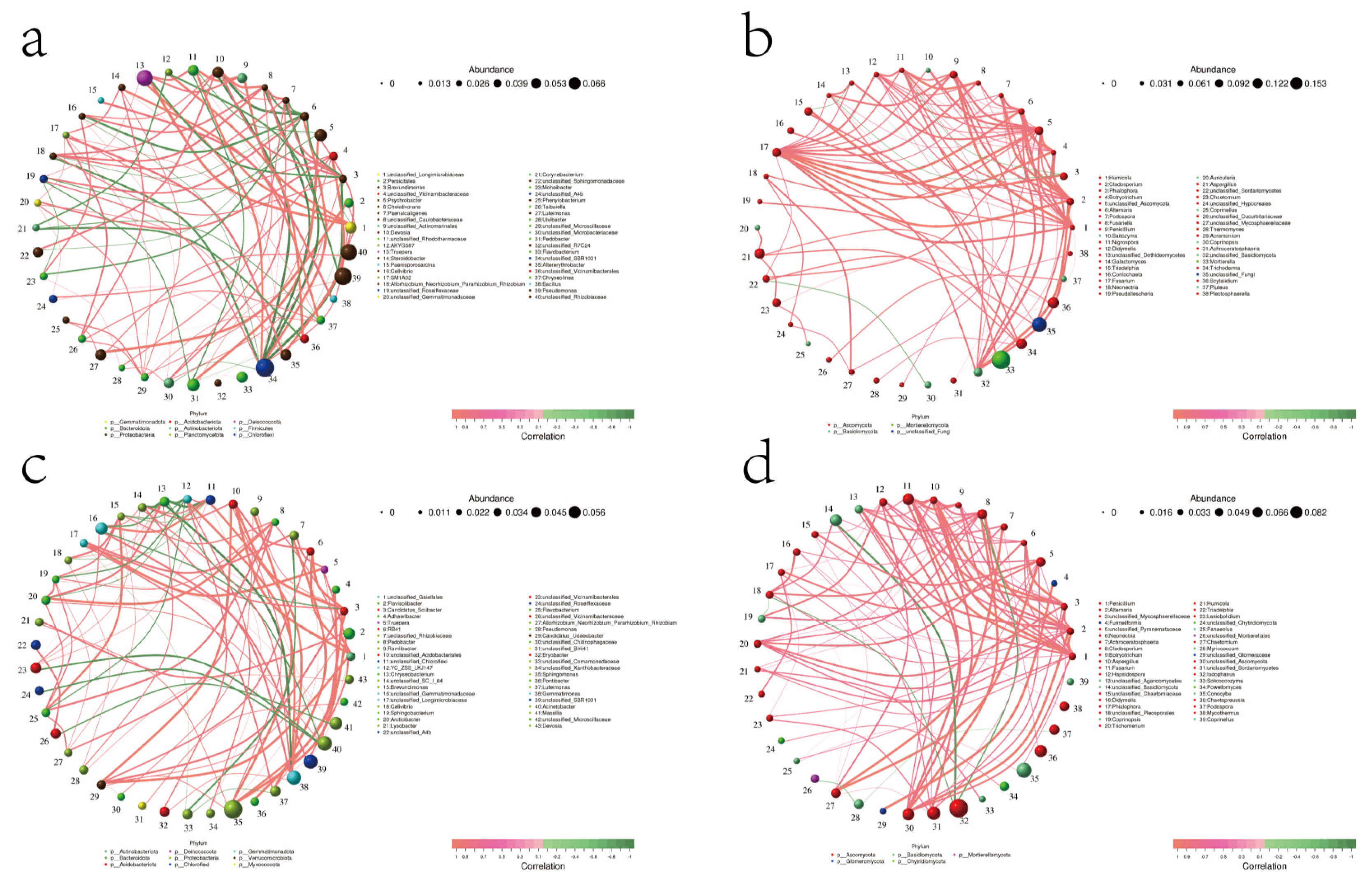
3.7.7. Microbial Interaction Network Diagrams
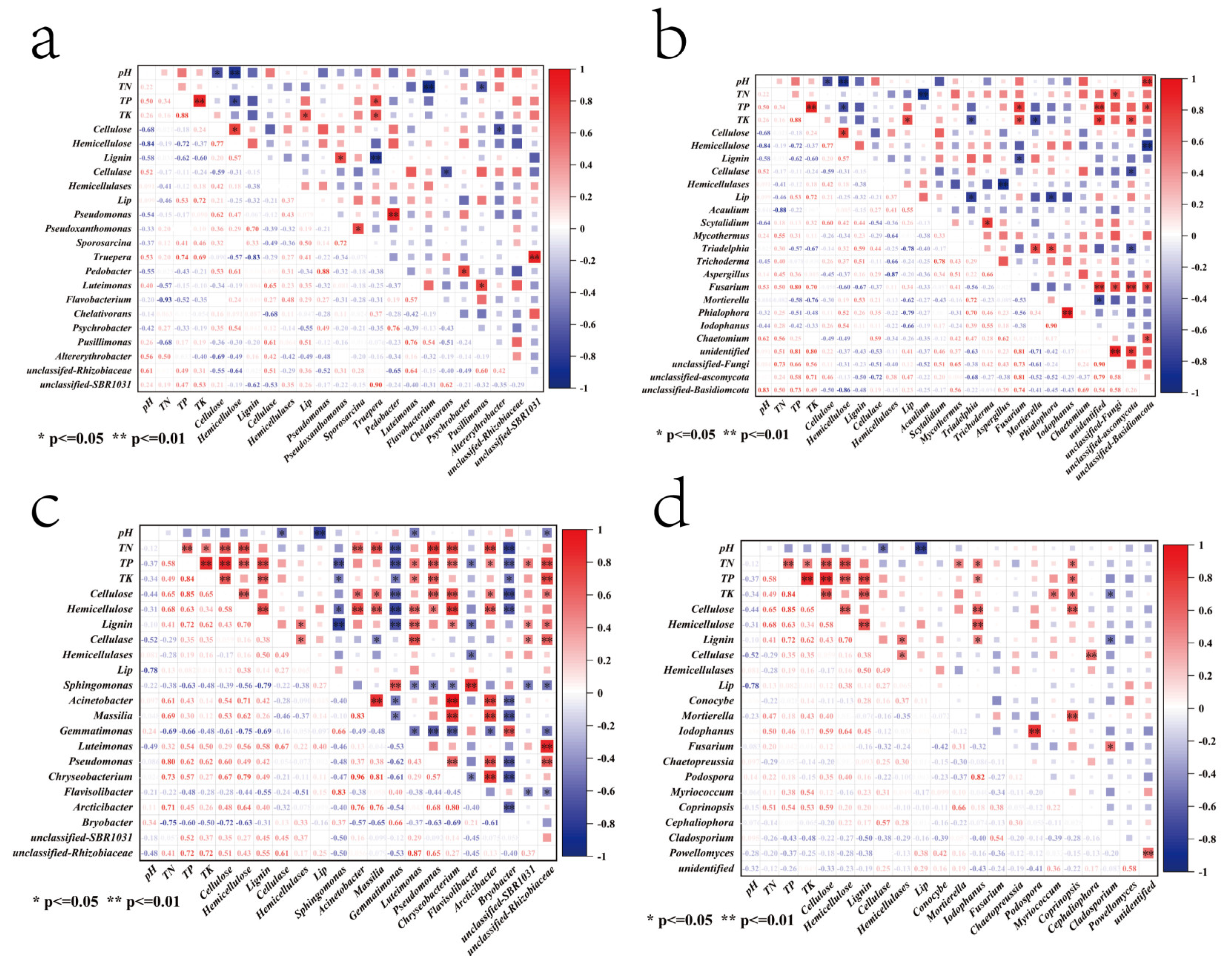
3.8. Relationship Between Microbial Bacterial and Fungal Communities and Soil Environmental Factors
3.9. Relationships Between Genus Levels of Microbial Bacterial and Fungal Communities and Lignocellulose, Lignocellulase, and Environmental Factors
4. Discussion
4.1. Effect of Composting of Different Proportions of Mycorrhizal Residues to the Field on Soil Parameters
4.2. The Impact of Varying Proportions of Composting Fungus Bran on the Composition and Diversity of Microbial Communities
4.3. Relationship Between Microbial Communities and Soil Parameters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Van Tam, N.; Wang, C.H. Use of Spent Mushroom Substrate and Manure Compost for Honeydew Melon Seedlings. J. Plant Growth Regul. 2015, 34, 417–424. [Google Scholar] [CrossRef]
- Okuda, Y. Sustainability perspectives for future continuity of mushroom production: The bright and dark sides. Front. Sustain. Food Syst. 2022, 6, 1026508. [Google Scholar] [CrossRef]
- Patel, S.S.; Bains, A.; Sridhar, K. Approaches and challenges for a sustainable low-carbon mushroom industry. Renew. Sustain. Energy Rev. 2025, 212, 115338. [Google Scholar] [CrossRef]
- He, T.; Zhang, W.; Zhang, H.; Sheng, J. Estimation of Manure Emissions Issued from Different Chinese Livestock Species: Potential of Future Production. Agriculture 2023, 13, 2143. [Google Scholar] [CrossRef]
- Zeng, Q.; Shi, G.Y.; Su, L. Organic Fertilizer Production with Fermentation and Composting of Chicken Manure Promoted by Flammulina Chaff. Chin. Agric. Sci. Bull. 2022, 38, 44–50. [Google Scholar]
- Dong, Q.; Cheng, H.Y.; Zhang, J.G.; Oh Kokyo Meng, L.J.; Wang, T.; Wang, Q.; Tian, Y. Effect of fungus chaff on soil microbe population and enzyme activity of three crop soils. Chin. J. Eco-Agric. 2016, 24, 1655–1662. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, L.; Zhou, X.; Tian, T.; Xu, S.; Li, D.; Li, C.; Li, Y. Optimizing Straw-Rotting Cultivation for Sustainable Edible Mushroom Production: Composting Spent Mushroom Substrate with Straw Additions. J. Fungi 2023, 9, 925. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Du, X.; Yin, R.; Sun, H.; Song, B.; Han, Q.; Wang, J.; Huang, Y. Performance of co-composting Pholiota nameko spent mushroom substrate and pig manure at different proportions: Chemical properties and humification process. J. Environ. Manag. 2024, 372, 123325. [Google Scholar] [CrossRef] [PubMed]
- Mahato, D.; Khamari, B.; Sahoo, J. Valorization of spent Oyster mushroom substrate with Trichoderma asperellum for suppression of sesame root rot in vitro. Waste Biomass Valorization 2024, 15, 755–769. [Google Scholar] [CrossRef]
- Lu, L.; Li, W.; Wu, X. Status of comprehensive utilization of Spent mushroom substrate. Edible Fungi 2022, 44, 1000–8357. [Google Scholar]
- Zhang, X.; Wang, Y.; Li, H. Response of soil microbial network complexity to mushroom residue amendment in black soils of Northeast China. Appl. Soil Ecol. 2023, 185, 104812. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Y.; Liu, H. Mushroom residue amendment reduces N2O emissions by altering nitrifier and denitrifier communities in a maize field. Sci. Total Environ. 2023, 856, 159102. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Z.; Sun, K. Co-application of mushroom residue and biochar improves nitrogen retention and microbial network stability in degraded soils of Northeast China. Agric. Ecosyst. Environ. 2022, 324, 107715. [Google Scholar] [CrossRef]
- Bernal, M.P. Composting of animal manures and chemical criteria for compost maturity assessment. Bioresour. Technol. 2009, 100, 5444–5453. [Google Scholar] [CrossRef]
- Wang, X. Reducing heavy metal bioavailability in chicken manure compost by adding biochar and spent mushroom substrate. Environ. Pollut. 2021, 268, 115845. [Google Scholar]
- Zhang, L. Effects of spent mushroom substrate compost on soil fertility and crop yield. J. Clean. Prod. 2018, 172, 2012–2020. [Google Scholar]
- Chen, Y. Composting of spent mushroom substrate and chicken manure: Effects on microbial community and nutrient transformation. Bioresour. Technol. 2022, 297, 122473. [Google Scholar]
- Li, R. Effects of different ratios of spent mushroom substrate to chicken manure on compost quality and soil properties. Agric. Ecosyst. Environ. 2019, 279, 1–10. [Google Scholar]
- Zhou, H. Spent mushroom substrate compost enhances tomato growth by improving rhizosphere microbiome. Front. Microbiol. 2022, 13, 825394. [Google Scholar]
- Yang, F. Integrated use of spent mushroom substrate and chicken manure improves soil fertility and crop yield in a wheat-maize rotation system. Soil Tillage Res. 2022, 199, 104592. [Google Scholar]
- Ji, Q.; Wu, Y.; Zhang, D. Effects of the fungi chaff of Pleurotus geesteranus on vegetable and soil quality in greenhouse. J. Hubei Agric. Sci. 2020, 59, 42–48. [Google Scholar] [CrossRef]
- Jiang, Q.; Lu, Z.; Ren, C. Analysis and evaluation of fertilizer fermented from spent mushroom substrate of Pleurotus eryngii Pholiotanameko. J. Hubei Agric. Sci. 2020, 59, 64–67. [Google Scholar] [CrossRef]
- Wang, M. Effect of edible fungus residue on rice yield increase. China Rice 2006, 2, 44–45. [Google Scholar]
- Chen, Z.; Zhang, X.; Yan, F. Effects of fungus bran bio-organic fertilizer on yield and quality of potato. Vegetables 2020, 11, 20–23. [Google Scholar]
- Cely-Vargas L, Y.; Zhang, W.; Cheema A, I. Effects of Compost-based Amendments from Sewage Sludge and Food Waste on Sandy Soil and Rosette Bok Choy’s Growth. Water Air Soil Pollut. 2024, 235, 1–14. [Google Scholar] [CrossRef]
- Lin, Y. A soil sampling method based on representativeness gradeof sampling points. Acta Pedol. Sin. 2011, 48, 938–946. [Google Scholar]
- Zech, W.; Guggenberger, G.; Haumaier, L. Lignin biogeochemistry of forest humus layers. Sci. Total Environ. 1994, 152, 191–198. [Google Scholar]
- Scott, R.W. Colorimetric determination of hexuronic acids in plant materials. Anal. Biochem. 1979, 99, 320–326. [Google Scholar] [CrossRef]
- Sun, R.; Lawther, J.; Banks, W.B. Fractional characterization of wheat straw lignin and cellulose. J. Agric. Food Chem. 1996, 44, 3724–3729. [Google Scholar]
- Yadav, M.; Singh, S.K.; Yadava, S. Purification, characterisation and coal depolymerisation activity of lignin peroxidase from Lenzitus betulina MTCC-1183. Appl. Biochem. Microbiol. 2012, 48, 583–589. [Google Scholar] [CrossRef]
- Ghose, T.K. Measurement of cellulase activities. Pure Appl. Chem. 1987, 59, 257–268. [Google Scholar] [CrossRef]
- Six, J.; Elliott, E.; Paustian, K. Response of soil microbial biomass, urease, and xylanase within particle size fractions to long-term soil management. Soil Biol. Biochem. 1999, 31, 1567–1574. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, Y.; Gao, J.; Peng, F.; Gao, P. Long-term combined application of manure and chemical fertilizer sustained higher nutrient status and rhizospheric bacterial diversity in reddish paddy soil of Central South China. Sci. Rep. 2018, 8, 16554. [Google Scholar] [CrossRef] [PubMed]
- Kulcu, R.; Yaldiz, O. Composting of goat manure and wheat straw using pine cones as a bulking agent. Bioresour. Technol. 2007, 98, 2700–2704. [Google Scholar] [CrossRef]
- Song, Y.; Wang, Y.; Li, R. Effects of common microplastics on aerobic composting of cow manure: Physiochemical characteristics, humification, and microbial community. J. Environ. Chem. Eng. 2022, 10, 108681. [Google Scholar] [CrossRef]
- Wang, M.; Wang, X.; Wu, Y.; Wang, X.; Zhao, J.; Liu, Y.; Chen, Z.; Jiang, Z.; Tian, W.; Zhang, J. Effects of thermophiles inoculation on the efficiency and maturity of rice straw composting. Bioresour. Technol. 2022, 354, 127195. [Google Scholar] [CrossRef]
- Ge, M.; Zhou, H.; Shen, Y.; Meng, H.; Li, R.; Zhou, J.; Cheng, H.; Zhang, X.; Ding, J.; Wang, J.; et al. Effect of aeration rates on enzymatic activity and bacterial community succession during cattle manure composting. Bioresour. Technol. 2020, 304, 122928. [Google Scholar] [CrossRef]
- Sánchez-Monedero, M.A.; Roig, A.; Paredes, C.; Bernal, M.P. Nitrogen transformation during organic waste composting by the Rutgers system and its effects on pH, EC and maturity of the composting mixtures. Bioresour. Technol. 2001, 78, 301–308. [Google Scholar] [CrossRef]
- Mao, H.; Zhang, H.; Fu, Q.; Zhong, M.; Li, R.; Zhai, B.; Wang, Z.; Zhou, L. Effects of four additives in pig manure composting on greenhouse gas emission reduction and bacterial community change. Bioresour. Technol. 2019, 292, 121896. [Google Scholar] [CrossRef]
- Roca-Pérez, L.; Martínez, C.; Marcilla, P.; Boluda, R. Composting rice straw with sewage sludge and compost effects on the soil-plant system. Chemosphere 2009, 75, 781–787. [Google Scholar] [CrossRef]
- Gmach, M.R.; Cherubin, M.R.; Kaiser, K.; Cerri, C.E.P. Processes that influence dissolved organic matter in the soil: A review. Sci. Agric. 2019, 77, e20180164. [Google Scholar] [CrossRef]
- Afreh, D.; Zhang, J.; Guan, D.; Liu, K.; Song, Z.; Zheng, C.; Deng, A.; Feng, X.; Zhang, X.; Wu, Y. Long-term fertilization on nitrogen use efficiency and greenhouse gas emissions in a double maize cropping system in subtropical China. Soil Tillage Res. 2018, 180, 259–267. [Google Scholar] [CrossRef]
- Amini, S.; Asoodar, M.A. The effect of conservation tillage on crop yield production (The Review). N. Y. Sci. J. 2015, 8, 25–29. [Google Scholar] [CrossRef]
- Tesfay, T.; Godifey, T.; Mesfin, R.; Kalayu, G. Evaluation of waste paper for cultivation of oyster mushroom (Pleurotus ostreatus) with some added supplementary materials. AMB Express 2020, 10, 15. [Google Scholar] [CrossRef]
- Yang, C.; Du, W.; Zhang, L.; Dong, Z. Effects of sheep manure combined with chemical fertilizers on maize yield and quality and spatial and temporal distribution of soil inorganic nitrogen. Complexity 2021, 2021, 4330666. [Google Scholar] [CrossRef]
- Chen, X.; Opoku-Kwanowaa, Y.; Li, J.; Wu, J. Application of organic wastes to primary saline-alkali soil in Northeast China: Effects on soil available nutrients and salt ions. Commun. Soil. Sci. Plant Anal. 2020, 51, 1238–1252. [Google Scholar] [CrossRef]
- Guo, J.; Liu, W.; Zhu, C. Bacterial rather than fungal community composition is associated with microbial activities and nutrient-use efficiencies in a paddy soil with short-term organic amendments. Plant Soil 2018, 424, 335–349. [Google Scholar] [CrossRef]
- Shang, L.; Wan, L.; Zhou, X.; Li, S.; Li, X. Effects of organic fertilizer on soil nutrient status, enzyme activity, and bacterial community diversity in Leymus chinensis steppe in Inner Mongolia, China. PLoS ONE 2020, 15, e0240559. [Google Scholar] [CrossRef]
- Zhang, L.; Guan, J. Im pact of Tourism Disturbance on Vegetation and Soil Microecological Environment of Jiulongshan Park. J. Southwest Univ. (Nat. Sci. Ed.) 2024, 46, 117–126. [Google Scholar] [CrossRef]
- Qiao, C.; Penton, C.R.; Xiong, W. Reshaping the rhizosphere microbiome by bio-organic amendment to enhance crop yield in a maize-cabbage rotation system. Appl. Soil. Ecol. 2019, 142, 136–146. [Google Scholar] [CrossRef]
- Wang, J.; Lu, T.; Liang, Z. Effects of Microorganisms from Different Sources on the Composting Process of Grape Branches and Pig Manure. J. Agric. Sci. Technol. 2024, 26, 224–233. [Google Scholar] [CrossRef]
- Kang, E.; Li, Y.; Zhang, X.; Yan, Z.; Wu, H.; Li, M.; Yan, L.; Zhang, K.; Wang, J.; Kang, X. Soil pH and nutrients shape the vertical distribution of microbial communities in an alpine wetland. Sci. Total Environ. 2021, 774, 145780. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Ma, Y. Research progress on the evolution of humic acid and the effect of composting process on HA. Appl. Chem. Ind. 2023, 52, 2865–2869. [Google Scholar]
- Koechli, C.; Campbell, A.N.; Pepe-Ranney, C.; Buckley, D.H. Aesesing fumgal contributiona to cellulose degradation in soil by using high-through put 487stable isotope probing. Soil. Biol. Biochem. 2019, 130, 150–158. [Google Scholar] [CrossRef]
- Lin, P.; Yan, Z.F.; Li, C.T. Luteimonas cellulosilyticus sp. nov., Cellulose-Degrading Bacterium Isolated from Soil in Changguangxi National Wetland Park, China. Curr. Microbiol. 2020, 77, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhao, J.; Yuan, J.; Hale, L.; Wen, T.; Huang, Q.; Vivanco, J.M.; Zhou, J.; Kowalchuk, G.A.; Shen, Q. Root exudates drive soil-microbe-nutrient feedbacks in response to plant growth. Plant Cell Environ. 2021, 44, 613–628. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, H.; Xie, W.; Yang, Z.; Lv, Q. Long-term effects of maize straw return and manure on the microbial community in cinnamon soil in Northern China using 16S rRNA sequencing. PLoS ONE 2021, 16, e0249884. [Google Scholar] [CrossRef]
- Pan, M.; Gan, X.; Mei, C.; Liang, Y. Structural analysis and transformation of biosilica during lignocellulose fractionation of rice straw. J. Mol. Struct. 2017, 1127, 575–582. [Google Scholar] [CrossRef]
- Zhang, X.; Dou, S.; Ndzelu, B.S.; Guan, X.W.; Zhang, B.Y.; Bai, Y. Effects of different corn straw amendments on humus composition and structural characteristics of humic acid in black soil. Commun. Soil. Sci. Plant Anal. 2020, 51, 107–117. [Google Scholar] [CrossRef]
- Wang, P. Effects of long-term different straw returning combinations on soil physical and chemical properties and bacterial community. Master’s Thesis, Jilin University, Changchun, China, 2024. [Google Scholar] [CrossRef]
- Jin, L.; Lyu, J.; Jin, N.; Xie, J.; Wu, Y.; Zhang, G.; Feng, Z.; Tang, Z.; Liu, Z.; Luo, S. Effects of different vegetable rotations on the rhizosphere bacterial community and tomato growth in a continuous tomato cropping substrate. PLoS ONE 2021, 16, e257432. [Google Scholar] [CrossRef]
- Tie, J.; Qiao, Y.; Jin, N.; Gao, X.; Liu, Y.; Lyu, J.; Zhang, G.; Hu, L.; Yu, J. Yield and Rhizosphere Soil Environment of Greenhouse Zucchini in Response to Different Planting and Breeding Waste Composts. Microorganisms 2023, 11, 1026. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Sun, T.; Sun, T. Effects of application of chicken manure on soil microbial community structure diversity of young rapeseed. J. Agric. Environ. Sci. 2019, 39, 2316–2324. [Google Scholar]
- Chen, L.; Zhou, W.; Luo, L.; Li, Y.; Chen, Z.; Gu, Y.; Chen, Q.; Deng, O.; Xu, X.; Lan, T.; et al. Short-term responses of soil nutrients, heavy metals, and microbial community to partial substitution of chemical fertilizer with spent mushroom substrates (SMS). Sci. Total Environ. 2022, 844, 157064. [Google Scholar] [CrossRef]
- Yang, Y.; Qiu, K.; Xie, Y.; Li, X.; Zhang, S.; Liu, W.; Huang, Y.; Cui, L.; Wang, S.; Bao, P. Geographical, climatic, and soil factors control the altitudinal pattern of rhizosphere microbial diversity and its driving effect on root zone soil multifunctionality in mountain ecosystems. Sci. Total Environ. 2023, 904, 166932. [Google Scholar] [CrossRef] [PubMed]
- Luan, H.; Gao, W.; Huang, S.; Tang, J.; Li, M.; Zhang, H.; Chen, X. Partial substitution of chemical fertilizer with organic amendments affects soil organic carbon composition and stability in a greenhouse vegetable production system. Soil Tillage Res. 2019, 191, 185–196. [Google Scholar] [CrossRef]
- Xue, G.; Bai, H.; Du, J. Effects of Different Treatments on Structure and Diversity of Soil Bacterial Community in Pepper Continuous Cropping Soil. China Veg. 2023, 3, 78–84. [Google Scholar] [CrossRef]
- Yang, Y.; Li, H.; Ma, K. Effect of Continuous Cropping on the Physicochemical Properties, Microbial Activity, and Community Characteristics of the Rhizosphere Soil of Codonopsis pilosula. China Environ. Sci. 2023, 44, 6387–6398. [Google Scholar] [CrossRef]
- Meng, Q.; Yang, W.; Men, M.; Bello, A.; Xu, X.; Xu, B.; Deng, L.; Jiang, X.; Sheng, S.; Wu, X.; et al. Microbial Community Succession and Response to Environmental Variables During Cow Manure and Corn Straw Composting. Front. Microbiol. 2019, 10, 529. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Basic Physical Property | CM | Auricularia heimuer Residue | Soil |
|---|---|---|---|
| pH | 7.87 | 8.27 | 8.17 |
| TN (g/kg) | 26.678 | 4.308 | 1.413 |
| TP (g/kg) | 19.52 | 1.85 | 1.3 |
| TK (g/kg) | 17.47 | 11.7 | 16.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wang, J.; Qian, K.; Feng, Y.; Ao, J.; Zhai, Y.; Li, Y.; Li, X.; Zhang, B.; Yu, H. Effects of Auricularia heimuer Residue Amendment on Soil Quality, Microbial Communities, and Maize Growth in the Black Soil Region of Northeast China. Agriculture 2025, 15, 879. https://doi.org/10.3390/agriculture15080879
Wang Y, Wang J, Qian K, Feng Y, Ao J, Zhai Y, Li Y, Li X, Zhang B, Yu H. Effects of Auricularia heimuer Residue Amendment on Soil Quality, Microbial Communities, and Maize Growth in the Black Soil Region of Northeast China. Agriculture. 2025; 15(8):879. https://doi.org/10.3390/agriculture15080879
Chicago/Turabian StyleWang, Ying, Jionghua Wang, Keqing Qian, Yuting Feng, Jiangyan Ao, Yinzhen Zhai, Yu Li, Xiao Li, Bo Zhang, and Han Yu. 2025. "Effects of Auricularia heimuer Residue Amendment on Soil Quality, Microbial Communities, and Maize Growth in the Black Soil Region of Northeast China" Agriculture 15, no. 8: 879. https://doi.org/10.3390/agriculture15080879
APA StyleWang, Y., Wang, J., Qian, K., Feng, Y., Ao, J., Zhai, Y., Li, Y., Li, X., Zhang, B., & Yu, H. (2025). Effects of Auricularia heimuer Residue Amendment on Soil Quality, Microbial Communities, and Maize Growth in the Black Soil Region of Northeast China. Agriculture, 15(8), 879. https://doi.org/10.3390/agriculture15080879