

Combined Transcriptome and Proteome Analysis Reveals the Molecular Mechanism by Which ZmPDI Improves Salt Resistance in Rice (Oryza sativa)

Abstract
1. Introduction
2. Materials and Methods
2.1. ZmPDI Genetic Heterologous Transformation
2.2. Salt Treatment and Physiological Index Measurement
2.3. RNA-Seq
2.4. Tandem Mass Tag-Based Protein Quantification
2.5. Bioinformatics Analysis and qRT-PCR Validation
3. Results
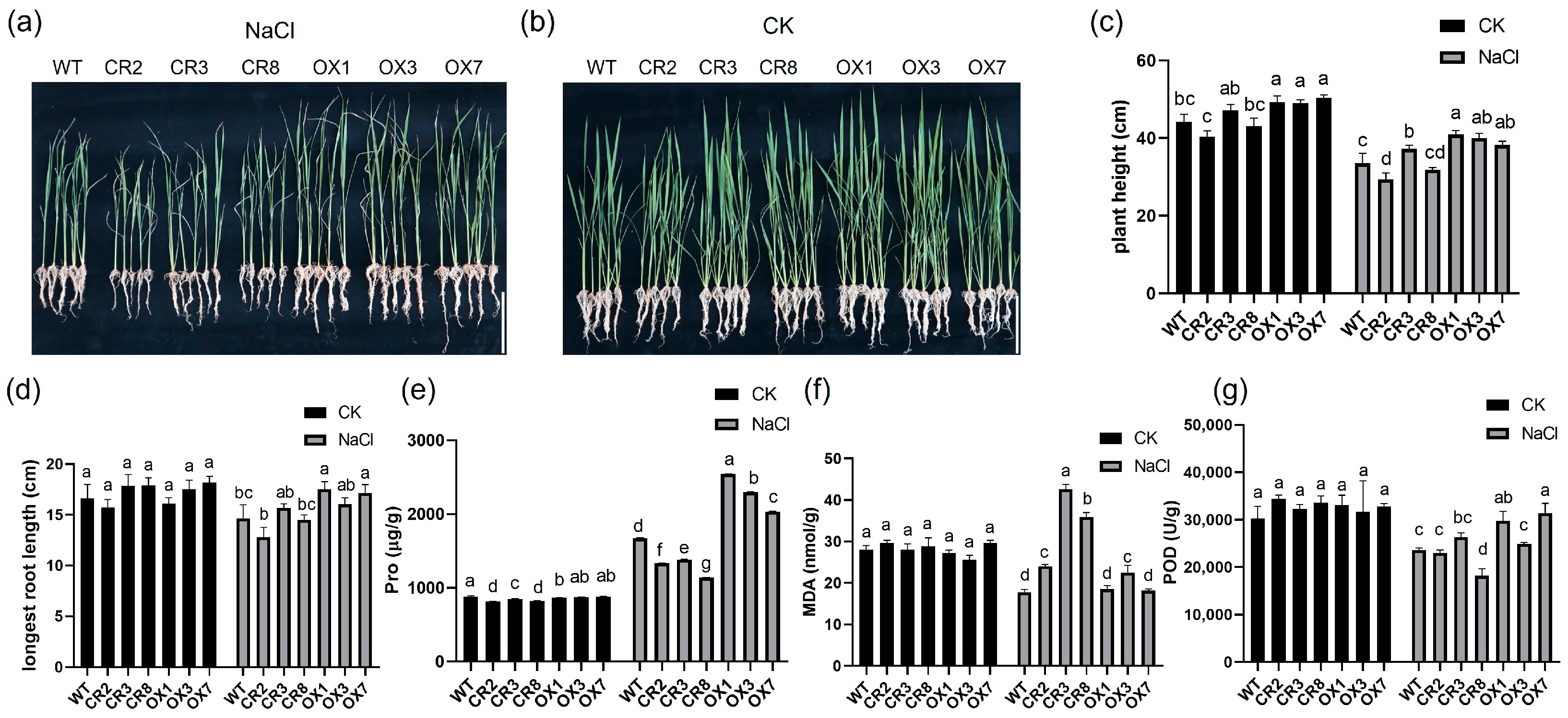
3.1. The Physiological Response of the Transgenic Plants and WT to Salt Stress
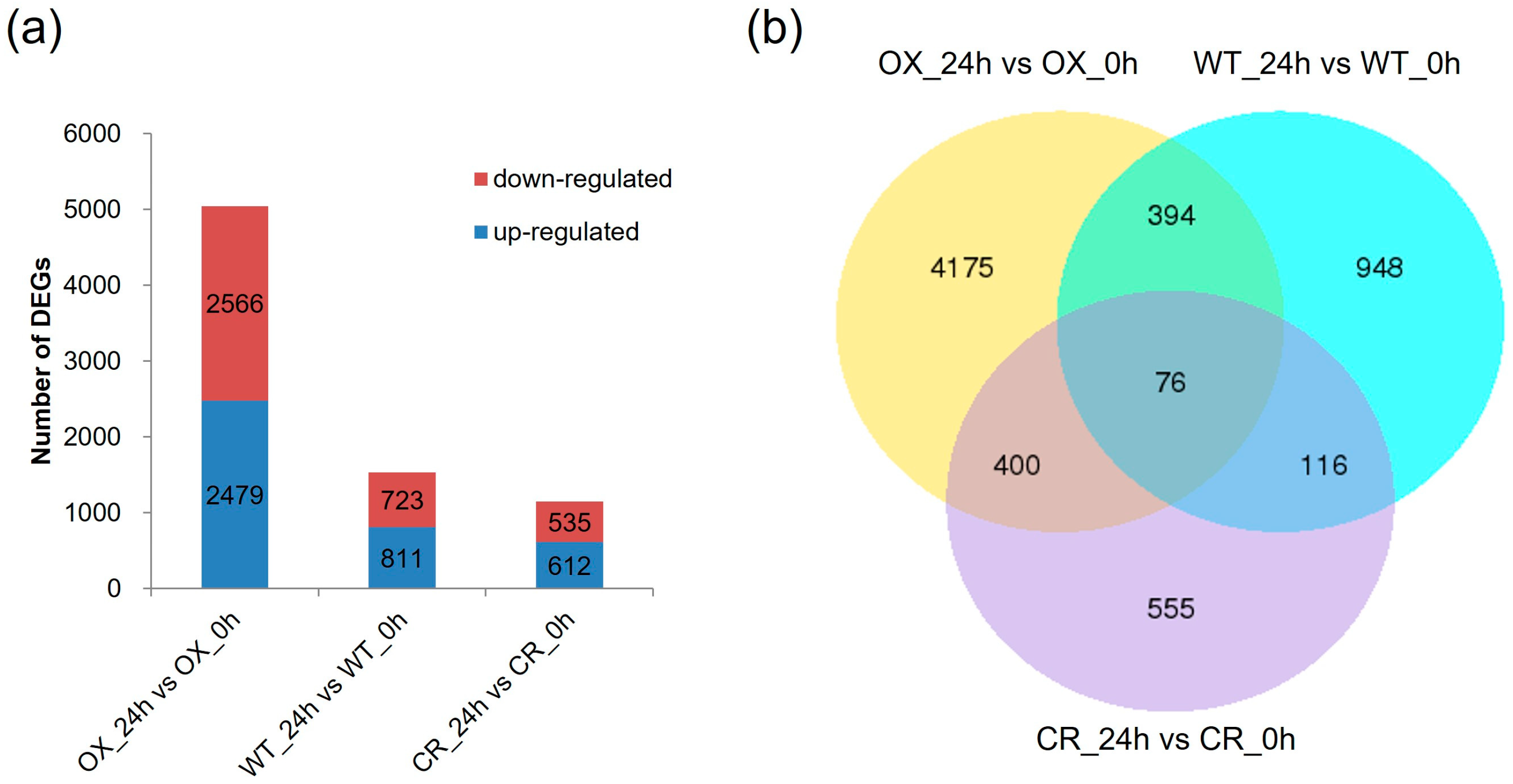
3.2. Transcriptome Sequencing of the ZmPDI-Overexpressing Rice, OSPDI Knock-Out Rice, and WT Rice
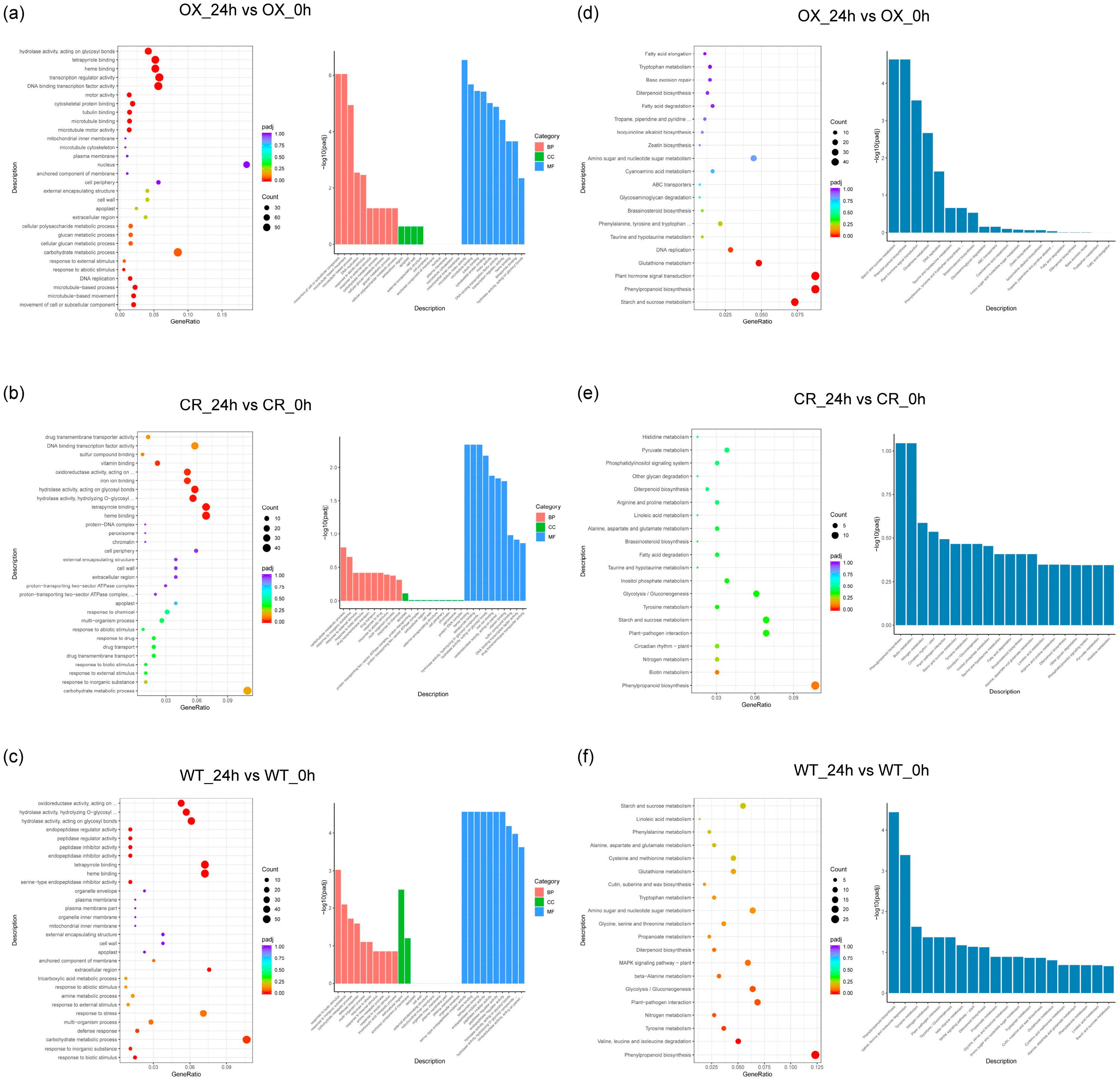
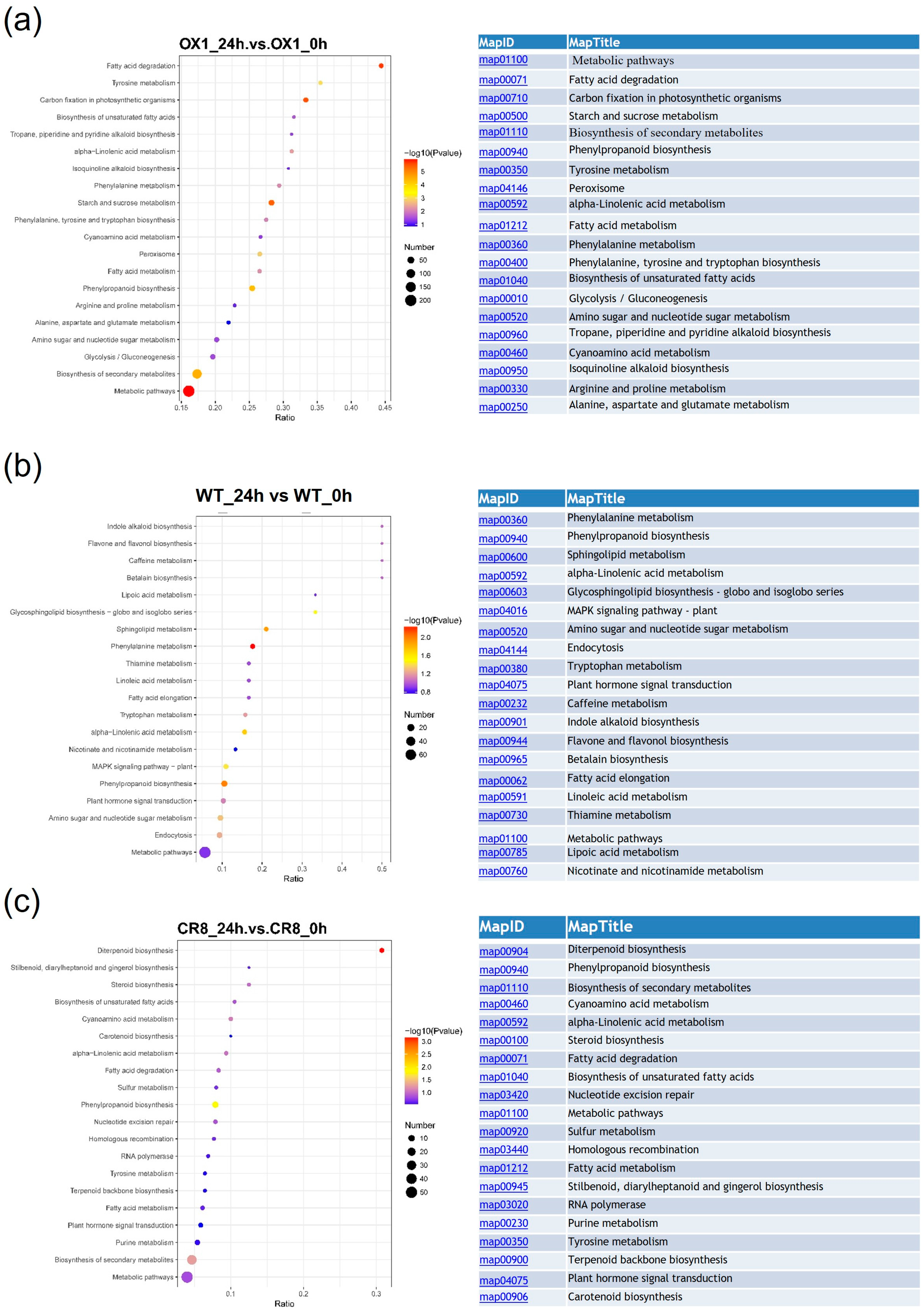
3.3. GO and KEGG Analysis of DEGs in ZmPDI-Overexpressing Rice, OsPDI Knock-Out Rice, and WT Rice in Response to Salt Stress
3.4. Proteome Sequencing of the ZmPDI-Overexpressing Rice, OSPDI Knock-Out Rice and WT
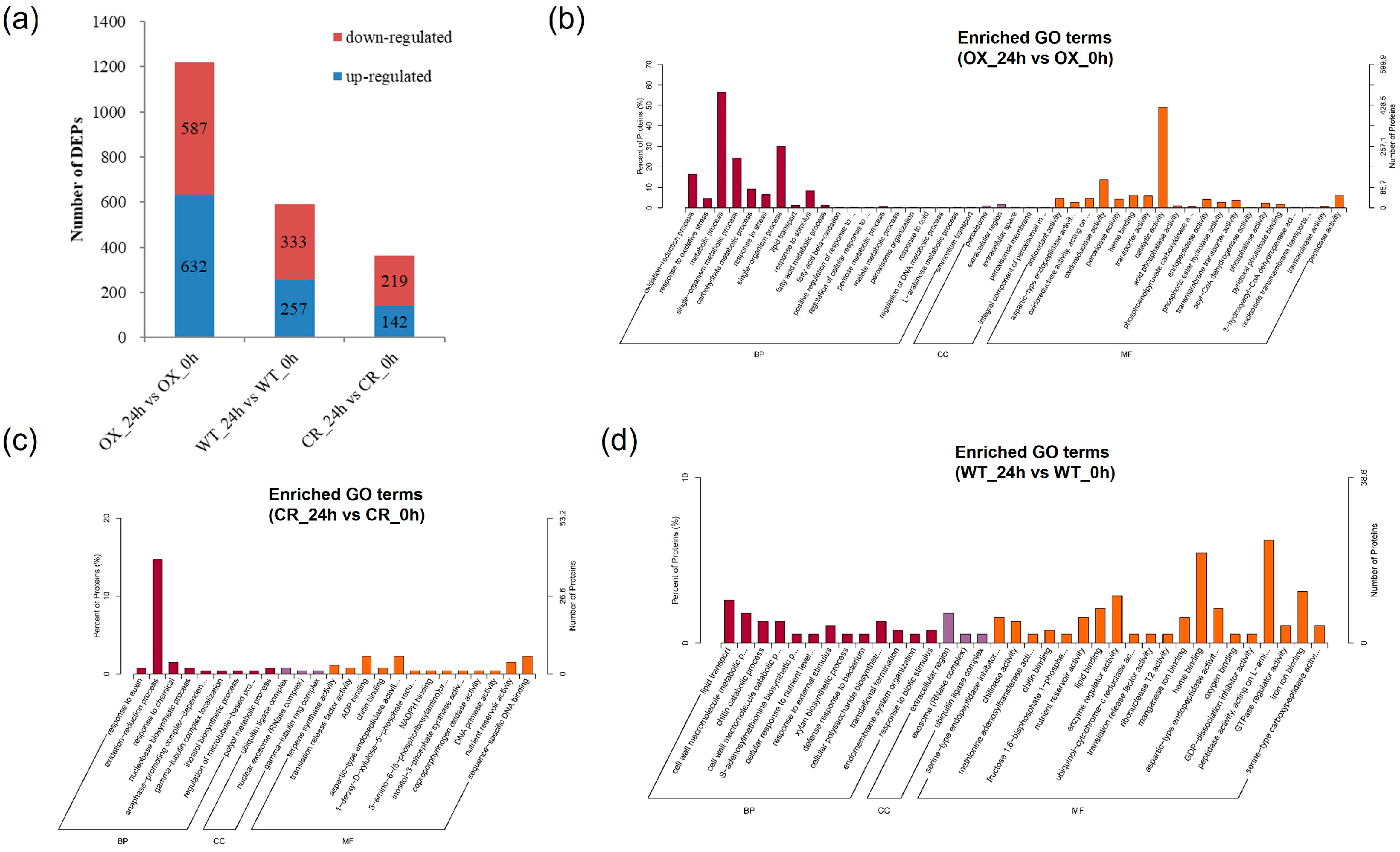
3.5. GO and KEGG Analysis of Different Expression Proteins in ZmPDI-Overexpressing Rice, OsPDI Knock-Out Rice, and WT in Response to Salt Stress
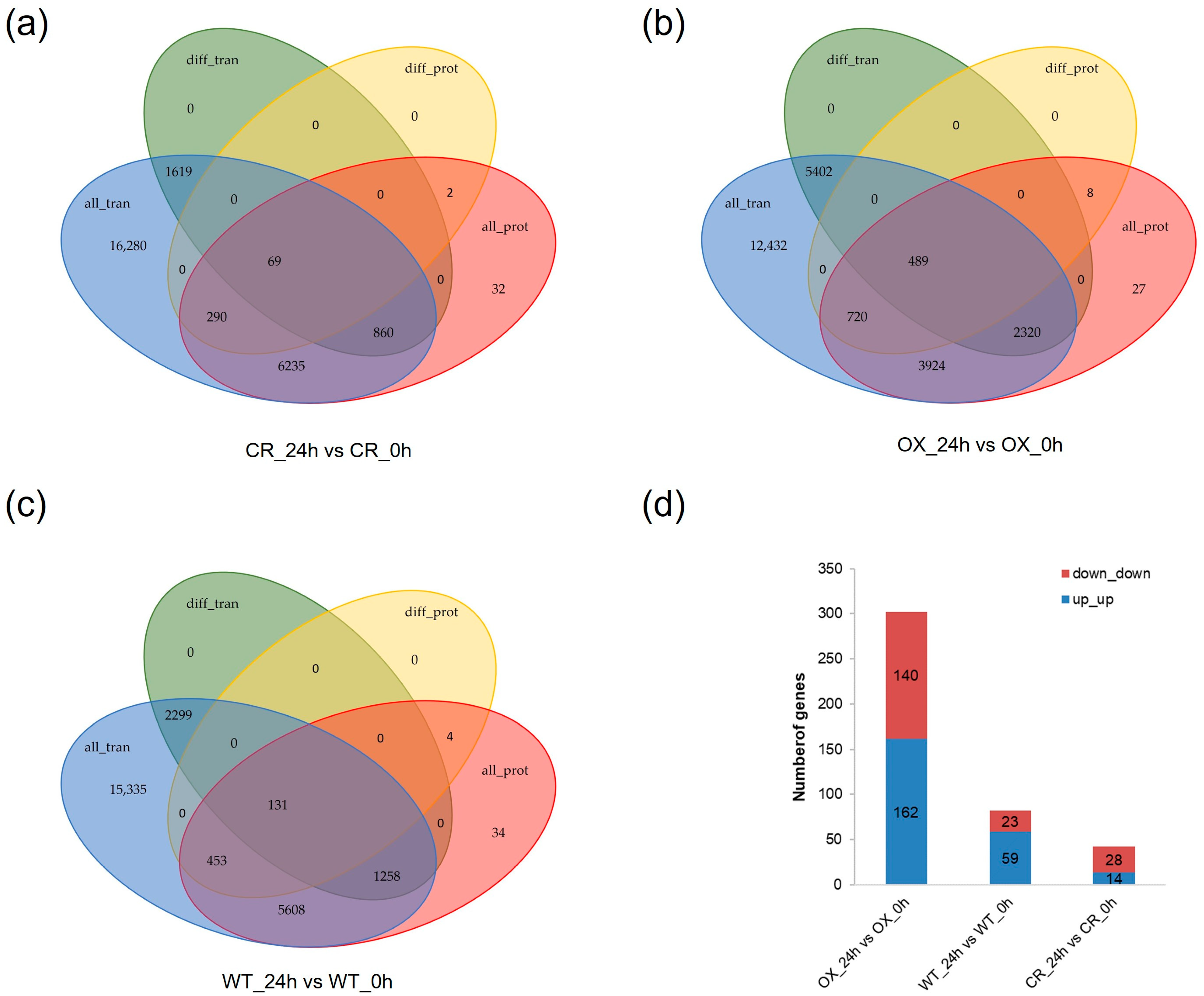
3.6. Combination Analysis of Transcriptome and Proteome Sequencing
4. Discussion
4.1. Overexpression of ZmPDI Can Enhance the Salt Tolerance of Rice, While Loss-of-Function of OsPDI in Rice Leads to Reduced Salt Tolerance
4.2. ZmPDI May Enhance Salt Tolerance in Rice by Regulating Laccase-6, Zingipain-2, WIP3, FKBP65, AKR4C10, GBSSII, Pho1, and TRXf1
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Muchate, N.S.; Nikalje, G.C.; Rajurkar, N.S.; Suprasanna, P.; Nikam, T.D. Plant salt stress: Adaptive responses, tolerance mechanism and bioengineering for salt tolerance. Bot. Rev. 2016, 82, 371–406. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, H.; Song, C.; Zhu, J.K.; Shabala, S. Mechanisms of Plant Responses and Adaptation to Soil Salinity. Innovation 2020, 1, 100017. [Google Scholar] [CrossRef]
- Shi, H.; Ye, T.; Chan, Z. Comparative proteomic and physiological analyses reveal the protective effect of exogenous polyamines in the bermudagrass (Cynodon dactylon) response to salt and drought stresses. J. Proteome Res. 2013, 12, 4951–4964. [Google Scholar] [CrossRef]
- Li, S.; Chen, J.; Guo, H.; Zong, J.; Zhang, F.; Chu, X.; Jiang, Q.; Ding, W.; Liu, J. Salinity tolerance evaluation of Zoysia turfgrass germplasm. Acta Pratacult. Sin. 2012, 4, 43–51. [Google Scholar]
- Marcum, K.B.; Murdoch, C.L. Salinity tolerance mechanisms of six C4 turfgrasses. J. Am. Soc. Hortic. Sci. 1994, 119, 779–784. [Google Scholar] [CrossRef]
- Du, Y.; Hei, Q.; Liu, Y.; Zhang, H.; Xu, K.; Xia, T. Isolation and characterization of a putative vacuolar Na+/H+ antiporter gene from Zoysia japonica L. J. Plant Biol. 2010, 53, 251–258. [Google Scholar] [CrossRef]
- Teng, K.; Tan, P.; Guo, W.; Yue, Y.; Fan, X.; Wu, J. Heterologous expression of a novel Zoysia japonica C2H2 Zinc finger gene, ZjZFN1, improved salt tolerance in Arabidopsis. Front. Plant Sci. 2018, 9, 1159. [Google Scholar] [CrossRef]
- Teng, K.; Tan, P.; Xiao, G.; Han, L.; Chang, Z.; Chao, Y. Heterologous expression of a novel Zoysia japonica salt-induced glycine-rich RNA-binding protein gene, ZjGRP, caused salt sensitivity in Arabidopsis. Plant Cell Rep. 2017, 36, 179–191. [Google Scholar] [CrossRef]
- Guo, H.; Ding, W.; Chen, J.; Chen, X.; Zheng, Y.; Wang, Z.; Liu, J. Genetic linkage map construction and QTL mapping of salt tolerance traits in Zoysiagrass (Zoysia japonica). PLoS ONE 2014, 9, e107249. [Google Scholar] [CrossRef]
- Chen, Y.; Zong, J.; Tan, Z.; Li, L.; Hu, B.; Chen, C.; Chen, J.; Liu, J. Systematic mining of salt-tolerant genes in halophyte-Zoysia matrella through cDNA expression library screening. Plant Physiol. Biochem. 2015, 89, 44–52. [Google Scholar] [CrossRef]
- Xie, Q.; Niu, J.; Xu, X.; Xu, L.; Zhang, Y.; Fan, B.; Liang, X.; Zhang, L.; Yin, S.; Han, L. De novo assembly of the Japanese lawngrass (Zoysia japonica Steud.) root transcriptome and identification of candidate unigenes related to early responses under salt stress. Front. Plant Sci. 2015, 6, 610. [Google Scholar]
- Wang, J.; An, C.; Guo, H.; Yang, X.; Chen, J.; Zong, J.; Li, J.; Liu, J. Physiological and transcriptomic analyses reveal the mechanisms underlying the salt tolerance of Zoysia japonica Steud. BMC Plant Biol. 2020, 20, 114. [Google Scholar] [CrossRef]
- Ali Khan, H.; Mutus, B. Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front. Chem. 2014, 2, 70. [Google Scholar] [CrossRef]
- Wang, L.; Yu, J.; Wang, C.C. Protein disulfide isomerase is regulated in multiple ways: Consequences for conformation, activities, and pathophysiological functions. BioEssays 2020, 43, e2000147. [Google Scholar] [CrossRef]
- Yuen, C.Y.L.; Shek, R.; Kang, B.-H.; Matsumoto, K.; Cho, E.J.; Christopher, D.A. Arabidopsis protein disulfide isomerase-8 is a type I endoplasmic reticulum transmembrane protein with thiol-disulfide oxidase activity. BMC Plant Biol. 2016, 16, 181. [Google Scholar] [CrossRef]
- Lu, Y.; Yuan, L.; Zhou, Z.; Wang, M.; Wang, X.; Zhang, S.; Sun, Q. The thiol-disulfide exchange activity of AtPDI1 is involved in the response to abiotic stresses. BMC Plant Biol. 2021, 21, 557. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Li, R.; Yuan, L.; Dai, Y.; Wang, X. Identification and functional analysis of a protein disulfide isomerase (AtPDI1) in Arabidopsis thaliana. Front. Plant Sci. 2018, 9, 913. [Google Scholar] [CrossRef]
- Fan, F.; Zhang, Y.; Wang, S.; Han, Y.; Wang, L.; Lu, D. Characterization of the oxidative protein folding activity of a unique plant oxidoreductase, Arabidopsis protein disulfide isomerase-11. Biochem. Biophy. Res. Commun. 2018, 495, 1041–1047. [Google Scholar] [CrossRef]
- Tominaga, J.; Nakahara, Y.; Horikawa, D.; Tanaka, A.; Kondo, M.; Kamei, Y.; Takami, T.; Sakamoto, W.; Unno, K.; Sakamoto, A.; et al. Overexpression of the protein disulfide isomerase AtCYO1 in chloroplasts slows dark-induced senescence in Arabidopsis. BMC Plant Biol 2018, 18, 80. [Google Scholar] [CrossRef]
- Kayum, M.A.; Park, J.-I.; Nath, U.K.; Saha, G.; Biswas, M.K.; Kim, H.-T.; Nou, I.-S. Genome-wide characterization and expression profiling of PDI family gene reveals function as abiotic and biotic stress tolerance in Chinese cabbage (Brassica rapa ssp. pekinensis). BMC Genomics. 2017, 18, 885. [Google Scholar] [CrossRef]
- Ming, Q.; Wang, K.; Wang, J.; Liu, J.; Li, X.; Wei, P.; Guo, H.; Chen, J.; Zong, J. The combination of RNA-seq transcriptomics and data-independent acquisition proteomics reveals the mechanisms underlying enhanced salt tolerance by the ZmPDI gene in Zoysia matrella [L.] Merr. Front. Plant Sci. 2022, 13, 970651. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.; Qu, A.; Wang, R.; Guo, H.; Li, X.; She, J.; Zong, J.; Li, J.; Liu, J. Establishment of genetic transformation system for Zoysia matrella. Chin. J. Tropical Crops. 2020, 41, 1566–1573. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R Package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Yang, Y.; Xiong, J.; Chen, R.; Fu, G.; Chen, T.; Tao, L. Excessive nitrate enhances cadmium (Cd) uptake by up-regulating the expression of OsIRT1 in rice (Oryza sativa). Environ. Exp. Bot. 2016, 122, 141–149. [Google Scholar] [CrossRef]
- Liang, W.; Ma, X.; Wan, P.; Liu, L. Plant salt-tolerance mechanism: A review. Biochem. Biophy. Res. Commun. 2018, 495, 286–291. [Google Scholar] [CrossRef]
- Pommerrenig, B.; Papini-Terzi, F.S.; Sauer, N. Differential regulation of sorbitol and sucrose loading into the phloem of plantago major in response to salt stress. Plant Physiol. 2007, 144, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.; Bledsoe, S.W.; Griffiths, C.A.; Kollman, A.; Paul, M.J.; Sakr, S.; Lagrimini, L.M. Differential role for trehalose metabolism in salt-stressed maize. Plant Physiol. 2015, 169, 1072–1089. [Google Scholar] [CrossRef] [PubMed]
- Munns, R. Comparative physiology of salt and water stress. Plant Cell Environ. 2002, 25, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.M.F.; Ali, E.F. Evaluation of proline functions in saline conditions. Phytochemistry 2017, 140, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Verbruggen, N.; Hermans, C. Proline accumulation in plants: A review. Amino Acids 2008, 35, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Dhindsa, R.S.; Plumb-Dhiiidsa, P.L.; Reid, D.M. Leaf senescence and lipid peroxidation: Effects of some phytohormones, and scavengers of free radicals and singlet oxygen. Physiol. Plantarum. 1982, 56, 453–457. [Google Scholar] [CrossRef]
- Stadtman, E.R. Protein oxidation and aging. Science 1992, 257, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, H.; Sivakumar, P.; Chakrabarty, R.; Thomas, G. Scavenging of reactive oxygen species in NaCl-stressed rice (Oryza sativa L.)—Differential response in salt-tolerant and sensitive varieties. Plant Sci. 2003, 165, 1411–1418. [Google Scholar] [CrossRef]
- Barros, J.; Serk, H.; Granlund, I.; Pesquet, E. The cell biology of lignification in higher plants. Ann. Bot. 2015, 115, 1053–1074. [Google Scholar] [CrossRef]
- Liu, Q.; Luo, L.; Wang, X.; Shen, Z.; Zheng, L. Comprehensive analysis of rice laccase gene (OsLAC) family and ectopic expression of OsLAC10 enhances tolerance to copper stress in Arabidopsis. Int. J. Mol. Sci. 2017, 18, 209. [Google Scholar] [CrossRef]
- Liu, M.; Dong, H.; Wang, M.; Liu, Q. Evolutionary divergence of function and expression of laccase genes in plants. J. Genet. 2020, 99, 23. [Google Scholar] [CrossRef]
- Zhao, Y.; Lin, S.; Qiu, Z.; Cao, D.; Wen, J.; Deng, X.; Wang, X.; Lin, J.; Li, X. MicroRNA857 is involved in the regulation of secondary growth of vascular tissues in Arabidopsis. Plant Physiol. 2015, 169, 2539–2552. [Google Scholar] [CrossRef]
- Qin, R.; Hu, Y.; Chen, H.; Du, Q.; Yang, J.; Li, W.X. MicroRNA408 negatively regulates salt tolerance by affecting secondary cell wall development in maize. Plant Physiol. 2023, 192, 1569–1583. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, X.M.; Xiong, H.X.; Mao, W.Y.; Zhao, P.; Sun, M.X. Papain-like and legumain-like proteases in rice: Genome-wide identification, comprehensive gene feature characterization and expression analysis. BMC Plant Biol. 2018, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Li, H.; Yin, B.; Zhang, Y.; Liu, Y.; Cheng, Z.; Liu, D.; Lu, H. The papain-like cysteine protease CEP1 is involved in programmed cell death and secondary wall thickening during xylem development in Arabidopsis. J. Exp. Bot. 2019, 70, 205–215. [Google Scholar] [CrossRef]
- Shukla, P.; Gautam, R.; Singh, N.K.; Ahmed, I.; Kirti, P.B. A proteomic study of cysteine protease induced cell death in anthers of male sterile tobacco transgenic plants. Physiol. Mol. Biol. Plants 2019, 25, 1073–1082. [Google Scholar] [CrossRef]
- Niño, M.C.; Kang, K.K.; Cho, Y.-G. Genome-wide transcriptional response of papain-like cysteine protease-mediated resistance against Xanthomonas oryzae pv. oryzae in rice. Plant Cell Rep. 2020, 39, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Pechan, T.; Ye, L.; Chang, Y.M.; Mitra, A.; Lin, L.; Davis, F.M.; Williams, W.P.; Luthe, D.S. A unique 33-kD cysteine proteinase accumulates in response to larval feeding in maize genotypes resistant to fall armyworm and other Lepidoptera. Plant Cell 2000, 12, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Khanna-Chopra, R.; Srivalli, B.; Ahlawat, Y.S. Drought induces many forms of cysteine proteases not observed during natural senescence. Biochem. Biophy. Res. Commun. 1999, 255, 324–327. [Google Scholar] [CrossRef]
- Lu, H.; Chandrasekar, B.; Oeljeklaus, J.; Misas-Villamil, J.C.; Wang, Z.; Shindo, T.; Bogyo, M.; Kaiser, M.; Van der Hoorn, R.A.L. Subfamily-specific fluorescent probes for cysteine proteases display dynamic protease activities during seed germination. Plant Physiol. 2015, 168, 1462–1475. [Google Scholar] [CrossRef]
- Martinez, M.; Cambra, I.; Carrillo, L.; Diaz-Mendoza, M.; Diaz, I. Characterization of the entire cystatin gene family in barley and their target cathepsin L-like cysteine-proteases, partners in the hordein mobilization during seed germination. Plant Physiol. 2009, 151, 1531–1545. [Google Scholar] [CrossRef]
- Kiyosaki, T.; Matsumoto, I.; Asakura, T.; Funaki, J.; Kuroda, M.; Misaka, T.; Arai, S.; Abe, K. Gliadain, a gibberellin-inducible cysteine proteinase occurring in germinating seeds of wheat, Triticum aestivum L., specifically digests gliadin and is regulated by intrinsic cystatins. FEBS J. 2007, 274, 1908–1917. [Google Scholar] [CrossRef]
- Zhang, X.M.; Wang, Y.; Lv, X.M.; Li, H.; Sun, P.; Lu, H.; Li, F.L. NtCP56, a new cysteine protease in Nicotiana tabacum L., involved in pollen grain development. J. Exp. Bot. 2009, 60, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liu, D.; Lv, X.; Wang, Y.; Xun, Z.; Liu, Z.; Li, F.; Lu, H. The cysteine protease CEP1, a key executor involved in tapetal programmed cell death, regulates pollen development in Arabidopsis. Plant Cell 2014, 26, 2939–2961. [Google Scholar] [CrossRef]
- Li, N.; Zhang, D.S.; Liu, H.S.; Yin, C.S.; Li, X.-X.; Liang, W.Q.; Yuan, Z.; Xu, B.; Chu, H.W.; Wang, J.; et al. The rice tapetum degeneration retardation gene is required for tapetum degradation and anther development. Plant Cell 2006, 18, 2999–3014. [Google Scholar] [CrossRef]
- Li, Y.; Suen, D.F.; Huang, C.Y.; Kung, S.Y.; Huang, A.H.C. The maize tapetum employs diverse mechanisms to synthesize and store proteins and flavonoids and transfer them to the pollen surface. Plant Physiol. 2012, 158, 1548–1561. [Google Scholar] [CrossRef]
- Koizumi, M.; Yamaguchi-Shinozaki, K.; Tsuji, H.; Shinozaki, K. Structure and expression of two genes that encode distinct drought-inducible cysteine proteinases in Arabidopsis thaliana. Gene 1993, 129, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.T.; Mullet, J.E. A salt- and dehydration-inducible pea gene, Cyp15a, encodes a cell-wall protein with sequence similarity to cysteine proteases. Plant Mol. Biol. 1995, 28, 1055–1065. [Google Scholar] [CrossRef]
- Zang, Q.W.; Wang, C.X.; Li, X.Y.; Guo, Z.A.; Jing, R.L.; Zhao, J.; Chang, X.P. Isolation and characterization of a gene encoding a polyethylene glycol-induced cysteine protease in common wheat. J. Biosci. 2010, 35, 379–388. [Google Scholar] [CrossRef]
- Zhou, L.; Li, M.; Du, Q.; Yang, S.; Li, J.; Fan, Y.; Mao, K.; Zhang, J.; Xiao, H.; Wang, J. Genome-wide identification of PLCPs in pepper and the functional characterization of CaCP34 in resistance to salt- and osmotic-induced leaf senescence. Sci. Horticult. 2023, 309, 111624. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, S.; Chen, C.; Li, M.; Du, Q.; Wang, J.; Yin, Y.; Xiao, H. CaCP15 gene negatively regulates salt and osmotic stress responses in Capsicum annuum L. Genes 2023, 14, 1409. [Google Scholar] [CrossRef]
- Vasudevan, D.; Gopalan, G.; Kumar, A.; Garcia, V.J.; Luan, S.; Swaminathan, K. Plant immunophilins: A review of their structure-function relationship. Biochim. Biophys. Acta 2015, 1850, 2145–2158. [Google Scholar] [CrossRef]
- Yang, D.; Li, Y.; Zhu, M.; Cui, R.; Gao, J.; Shu, Y.; Lu, X.; Zhang, H.; Zhang, K. Genome-wide identification and expression analysis of the cucumber FKBP gene family in response to abiotic and biotic stresses. Genes 2023, 14, 2006. [Google Scholar] [CrossRef]
- He, Z.; Li, L.; Luan, S. Immunophilins and parvulins. superfamily of peptidyl prolyl isomerases in Arabidopsis. Plant Physiol. 2004, 134, 1248–1267. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, H.; Li, W.; Mu, C.; Zhang, F.; Wang, L.; Meng, Z. Genome-wide analysis and environmental response profiling of the FK506-binding protein gene family in maize (Zea mays L.). Gene 2012, 498, 212–222. [Google Scholar] [CrossRef]
- Gollan, P.J.; Bhave, M. Genome-wide analysis of genes encoding FK506-binding proteins in rice. Plant Mol. Biol. 2010, 72, 1–16. [Google Scholar] [CrossRef]
- Gollan, P.J.; Bhave, M.; Aro, E.M. The FKBP families of higher plants: Exploring the structures and functions of protein interaction specialists. FEBS Lett. 2012, 586, 3539–3547. [Google Scholar] [CrossRef]
- Alavilli, H.; Lee, H.; Park, M.; Yun, D.J.; Lee, B.-H. Enhanced multiple stress tolerance in Arabidopsis by overexpression of the polar moss peptidyl prolyl isomerase FKBP12 gene. Plant Cell Rep. 2017, 37, 453–465. [Google Scholar] [CrossRef]
- Subin, C.S.; Pradeep, M.A.; Vijayan, K.K. FKBP-type peptidyl-prolyl cis-trans isomerase from thermophilic microalga, Scenedesmus sp.: Molecular characterisation and demonstration of acquired salinity and thermotolerance in E. coli by recombinant expression. J. Appl. Phycol. 2016, 28, 3307–3315. [Google Scholar] [CrossRef]
- Cheung, M.Y.; Auyeung, W.-K.; Li, K.P.; Lam, H.M. A rice immunophilin homolog, OsFKBP12, is a negative regulator of both biotic and abiotic stress responses. Int. J. Mol. Sci. 2020, 21, 8791. [Google Scholar] [CrossRef]
- Yang, H.; Karali, D.; Oxley, D.; Runions, J.; Ktistakis, N.; Farmaki, T. The Arabidopsis thaliana immunophilin ROF1 directly interacts with PI(3)P and PI(3,5)P2 and affects germination under osmotic stress. PLoS ONE 2012, 7, e48241. [Google Scholar]
- Meiri, D.; Tazat, K.; Cohen-Peer, R.; Farchi-Pisanty, O.; Aviezer-Hagai, K.; Avni, A.; Breiman, A. Involvement of Arabidopsis ROF2 (FKBP65) in thermotolerance. Plant Mol. Biol. 2009, 72, 191–203. [Google Scholar] [CrossRef]
- Smith, D.F.; Toft, D.O. Minireview: The intersection of steroid receptors with molecular chaperones: Observations and questions. Mol. Endocrinol. 2008, 22, 2229–2240. [Google Scholar] [CrossRef]
- Pratt, W.B.; Toft, D.O. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997, 18, 306–360. [Google Scholar]
- Miller, G.A.D.; Mittler, R.O.N. Could heat shock transcription factors function as hydrogen peroxide sensors in plants? Ann. Bot. 2006, 98, 279–288. [Google Scholar] [CrossRef]
- Panchuk, I.I.; Volkov, R.A.; Schöffl, F. Heat stress- and heat shock transcription factor-dependent expression and activity of ascorbate peroxidase in Arabidopsis. Plant Physiol. 2002, 129, 838–853. [Google Scholar] [CrossRef]
- Davletova, S.; Rizhsky, L.; Liang, H.; Shengqiang, Z.; Oliver, D.J.; Coutu, J.; Shulaev, V.; Schlauch, K.; Mittler, R. Cytosolic ascorbate peroxidase 1 is a central component of the reactive oxygen gene network of Arabidopsis. Plant Cell 2005, 17, 268–281. [Google Scholar] [CrossRef]
- Li, C.; Chen, Q.; Gao, X.; Qi, B.; Chen, N.; Xu, S.; Chen, J.; Wang, X. AtHsfA2 modulates expression of stress responsive genes and enhances tolerance to heat and oxidative stress in Arabidopsis. Sci. China Ser. C 2005, 48, 540–550. [Google Scholar] [CrossRef]
- Ogawa, D.; Yamaguchi, K.; Nishiuchi, T. High-level overexpression of the Arabidopsis HsfA2 gene confers not only increased themotolerance but also salt/osmotic stress tolerance and enhanced callus growth. J. Exp. Bot. 2007, 58, 3373–3383. [Google Scholar] [CrossRef]
- Vaghela, B.; Vashi, R.; Rajput, K.; Joshi, R. Plant chitinases and their role in plant defense: A comprehensive review. Enzyme Microl. Technol. 2022, 159, 110055. [Google Scholar] [CrossRef]
- Bravo, J.M.; Campo, S.; Murillo, I.; Coca, M.I.; Segundo, B.S. Fungus- and wound-induced accumulation of mRNA containing a class II chitinase of the pathogenesis-related protein 4 (PR-4) family of maize. Plant Mol. Biol. 2003, 52, 745–759. [Google Scholar] [CrossRef]
- Bokmaa, E.; Barends, T.; Terwisscha van Scheltingab, A.C.; Dijkstrab, B.W.; Beintema, J.J. Enzyme kinetics of hevamine, a chitinase from the rubber tree Hevea brasiliensis. FEBS Lett. 2000, 478, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Passarinho, P.A.; de Vries, S.C. Arabidopsis chitinases: A genomic survey. Arab. Book 2002, 1, e0023. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Xu, L.; Fu, Z.; Yang, Y.; Guo, J.; Wang, S.; Que, Y. ScChi, encoding an acidic class III chitinase of sugarcane, confers positive responses to biotic and abiotic stresses in sugarcane. Int. J. Mol. Sci. 2014, 15, 2738–2760. [Google Scholar] [CrossRef] [PubMed]
- Dana, M.d.l.M.; Pintor-Toro, J.A.; Cubero, B. Transgenic tobacco plants overexpressing chitinases of fungal origin show enhanced resistance to biotic and abiotic stress agents. Plant Physiol. 2006, 142, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, H.; Liu, C.; Li, L.; Liu, L.; Han, X.; Wan, Z.; Sha, A. Transcriptome profile analysis of two Vicia faba cultivars with contrasting salinity tolerance during seed germination. Sci. Rep. 2020, 10, 7250. [Google Scholar] [CrossRef] [PubMed]
- Seo, P.J.; Lee, A.K.; Xiang, F.; Park, C.M. Molecular and functional profiling of Arabidopsis pathogenesis-related genes: Insights into their roles in salt response of seed germination. Plant Cell Physiol. 2008, 49, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, Y.; Li, Z.; Shi, W.; Gao, F.; Zhou, Y.; Zhang, G.; Feng, J. Genome-wide identification and expression analyses of the chitinases under cold and osmotic stress in Ammopiptanthus nanus. Genes 2019, 10, 472. [Google Scholar] [CrossRef]
- Chen, H.J.; Chen, J.Y.; Wang, S.J. Molecular regulation of starch accumulation in rice seedling leaves in response to salt stress. Acta Physiol. Plant. 2007, 30, 135–142. [Google Scholar] [CrossRef]
- Pattanagul, W.; Thitisaksakul, M. Effect of salinity stress on growth and carbohydrate metabolism in three rice (Oryza sativa L.) cultivars differing in salinity tolerance. Indian J. Exp. Biol. 2008, 46, 736–742. [Google Scholar]
- Yuenyong, W.; Chinpongpanich, A.; Comai, L.; Chadchawan, S.; Buaboocha, T. Downstream components of the calmodulin signaling pathway in the rice salt stress response revealed by transcriptome profiling and target identification. BMC Plant Biol. 2018, 18, 335. [Google Scholar] [CrossRef]
- Baroja-Fernández, E.; Francisco José Muñoz, T.S.; Rodríguez-López, M.; Akazawa, T.; Pozueta-Romero, J. Sucrose synthase catalyzes the de novo production of ADPglucose linked to starch biosynthesis in heterotrophic tissues of plants. Plant Cell Physiol. 2003, 44, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.K.; Koper, K.; Satoh, H.; Okita, T.W. Rice endosperm starch phosphorylase (Pho1) assembles with disproportionating enzyme (Dpe1) to form a protein complex that enhances synthesis of malto-oligosaccharides. J. Biol. Chem. 2016, 291, 19994–20007. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Shibahara, K.; Tokunaga, T.; Nishi, A.; Tasaki, M.; Hwang, S.K.; Okita, T.W.; Kaneko, N.; Fujita, N.; Yoshida, M.; et al. Mutation of the plastidial alpha-glucan phosphorylase gene in rice affects the synthesis and structure of starch in the endosperm. Plant Cell 2008, 20, 1833–1849. [Google Scholar] [CrossRef] [PubMed]
- Dauvillee, D.; Chochois, V.; Steup, M.; Haebel, S.; Eckermann, N.; Ritte, G.; Ral, J.P.; Colleoni, C.; Hicks, G.; Wattebled, F.; et al. Plastidial phosphorylase is required for normal starch synthesis in Chlamydomonas reinhardtii. Plant J. 2006, 48, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, S.D.; Michelet, L.; Zaffagnini, M.; Massot, V.; Issakidis-Bourguet, E. Thioredoxins in chloroplasts. Curr. Genet. 2007, 51, 343–365. [Google Scholar] [CrossRef] [PubMed]
- Balsera, M.; Uberegui, E.; Schürmann, P.; Buchanan, B.B. Evolutionary development of redox regulation in chloroplasts. Antioxid. Redox. Sign. 2014, 21, 1327–1355. [Google Scholar] [CrossRef] [PubMed]
- Michelet, L.; Zaffagnini, M.; Morisse, S.; Sparla, F.; Perez-Perez, M.E.; Francia, F.; Danon, A.; Marchand, C.H.; Fermani, S.; Trost, P.; et al. Redox regulation of the Calvin-Benson cycle: Something old, something new. Front. Plant Sci. 2013, 4, 470. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, S.D.; Tedesco, D.; Crozet, P.; Michelet, L.; Fermani, S.; Zaffagnini, M.; Henri, J. Crystal structure of chloroplastic thioredoxin f2 from Chlamydomonas reinhardtii reveals distinct surface properties. Antioxidants 2018, 7, 171. [Google Scholar] [CrossRef]
- Née, G.; Zaffagnini, M.; Trost, P.; Issakidis-Bourguet, E. Redox regulation of chloroplastic glucose-6-phosphate dehydrogenase: A new role for f-type thioredoxin. FEBS Lett. 2009, 583, 2827–2832. [Google Scholar] [CrossRef]
- Kang, Z.; Qin, T.; Zhao, Z. Thioredoxins and thioredoxin reductase in chloroplasts: A review. Gene 2019, 706, 32–42. [Google Scholar] [CrossRef]
- Garcia-Canas, R.; Florencio, F.J.; Lopez-Maury, L. Back to the future: Transplanting the chloroplast TrxF-FBPase-SBPase redox system to cyanobacteria. Front. Plant Sci. 2022, 13, 1052019. [Google Scholar] [CrossRef] [PubMed]
- Belin, C.; Bashandy, T.; Cela, J.; Delorme-Hinoux, V.; Riondet, C.; Reichheld, J.P. A comprehensive study of thiol reduction gene expression under stress conditions in Arabidopsis thaliana. Plant Cell Environ. 2015, 38, 299–314. [Google Scholar] [CrossRef]
- Fernandez-Trijueque, J.; Barajas-Lopez Jde, D.; Chueca, A.; Cazalis, R.; Sahrawy, M.; Serrato, A.J. Plastid thioredoxins f and m are related to the developing and salinity response of post-germinating seeds of Pisum sativum. Plant Sci 2012, 188–189, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhao, H.; Cao, K.; Hu, L.; Du, T.; Baluska, F.; Zou, Z. Beneficial roles of melatonin on redox regulation of photosynthetic electron transport and synthesis of D1 protein in tomato seedlings under salt stress. Front. Plant Sci. 2016, 7, 1823. [Google Scholar] [CrossRef]
- Han, X.; Wang, Y.; Liu, X.; Jiang, L.; Ren, Y.; Liu, F.; Peng, C.; Li, J.; Jin, X.; Wu, F.; et al. The failure to express a protein disulphide isomerase-like protein results in a floury endosperm and an endoplasmic reticulum stress response in rice. J. Exp. Bot. 2012, 63, 121–130. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Pothiraj, R.; Suthanthiram, B.; Somasundaram, S.M.; Subbaraya, U. Phylogenomic classification and synteny network analyses deciphered the evolutionary landscape of aldo-keto reductase (AKR) gene superfamily in the plant kingdom. Gene 2022, 816, 146169. [Google Scholar] [CrossRef]
- Sengupta, D.; Naik, D.; Reddy, A.R. Plant aldo-keto reductases (AKRs) as multi-tasking soldiers involved in diverse plant metabolic processes and stress defense: A structure-function update. J. Plant Physiol. 2015, 179, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Sun, H.; Zhang, J.; Hou, Y.; Zhang, T.; Kang, J.; Wang, Z.; Yang, Q.; Long, R. Analysis of aldo-keto reductase gene family and their responses to salt, drought, and abscisic acid stresses in Medicago truncatula. Int. J. Mol. Sci. 2020, 21, 754. [Google Scholar] [CrossRef]
- Penning, T.M. The aldo-keto reductases (AKRs): Overview. Chem. Biol. Interact. 2015, 234, 236–246. [Google Scholar] [CrossRef]
- Simpson, P.J.; Tantitadapitak, C.; Reed, A.M.; Mather, O.C.; Bunce, C.M.; White, S.A.; Ride, J.P. Characterization of two novel aldo-keto reductases from Arabidopsis: Expression patterns, broad substrate specificity, and an open active-site structure suggest a role in toxicant metabolism following stress. J. Mol. Biol. 2009, 392, 465–480. [Google Scholar] [CrossRef]
- Saito, R.; Shimakawa, G.; Nishi, A.; Iwamoto, T.; Sakamoto, K.; Yamamoto, H.; Amako, K.; Makino, A.; Miyake, C. Functional analysis of the AKR4C subfamily of Arabidopsis thaliana: Model structures, substrate specificity, acrolein toxicity, and responses to light and [CO2]. Biosci. Biotech. Bioch. 2014, 77, 2038–2045. [Google Scholar] [CrossRef]
- Éva, C.; Zelenyánszki, H.; Tömösközi-Farkas, R.; Tamás, L. Transgenic barley expressing the Arabidopsis AKR4C9 aldo-keto reductase enzyme exhibits enhanced freezing tolerance and regenerative capacity. S. Afr. J. Bot. 2014, 93, 179–184. [Google Scholar] [CrossRef]
- Bartels, D.; Engelhardt, K.; Roncarati, R.; Schneider, K.; Rotter, M.; Salamini, F. An ABA and GA modulated gene expressed in the barley embryo encodes an aldose reductase related protein. EMBO J. 1991, 10, 1037–1043. [Google Scholar] [CrossRef]
- Lee, S.P.; Chen, T.H.H. Molecular cloning of abscisic acid-responsive mRNAs expressed during the induction of freezing tolerance in bromegrass (Bromus inermis Leyss) suspension culture. Plant Physiol. 1993, 101, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Foley, M.E. Cloning and characterization of differentially expressed genes in imbibed dormant and afterripened Avena fatua embryos. Plant Mol. Biol. 1995, 29, 823–831. [Google Scholar] [CrossRef]
- Mundree, S.G.; Whittaker, A.; Thomson, J.A.; Farrant, J.M. An aldose reductase homolog from the resurrection plant Xerophyta viscosa Baker. Planta 2000, 211, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Gavidia, I.; Perez-Bermudez, P.; Seitz, H.U. Cloning and expression of two novel aldo-keto reductases from Digitalis purpurea leaves. Eur. J. Biochem. 2002, 269, 2842–2850. [Google Scholar] [CrossRef]
- Songsiriritthigul, C.; Narawongsanont, R.; Tantitadapitak, C.; Guan, H.H.; Chen, C.J. Structure-function study of AKR4C14, an aldo-keto reductase from Thai jasmine rice (Oryza sativa L. ssp. indica cv. KDML105). Acta Crystallogr. Sect. D Struct. Biol. 2020, 76, 472–483. [Google Scholar] [CrossRef]
- Narawongsanont, R.; Kabinpong, S.; Auiyawong, B.; Tantitadapitak, C. Cloning and characterization of AKR4C14, a rice aldo-keto reductase, from Thai Jasmine rice. Protein J. 2012, 31, 35–42. [Google Scholar] [CrossRef]
- Auiyawong, B.; Narawongsanont, R.; Tantitadapitak, C. Characterization of AKR4C15, a novel member of aldo-keto reductase, in comparison with other rice AKR(s). Protein J. 2017, 36, 257–269. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GeneID | ProteinID | RNA-seq | Proteome | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| log2 FC(WT_24h/0h) | log2 FC(OX_24h/0h) | log2 FC(CR_24h/0h) | log2 [FC(OX_24h/0h)/FC(CK_24h/0h)] | log2 [FC(CR_24h/0h)/FC(CK_24h/0h)] | log2 FC(WT_24h/0h) | log2 FC(OX_24h/0h) | log2 FC(CR_24h/0h) | log2 [FC(OX_24h/0h)/FC(CK_24h/0h)] | log2 [FC(CR_24h/0h)/FC(CK_24h/0h)] | Gene Name | Description | ||
| 4324320 | XP_015616604.1 | 0.3001627 | −0.966663 | −0.633929656 | −1.26683 | −0.93409 | 0.312297 | −0.71909 | 0.05656 | −1.03139 | −0.25574 | TRXf1 | thioredoxin F, chloroplastic-like |
| 4327925 | XP_015621254.1 | 0.3169881 | −1.860921 | −1.994143945 | −2.17791 | −2.31113 | 0.13762 | −1.00638 | −0.37458 | −1.144 | −0.5122 | FKBP46 | peptidyl-prolyl cis-trans isomerase FKBP65 |
| 4334171 | XP_015631420.1 | −0.215354 | −1.722303 | −0.804273692 | −1.50695 | −0.58892 | −0.05071 | −1.05794 | −0.24896 | −1.00723 | −0.19824 | OsPho1 | alpha-1,4 glucan phosphorylase L isozyme, chloroplastic/amyloplastic |
| 4338987 | XP_015640121.1 | −0.519035 | −2.559227 | 0.140924626 | −2.04019 | 0.65996 | −0.07701 | −1.15113 | −0.29747 | −1.07412 | −0.22046 | AKR4C10 | aldo-keto reductase family 4 member C10 |
| 4343010 | XP_015647210.1 | −0.304685 | −2.559233 | 0.501779613 | −2.25455 | 0.806465 | −0.76549 | −1.77274 | −0.80999 | −1.00725 | −0.0445 | OsGBSSII | granule-bound starch synthase 1b, chloroplastic/amyloplastic |
| 4350821 | XP_015615550.1 | 2.9533801 | −0.138991 | 3.573679797 | −3.09237 | 0.6203 | 0.674683 | −0.4243 | 0.373637 | −1.09898 | −0.30105 | WIP3 | Wound-induced protein precursor |
| 107276064 | XP_015611357.1 | 2.0799684 | 3.3114153 | 2.212447716 | 1.231447 | 0.132479 | 0.314234 | 1.551538 | 0.611157 | 1.237304 | 0.296923 | zingipain-2 | putative cysteine proteinase |
| 107276446 | XP_015621471.1 | −0.911379 | 1.1211904 | 0.472168907 | 2.032569 | 1.383548 | −0.03991 | 1.381661 | −0.32439 | 1.421576 | −0.28448 | Oslaccase-6 | laccase-6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Wang, K.; Li, L.; Sun, Q.; Li, D.; Hao, D.; Chen, J.; Zong, J.; Liu, J.; Guo, H.; et al. Combined Transcriptome and Proteome Analysis Reveals the Molecular Mechanism by Which ZmPDI Improves Salt Resistance in Rice (Oryza sativa). Agriculture 2024, 14, 615. https://doi.org/10.3390/agriculture14040615
Wang J, Wang K, Li L, Sun Q, Li D, Hao D, Chen J, Zong J, Liu J, Guo H, et al. Combined Transcriptome and Proteome Analysis Reveals the Molecular Mechanism by Which ZmPDI Improves Salt Resistance in Rice (Oryza sativa). Agriculture. 2024; 14(4):615. https://doi.org/10.3390/agriculture14040615
Chicago/Turabian StyleWang, Jingjing, Kai Wang, Ling Li, Qixue Sun, Dandan Li, Dongli Hao, Jingbo Chen, Junqin Zong, Jianxiu Liu, Hailin Guo, and et al. 2024. "Combined Transcriptome and Proteome Analysis Reveals the Molecular Mechanism by Which ZmPDI Improves Salt Resistance in Rice (Oryza sativa)" Agriculture 14, no. 4: 615. https://doi.org/10.3390/agriculture14040615
APA StyleWang, J., Wang, K., Li, L., Sun, Q., Li, D., Hao, D., Chen, J., Zong, J., Liu, J., Guo, H., & Chen, R. (2024). Combined Transcriptome and Proteome Analysis Reveals the Molecular Mechanism by Which ZmPDI Improves Salt Resistance in Rice (Oryza sativa). Agriculture, 14(4), 615. https://doi.org/10.3390/agriculture14040615