
Fine-Mapping of qECL7.1, a Quantitative Trait Locus Contributing to Epicotyl Length in Adzuki Bean (Vigna angularis)

 ,
,
Abstract
1. Introduction
2. Materials and Methods
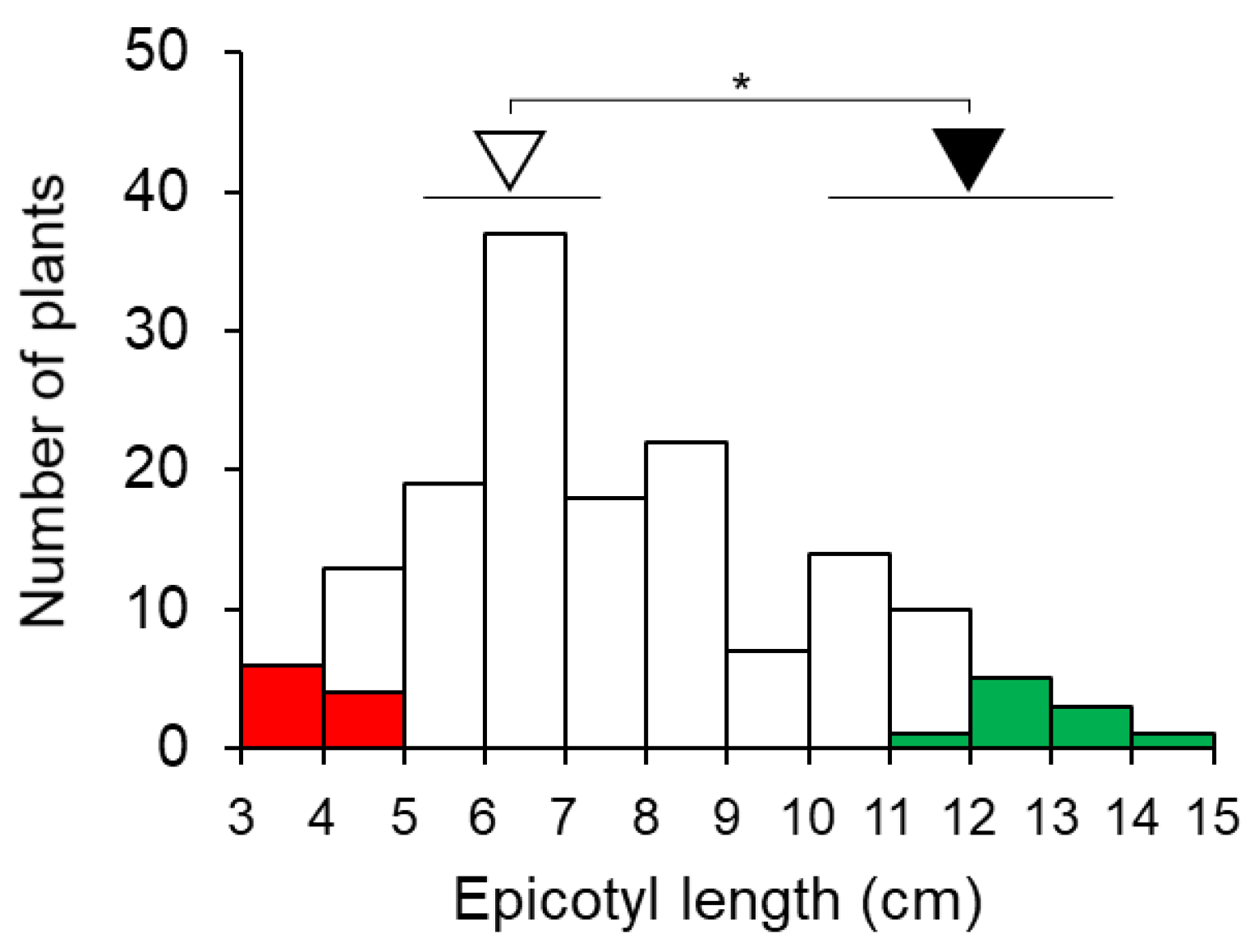
2.1. Plant Materials and Phenotypic Evaluation of RILs
2.2. Construction of Sequencing Libraries and Sequencing
2.3. Phenotypic Trait Evaluation of F2 Population
2.4. Genome-Wide Detection of SNPs and InDels, and Marker Analysis
2.5. QTL Analysis Using Linkage Mapping of the F2 Population
2.6. Interaction Assessment between Major QTLs
2.7. Fine Mapping of qECL7.1
2.8. Statistical Analysis
3. Results
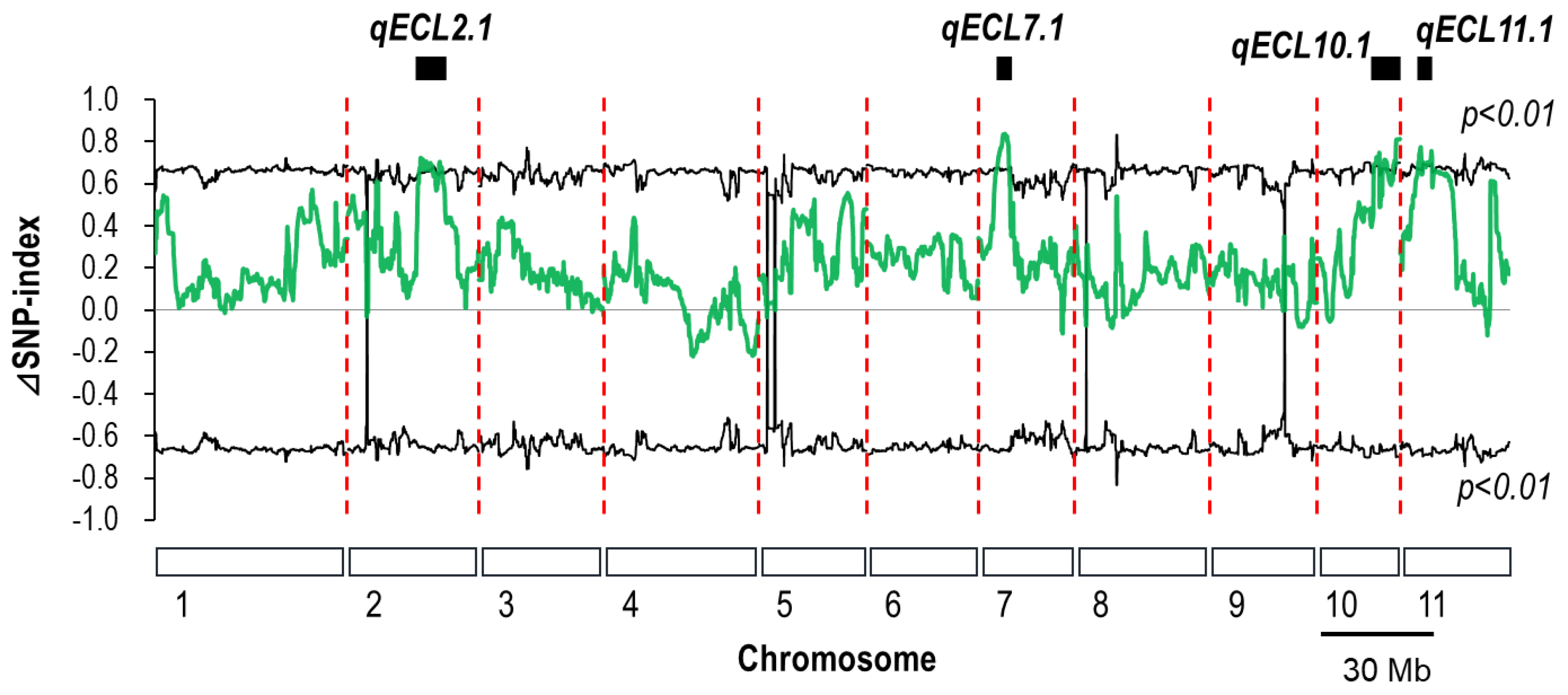
3.1. QTL-Seq Analysis for Epicotyl Length in F7 RILs
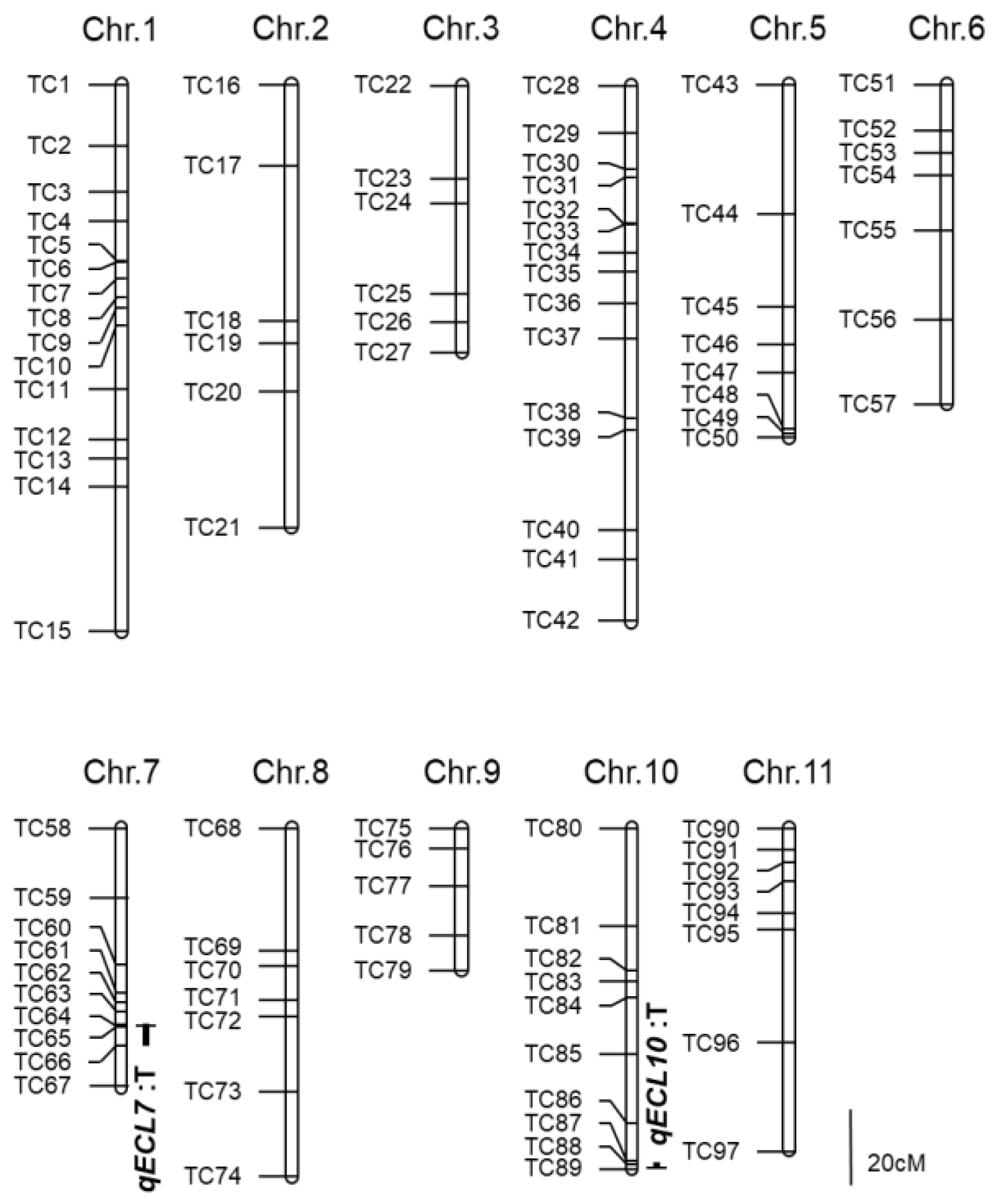
3.2. QTL Detection by Linkage Mapping
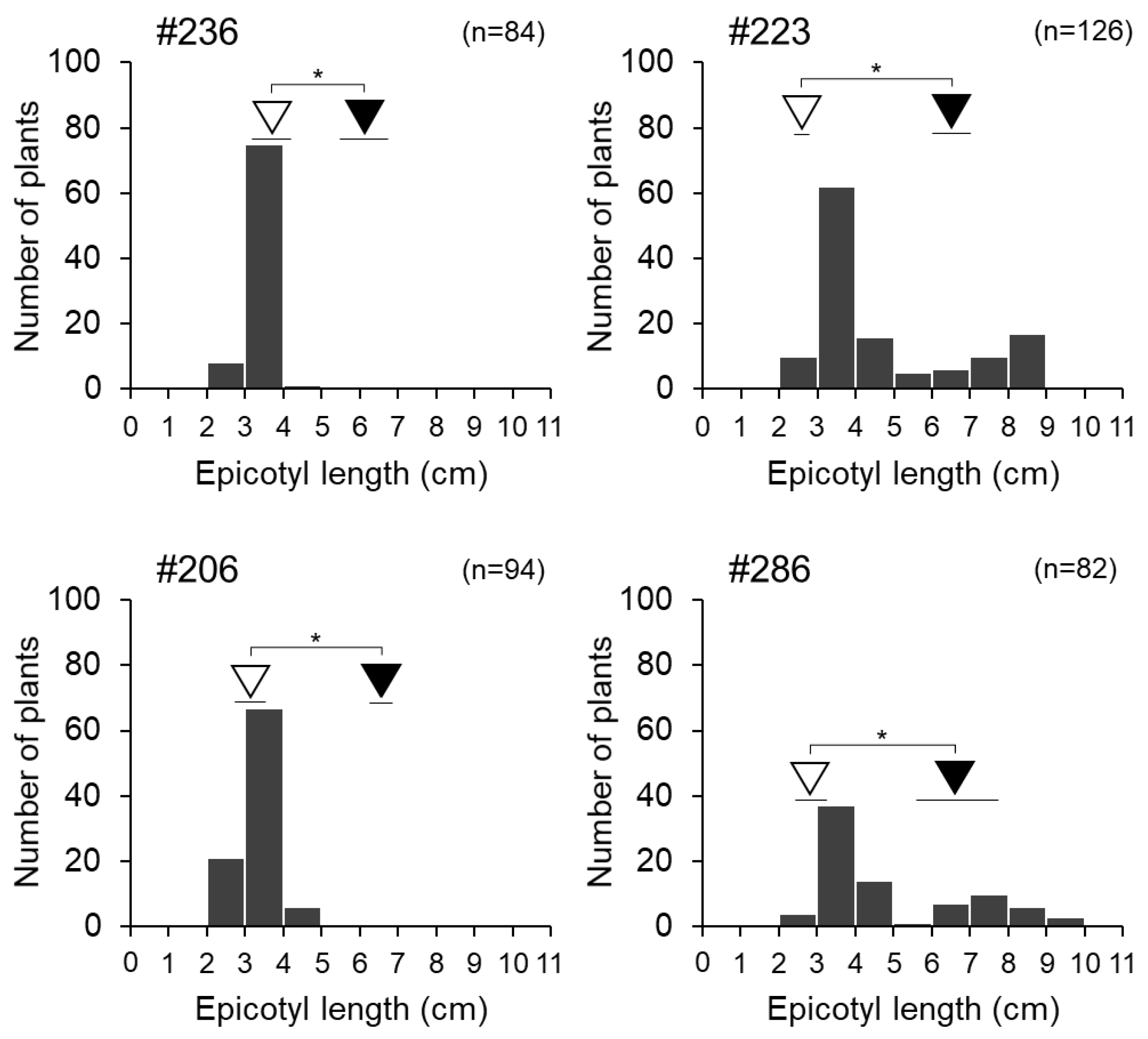
3.3. Interaction between qECL7.1 and qECL10.1
3.4. Fine Mapping of Epicotyl Length QTL qECL7.1 on Chromosome 7
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gontijo, W.D.R.; Sousa, P.H.S.; Matsuo, É.; Resende, J.C.; Barros, P.H.F.C.; Bomtempo, G.L. Epicotyl length in seedlings of soybean cultivars subjected to reduced inter-row spacing. Agron. Sci. Biotechnol. 2021, 7, 1–7. [Google Scholar] [CrossRef]
- Hisanori, S.; Tazawa, A.; Sato, H. Genetic Resources and Comparative Genomics of Legumes (Glycine and Vigna): Azuki Bean Cultivation in Hokkaido and Breeding at Tokachi Agricultural Experiment Station. In Proceedings of the 14th NIAS International Workshop on Genetic Resources, Tsukuba, Japan, 14 September 2009; pp. 87–90. [Google Scholar]
- Mori, M.; Maki, K.; Kawahata, T.; Kawahara, D.; Kato, Y.; Yoshida, T.; Nagasawa, H.; Sato, H.; Nagano, A.J.; Bethke, P.C.; et al. Mapping of qtls controlling epicotyl length in adzuki bean (Vigna angularis). Breed. Sci. 2021, 71, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Isemura, T.; Kaga, A.; Konishi, S.; Ando, T.; Tomooka, N.; Han, O.K.; Vaughan, D.A. Genome dissection of traits related to domestication in azuki bean (Vigna angularis) and comparison with other warm-season legumes. Ann. Bot. 2007, 100, 1053–1071. [Google Scholar] [CrossRef] [PubMed]
- Kaga, A.; Isemura, T.; Tomooka, N.; Vaughan, D.A. The genetics of domestication of the azuki bean (Vigna angularis). Genetics 2008, 178, 1013–1036. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Guo, N.; Zhang, Y.; Bu, Y.; Zhao, J.; Xing, H. Combining QTL-seq and linkage mapping to fine map a wild soybean allele characteristic of greater plant height. BMC Genom. 2018, 19, 226. [Google Scholar] [CrossRef]
- Kumar, K.; Purayannur, S.; Kaladhar, V.C.; Parida, S.K.; Verma, P.K. mQTL-seq and classical mapping implicates the role of an AT-HOOK MOTIF CONTAINING NUCLEAR LOCALIZED (AHL) family gene in Ascochyta blight resistance of chickpea. Plant Cell Environ. 2018, 41, 2128–2140. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. JoinMap 5®, Software for the Calculation of Genetic Linkage Maps in Experimental Populations of Diploid Species. Kyazma B.V., Wageningen, The Netherlands. 2019. Available online: www.kyazma.nl (accessed on 15 May 2023).
- Kosambi, D.D. The estimation of map distances from recombination values. Ann. Eugen. 1943, 12, 172–175. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. MapQTL 6® Software for the Mapping of Quantitative Trait Loci in Experimental Populations of Diploid Species. Kyazma B.V., Wageningen, The Netherlands. 2019. Available online: www.kyazma.nl (accessed on 15 May 2023).
- Dunnett, C.W. A Multiple Comparison Procedure for Comparing Several Treatments with a Control. J. Am. Stat. Assoc. 1955, 50, 1096–1121. [Google Scholar] [CrossRef]
- Hong, H.; Li, M.; Chen, Y.; Wang, H.; Wang, J.; Guo, B.; Gao, H.; Ren, H.; Yuan, M.; Han, Y.; et al. Genome-wide association studies for soybean epicotyl length in two environments using 3VmrMLM. Front. Plant Sci. 2022, 13, 1033120. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Chong, K. The novel functions of kinesin motor proteins in plants. Protoplasma 2012, 249, 95–100. [Google Scholar] [CrossRef]
- Yang, L.; Wang, J.; Han, Z.; Lei, L.; Liu, H.L.; Zheng, H.; Xin, W.; Zou, D. Combining QTL-seq and linkage mapping to fine map a candidate gene in qCTS6 for cold tolerance at the seedling stage in rice. BMC Plant. Biol. 2021, 21, 278. [Google Scholar] [CrossRef]
- Lei, L.; Zheng, H.; Bi, Y.; Yang, L.; Liu, H.; Wang, J.; Sun, J.; Zhao, H.; Li, X.; Li, J.; et al. Identification of a Major QTL and Candidate Gene Analysis of Salt Tolerance at the Bud Burst Stage in Rice (Oryza sativa L.) Using QTL-Seq and RNA-Seq. Rice 2020, 13, 55. [Google Scholar] [CrossRef]
- Das, S.; Upadhyaya, H.D.; Bajaj, D.; Kujur, A.; Badoni, S.; Laxmi Kumar, V.; Tripathi, S.; Gowda, C.L.L.; Sharma, S.; Singh, S.; et al. Deploying QTL-seq for rapid delineation of a potential candidate gene underlying major trait-associated QTL in chickpea. DNA Res. 2014, 22, 193–203. [Google Scholar] [CrossRef]
- Das, S.; Singh, M.; Srivastava, R.; Bajaj, D.; Saxena, M.S.; Rana, J.C.; Bansal, K.C.; Tyagi, A.K.; Parida, S.K. MQTL-seq delineates functionally relevant candidate gene harbouring a major QTL regulating pod number in chickpea. DNA Res. 2015, 23, 53–65. [Google Scholar] [CrossRef]
- Srivastava, R.; Upadhyaya, H.D.; Kumar, R.; Daware, A.; Basu, U.; Shimray, P.W.; Tripathi, S.; Bharadwaj, C.; Tyagi, A.K.; Parida, S.K. A multiple QTL-Seq strategy delineates potential genomic loci governing flowering time in chickpea. Front. Plant Sci. 2017, 8, 1105. [Google Scholar] [CrossRef]
- Singh, V.; Sinha, P.; Obala, J.; Khan, A.W.; Chitikineni, A.; Saxena, R.K.; Varshney, R.K. QTL-seq for the identification of candidate genes for days to flowering and leaf shape in pigeonpea. Heredity 2022, 128, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Clevenger, J.; Chu, Y.; Chavarro, C.; Botton, S.; Culbreath, A.; Isleib, T.G.; Holbrook, C.C.; Ozias-Akins, P. Mapping late leaf spot resistance in peanut (Arachis hypogaea) using QTL-seq reveals markers for marker-assisted selection. Front. Plant Sci. 2018, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sandhu, N.; Dixit, S.; Yadav, S.; Swamy, B.P.M.; Shamsudin, N.A.A. Marker-assisted selection strategy to pyramid two or more QTLs for quantitative trait-grain yield under drought. Rice 2018, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Assefa, T.; Otyama, P.I.; Brown, A.V.; Kalberer, S.R.; Kulkarni, R.S.; Cannon, S.B. Genome-wide associations and epistatic interactions for internode number, plant height, seed weight and seed yield in soybean. BMC Genom. 2019, 20, 527. [Google Scholar] [CrossRef]
- Cao, Y.; Li, S.; Chen, G.; Wang, Y.; Bhat, J.A.; Karikari, B.; Kong, J.; Gai, J.; Zhao, T. Deciphering the genetic architecture of plant height in soybean using two RIL populations sharing a common M8206 parent. Plants 2019, 8, 373. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Physical Interval | Δ(SNP-index) 1 Range |
|---|---|---|
| (Mb) | ||
| 2 | 25.30–25.60 | 0.64–0.67 |
| 2 | 25.75–26.15 | 0.64–0.66 |
| 2 | 27.10–27.25 | 0.64–0.65 |
| 2 | 27.55–27.85 | 0.64–0.65 |
| 2 | 28.10–29.05 | 0.64–0.68 |
| 2 | 31.65–32.75 | 0.63–0.70 |
| 7 | 6.50–10.65 | 0.65–0.83 |
| 10 | 19.75–21.75 | 0.66–0.76 |
| 10 | 22.75–24.35 | 0.66–0.73 |
| 10 | 26.55–28.85 | 0.66–0.80 |
| 11 | 6.30–7.45 | 0.65–0.75 |
| 11 | 7.80–11.10 | 0.65–0.73 |
| Chromosome | LOD Peak Position | Nearest Marker | Marker Interval | LOD 1 Value | AE 2 | DE 3 | PVE 4 |
|---|---|---|---|---|---|---|---|
| (cM) | (cm) | (cm) | (%) | ||||
| 7 | 51.1 | TC64 | TC63–TC66 | 6.7 | −0.8 | −0.1 | 21.2 |
| 10 | 88.4 | TC89 | TC87–TC89 | 4.4 | −0.6 | −0.5 | 13.4 |
| Line | Genotype of Marker in Recombinants | Epicotyl Length (cm) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TC64_9,479,153 | TC65_9,984,711 | TC98_10,131,117 | TC99_10,211,134 | TC100_10,305,602 | TC101_10,308,093 | TC102_10,628,880 | TC103_11,772,495 | TC66_13,081,534 | Mean | SD | Estimated Genotype of qECL7.1 | |||
| Chihayahime | A | A | A | A | A | A | A | A | A | 3.2 | ± | 0.5 | CH | |
| Toiku161 | B | B | B | B | B | B | B | B | B | 6.9 | ± | 0.7 | * | T161 |
| #223_105_7 | A | A | A | A | A | A | A | A | A | 3.4 | ± | 0.5 | --- | CH (Control) |
| #223_105_1 | B | B | B | B | B | B | B | B | B | 6.9 | ± | 0.7 | * | T161 (control) |
| #223_81_75 | B | B | B | B | B | B | B | A | A | 7.0 | ± | 0.7 | * | T161 |
| #223_112_347 | B | B | B | B | B | B | A | A | A | 7.3 | ± | 0.8 | * | T161 |
| #223_160_46 | B | B | B | B | A | A | A | A | A | 3.9 | ± | 0.6 | CH | |
| #223_95_178_193 | B | B | B | A | A | A | A | A | A | 4.0 | ± | 0.4 | CH | |
| #223_91_134_15 | A | A | A | A | A | A | A | B | B | 4.0 | ± | 0.5 | CH | |
| Gene | Position (bp) | Length (bp) | Description |
|---|---|---|---|
| Vigan.07G111100.01 | Chr07:10217067–10225123 (+ strand) | 8056 | Biological process: cell redox homeostasis (GO:0045454) |
| Vigan.07G111200.01 | Chr07:10225971–10227563 (− strand) | 1592 | Similar to early nodulin-like protein 2-like. [XP_006592879.1, Glycine max] |
| Vigan.07G111300.01 | Chr07:10245110–10253581 (+ strand) | 8471 | Biological process: regulation of Rab GTPase activity (GO:0032313) |
| Vigan.07G111400.01 | Chr07:10259315–10262198 (+ strand) | 2883 | cellulose microfibril organization |
| Vigan.07G111500.01 | Chr07:10267247–10267599 (+ strand) | 352 | Similar to integrase core domain containing protein. [I6ZTT9, Phaseolus vulgaris] |
| Vigan.07G111600.01 | Chr07:10270113–10274630 (− strand) | 4517 | Mitochondrial outer membrane translocase complex, subunit Tom22, plant (IPR017411) |
| Vigan.07G111700.01 | Chr07:10274131–10274487 (+ strand) | 356 | Hypothetical protein. |
| Vigan.07G111800.01 | Chr07:10278476–10286164 (− strand) | 7688 | DDRGK domain containing protein (IPR019153) |
| Vigan.07G111900.01 | Chr07:10287789–10289985 (− strand) | 2196 | Hypothetical protein. |
| Vigan.07G112000.01 | Chr07:10291534–10291999 (− strand) | 465 | Hypothetical protein. |
| Vigan.07G112100.01 | Chr07:10292828–10293475 (− strand) | 647 | Hypothetical protein. |
| Vigan.07G112300.01 | Chr07:10297927–10298963 (+ strand) | 1036 | Hypothetical protein. |
| Vigan.07G112400.01 | Chr07:10311827–10312393 (+ strand) | 566 | Hypothetical protein. |
| Vigan.07G112500.01 | Chr07:10349998–10352995 (+ strand) | 2997 | Biological process: oxidation-reduction process (GO:0055114) |
| Vigan.07G112600.01 | Chr07:10363325–10365026 (− strand) | 1701 | Hypothetical protein. |
| Vigan.07G112700.01 | Chr07:10444164–10448247 (+ strand) | 4083 | Biological process: oxidation-reduction process (GO:0055114) |
| Vigan.07G112800.01 | Chr07:10463548–10464907 (+ strand) | 1359 | Similar to uncharacterized protein. [D8WJ57, Glycine max] |
| Vigan.07G112900.01 | Chr07:10472813–10481791 (+ strand) | 8978 | Similar to uncharacterized protein. [K7LW88, Glycine max] |
| Vigan.07G113000.01 | Chr07:10482576–10483609 (− strand) | 1033 | Similar to Potassium/sodium hyperpolarization. [XP_007021055.1, Theobroma cacao] |
| Vigan.07G113100.01 | Chr07:10495792–10500041 (+ strand) | 4249 | WD40/YVTN repeat-like-containing domain (IPR015943) |
| Vigan.07G113200.01 | Chr07:10501026–509455 (− strand) | 8426 | UHRF1-binding protein 1-like (IPR026728) |
| Vigan.07G113300.01 | Chr07:10505128–10506155 (+ strand) | 1027 | Non-protein coding gene. |
| Vigan.07G113400.01 | Chr07:10537568–10544061 (+ strand) | 6493 | WD40/YVTN repeat-like-containing domain (IPR015943) |
| Vigan.07G113500.01 | Chr07:10558674–10564005 (+ strand) | 5331 | Glycosyl transferase, family 8 (IPR002495) |
| Vigan.07G113600.01 | Chr07:10562715–10563808 (− strand) | 1093 | Domain of unknown function DUF1995 (IPR018962) |
| Vigan.07G113700.01 | Chr07:10564235–10566639 (− strand) | 2404 | Myc-type, basic helix-loop-helix (bHLH) domain (IPR011598) |
| Vigan.07G113800.01 | Chr07:10578898–10580621 (− strand) | 1723 | Biological process: cell redox homeostasis (GO:0045454) |
| Vigan.07G113900.01 | Chr07:10584354–10585861 (+ strand) | 1507 | Cellular component: photosystem II oxygen evolving complex (GO:0009654) |
| Vigan.07G114000.01 | Chr07:10584354–10585924 (− strand) | 1570 | Non-protein coding gene. |
| Vigan.07G114100.01 | Chr07:10586027–0592438 (− strand) | 6411 | Molecular function: microtubule motor activity (GO:0003777) |
| Vigan.07G114200.01 | Chr07:10597826–10599550 (− strand) | 1724 | HSP20-like chaperone (IPR008978) |
| Vigan.07G114300.01 | Chr07:10599645–0600897 (− strand) | 1252 | Non-protein coding gene. |
| Vigan.07G114400.01 | Chr07:10602299–10606380 (+ strand) | 4081 | Cellular component: actin filament (GO:0005884) |
| Vigan.07G114500.01 | Chr07:10617379–10618239 (− strand) | 860 | Molecular function: DNA binding (GO:0003677) |
| Vigan.07G114600.01 | Chr07:10625237–10626614 (− strand) | 1377 | Molecular function: DNA binding (GO:0003677) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kachapila, M.; Horiuchi, Y.; Nagasawa, H.; Michihata, N.; Yoshida, T.; Kato, Y.; Bethke, P.C.; Kato, K.; Mori, M. Fine-Mapping of qECL7.1, a Quantitative Trait Locus Contributing to Epicotyl Length in Adzuki Bean (Vigna angularis). Agriculture 2023, 13, 1305. https://doi.org/10.3390/agriculture13071305
Kachapila M, Horiuchi Y, Nagasawa H, Michihata N, Yoshida T, Kato Y, Bethke PC, Kato K, Mori M. Fine-Mapping of qECL7.1, a Quantitative Trait Locus Contributing to Epicotyl Length in Adzuki Bean (Vigna angularis). Agriculture. 2023; 13(7):1305. https://doi.org/10.3390/agriculture13071305
Chicago/Turabian StyleKachapila, Modester, Yuki Horiuchi, Hidetaka Nagasawa, Noe Michihata, Toru Yoshida, Yuta Kato, Paul C. Bethke, Kiyoaki Kato, and Masahiko Mori. 2023. "Fine-Mapping of qECL7.1, a Quantitative Trait Locus Contributing to Epicotyl Length in Adzuki Bean (Vigna angularis)" Agriculture 13, no. 7: 1305. https://doi.org/10.3390/agriculture13071305
APA StyleKachapila, M., Horiuchi, Y., Nagasawa, H., Michihata, N., Yoshida, T., Kato, Y., Bethke, P. C., Kato, K., & Mori, M. (2023). Fine-Mapping of qECL7.1, a Quantitative Trait Locus Contributing to Epicotyl Length in Adzuki Bean (Vigna angularis). Agriculture, 13(7), 1305. https://doi.org/10.3390/agriculture13071305