Abstract
Cowpea is an important crop for small-scale farmers in poor areas but is also being developed for commercial agriculture as a possible substitute for commercial legumes. Endophytic fungi are omnipresent and play crucial but diverse roles in plants. This study characterized the endophyte component of the cowpea mycobiome from leaves, main and crown stems and roots using Illumina MiSeq of the ITS2 region of the ribosomal operon. Ascomycetes exhibited the highest diversity, with Molecular Operational Taxonomic Units (MOTUs) assigned as Macrophomina, Cladosporium, Phoma, Fusarium and Cryptococcus, among the most dominant genera. Certain MOTUS showed preferential colonization patterns for above or below ground tissues. Several MOTU generic groups known to include phytopathogenic species were found, with relative abundances ranging from high to very low. Phylogenetic analyses of reads for some MOTUs showed that a level of identification could be obtained to species level. It also confirmed the absences of other species, including phytopathogens. This is the first study that adopted a holistic metagenomic typing approach to study the fungal endophytes of cowpea from a single location, a crop that is so integral for low-income households of the world.
1. Introduction
Cowpea (Vigna unguiculata) is one of the most economically important indigenous African legume crops [1] and is cultivated in more than 60 countries of Asia, Oceania, the Middle East, southern Europe, Africa, southern United States of America and Central and South America [2]. Cowpeas are grown mostly for their edible beans, but the leaves, green seeds, dry seeds and pods can also be consumed. Cowpeas are usually cooked to make them edible, usually by boiling [3,4,5]. They are widely used as an inexpensive protein source in most rural and semi-urban areas and as animal feed and a cash crop [6,7]. Africa is a major area of production where the crop is crucial for low input agriculture that is the basic characteristic of most parts of the continent [8]. In South Africa, small-scale farmers form a large producer group of cowpea under dryland farming conditions [9], although there are no records with regard to the size of the areas under production and the quantities produced. Cowpea crop fixes 80% nitrogen for its growth demand from the atmosphere [10], thereby reducing nitrogen fertilizer demand and costs of its production. It is also an important companion crop in most cereal-legume cropping systems because of the benefit from the residual nitrogen originating from the decay of its leaf litter, roots and root nodules [11].
Cowpea yield can be low due to diseases such as damping-off [12] and stem rot which have been reported in many countries [13]. Damping-off and stem rot diseases are caused by many different species of fungi, including Pythium aphanidermatum, Rhizoctonia solani, Phytophthora sp., Fusarium solani and Sclerotium rolfsii [14,15]. Another important disease is Fusarium wilt caused by F. oxysporum f. sp. tracheiphilum [16,17,18]. In Brazil, this disease is responsible for heavy losses in cowpea production, while in Nigeria and the United States of America, plant mortality can reach levels above 50% [18]. In South Africa F. equiseti, F. graminearum, F. chlamydosporum, F. sambucinum and F. subglutinans [19] have been isolated from cowpea. These Fusarium species were mycotoxin producers. Other diseases of cowpea in South Africa are Colletotrichum stem disease caused by Colletotrichum dematium [20]. P. ultimum [15] and R. solani [12] also have been consistently isolated from cowpea seedlings with symptoms of damping-off. Cercospora leaf spot is caused by Mycosphaerella cruenta [21] and Alternaria cassiae also affects leaves [22].
Cowpea seed have been reported to be susceptible to Aspergillus infection and to aflatoxin production [23]. Fusarium toxins such as fumonisin B1, B2 and B3 have a major effect on the health of farm animals and humans [24]. Indeed, the same toxins produced by F. proliferatum, F. nygamaia and F. verticillioides isolates in South Africa have a major toxicological significance in animal and human health [19]. Fumonisin B1 causes equine leukoencephalomalacia in horses and pulmonary edema in pigs. High incidence of human esophageal cancer in the Transkei region, Southern Africa has been associated with incidence of F. verticillioides infection on home grown maize [19].
Culture-based and environmental sequencing studies of surface sterilized stems and leaves have revealed an astounding diversity of fungal species existing sub-cuticularly or deeper within the tissues of healthy plants [25,26]. Such fungi are designated as endophytes, a term long used to indicate fungal residence within plant tissues rather than on plant surface asymptomatically without causing visible disease symptoms [27]. They can be obligatory or facultative bio-trophic and have a continuum of ecological functions from mutualists to saprophytes to latent pathogens [28]. In fact, a number of known pathogens have cryptic endophytic life stages, thus evading detection and complicating disease management programmes [28]. Others have been shown to improve plant health [29,30].
Phytobiome research of agricultural plants aims to maximize sustainable food production by generating, optimizing and translating knowledge of all factors influencing the plant into practice [31,32,33,34,35,36]. The last two decades of research have increasingly highlighted mechanisms of microbial facilitation of plant nutrient use. For example, nutrients that are not readily mineralized in the absence of the plant can be available to plant-microbe interactions, including widespread priming effects on C and N mineralization and solubilization of phosphate in the rhizosphere [32,33,34]. The first step in phytobiome research is often to characterize the microbes associated with the particular crop [35,36].
With the appearance of next generation sequencing (NGS), metagenomic surveillance approaches have revealed greater microbial diversity than culture-based methods and promoted phytobiome research [31,37]. Furthermore, these environmental sequencing approaches can detect microorganisms that could be missed in culture-dependent approaches and rare taxa [38]. When focusing on endophytes in cowpea plants, their identification and quantification can provide a foundation towards understanding the interactions between cowpea plants and endophytes, including pathogenic and beneficial species. Furthermore, the ability to detect pathogens and their relative abundance will aid studies in the epidemiology of pathogens and could benefit disease management and monitoring. Characterizing the endophytic communities in the entire plant, including above and below ground tissues, will lead to a more holistic approach to improve plant health.
Cowpea is one of the most important crops for livelihood in Africa, including South Africa. The present study aims to characterize the composition of the fungal endophyte communities associated with different above and below ground cowpea tissues with a NGS approach, in this case using an Illumina MiSeq platform. We specifically focused on three main questions: (i) What is the composition of the fungal endophytic communities found in cowpea from a single location in South Africa at a single time? (ii) What is the fungal endophyte composition in the different plant tissues of cowpea at a single time? (iii) Do the plant parts harbour any latent pathogens or potentially beneficial fungi? The study represents the first high throughput sequencing based phytobiome characterization of cowpea not only in Africa, but also in the world, and will generate the first set of baseline knowledge of the fungal communities of cowpea plants in the field from this particular location. It also served to establish the technique and to illustrate its usefulness for the industry and growers in South Africa.
2. Materials and Methods
2.1. Field Sampling
Asymptomatic cowpea plants (6-weeks-old) were collected from a trial plot at the Small Grain Institute (Agricultural Research Council) in Potchefstroom, South Africa, in February 2016 (late summer). The plants were planted in three replicated blocks following standard cultivation processes, with the blocks randomly distributed between fallow plots and plots containing other crops, namely sorghum, soybean, dry bean and Bambara groundnut. The adjacent fields to the trial plot were sowed with sunflower and maize plants. From each block, five plants were randomly chosen and transported in a cool box to the laboratory of the Department of Genetics, University of the Free State, for further processing. The plants were not treated with fungicides at the time of sampling. All necessary permissions were obtained to sample and transport the plants in accordance with national and institutional regulations.
The roots, main stem, crown stem and leaves were the focus of this study. These plant parts were separated from the plants. Ten leaves from each plant were collected, cut into 1-cm-diam squares and 10 squares per leaf were randomly picked. The stems and roots per plant were cut into 1-cm-long pieces, and 10 random pieces were then selected. The plant material was placed in separate falcon tubes and surface sterilized in 3% sodium hypochlorite for 3 min, followed by rinsing in sterile distilled H2O for 1 min, immersion in 70% ethanol for 2 min and a final rinse with sterile distilled water for 1 min. The plant material was freeze dried and pulverized in a Qiagen Tissue Lyser II cell disrupter (Whitehead Scientific, Cape Town, South Africa) for the environmental DNA extraction.
2.2. Illumina Sequencing of the Metagenomic DNA
The ribosomal RNA Internal Transcribed Spacer 2 (ITS2) region has been targeted because its minimum length invariability lacks the problem of co-amplification with the SSU intron, and it is better represented in the environmental sequence databases in comparison to ITS1 gene sequences [39]. Metagenomic DNA from 0.1 g of each pulverized plant sample was extracted using the Nucleospin® Plant II mini Kit (Macherey Nagel, Düren, Germany). DNA concentrations were determined using a Nanodrop LITE spectrophotometer (Thermo Scientific, Waltham, MA, USA) and diluted to a standard 10 ng/µL for Polymerase Chain Reactions (PCR). The Internal Transcribed Spacer 2 region of the ribosomal operon was amplified using the primers ITS3F (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGCATCGATGAAGAACGCAGC3′) and ITS4R (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTCCTCCGCTTATTGATATG-3′), with Illumina MiSeq (Sigma Aldrich, St. Louis, MO, USA) specific adaptor sequences in bold [40]. Reactions (25 µL) consisted of 20 ng of template DNA, 0.6 µM of each of the primers and 2U of HiFi Ready Mix DNA Polymerase (KAPA Biosystems, Lasec, SA, Cape Town, South Africa). The PCRs were performed in a G-Storm GS04822 thermal cycler (Somerton Biotechnology Centre, Bristol, UK) with parameters set to an initial denaturation at 95 °C for 3 min, followed by 25 cycles of denaturation at 98 °C for 20 s, annealing at 65 °C for 30 s and extension at 72 °C for 30 s. A final extension was performed at 72 °C for 5 min. The PCR products were visualized on 2% agarose gel electrophoresis supplemented with GelRed (Biotium, Inc., Separations, Johannesberg, South Africa) fluorescent nucleic acid dye.
The PCR amplicons were sent for sequencing library construction and subsequent sequencing at the Next Generation Sequencing Facility, Department of Health Sciences, University of the Free State, South Africa. The quality and quantity assessment of the PCR products were performed using a 2100 Bioanalyzer using a DNA 12,000 Chip (Agilent Technologies, Santa Clara, CA, USA). The amplified PCR products were gel purified using the Agencourt AMPure XP Bead Clean-up kit (Beckman Coulter, Atlanta, Georgia, United States), normalized, pooled and denatured before being submitted to the MiSeq platform with paired 300 bp reads and MiSeq v3 reagents (Illumina Inc., San Diego, CA, USA).
2.3. Cluster Analysis
The obtained DNA sequence data were analysed using QIIME 1 as has been applied in a previous publication [41]. Briefly, before running the QIIME 1 pipeline, the quality of the sequencing was assessed and quality control performed using PrinSeq-lite v0.20.4 [42]. All datasets were pre-processed and trimmed to obtain an average quality score of ≥20 using a 5 nt window with a 3 nt step. All sequences shorter than 200 bp were filtered out and paired end reads merged using PEAR 0.9.6 [43]. The demultiplex and quality filtering script in QIIME 2 was run without any additional inputs to obtain a FASTA output file that could be analysed in the QIIME 2 pipeline. Chimeric sequences were identified, using usearch 6.1.544 as the chimera detection method [44], and filtered out of the quality trimmed reads by using identify_chimeric_seqs.py and filter_fasta.py commands, respectively, in QIIME 1. Fungal Principal Coordinate analysis (PCoA) was performed based on unweighted UniFrac distances.
Molecular Operational Taxonomic Unit (MOTU) was defined and tentative taxonomic names were assigned to representative MOTUs using the pick_open_reference_otus.py script. This was done at 99% sequence identity against the UNITE ITS reference database (alpha version 12_11) [45]. Fungal alpha diversities were analysed in QIIME 1 with alpha_rarefaction.py using the Shannon diversity metric. Beta-diversities were analysed using Principal Coordinates Analysis (PCoA) plots in R (www.r-project.org, accessed on 25 July 2021) using “plot_ordination” in the “Phyloseq” package using Bray–Curtis distance [46]. Prior to beta diversity analysis, the OTU-table was normalized using normalize_table.py in QIIME with the CSS normalization option [47]. The results for the PCoA and relative abundances were compared using 99% identity level. Sequence data were submitted to Genbank as BioProject ID PRJNA738463.
2.4. Phylogenetic Analysis
Phylogenetic analyses were carried out on sequence reads in order to refine the identities of selected representatives of genera that could be of possible importance as phytopathogens or possible biocontrol agents. Sequences obtained were queried using nucleotide BLAST searches against the National Center for Biotechnology Information (NCBI) database (Genbank). At least five representative sequences with a 99 to 100% level of similar identity and a high maximum coverage (higher than 95%) to respective query sequences were downloaded and aligned with the query sequence using the MAFFT server [48] and default parameters. In other cases, the query sequences were included in more comprehensive datasets for a genus, such as that of Fusarium [41]. The alignments were edited manually where needed. Maximum Likelihood (ML) phylogenetic analyses were performed with MEGA 7.2.2 [49] with a 1000 bootstrap replication performed to determine the support of branches [50]. Specific evolutionary models for the ML analyses were determined prior for each dataset using MEGA. Phylogenies were not produced for certain important genera, such as Alternaria, Phoma and Cladosporium, since it was previously shown that the ITS2 region, which is often used for Illumina deep sequencing, is not even adequate to distinguish the genera in these groups [41].
3. Results
3.1. Illumina Sequencing of the Metagenomic DNA
After quality checking and data filtering, a minimum of 11,417 to a maximum of 21,680 sequences were retained for the different plant parts (Table 1). Read lengths ranged from 200 to 251 bp. The sequences represented a minimum of 51 MOTUs to a maximum of 135 MOTUs for the different plant parts (Table 1), while there were 175 unique MOTUs. Richness and diversity were significantly greater in the roots while they were lowest in crown stem (Figure 1). Rarefaction curves showed that the sampling depth and sequencing coverage were adequate for all four plant parts (Supplementary Figure S1).

Table 1.
Statistical summary of reads and Molecular Operational Taxonomic Units (MOTs) for the four cowpea plant tissues investigated.
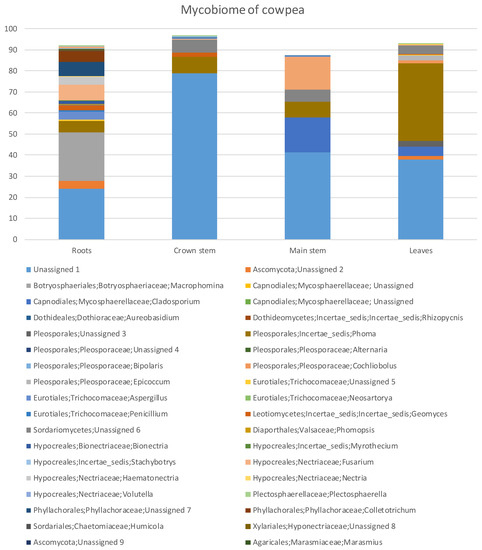
Figure 1.
Percentage fungal relative abundance of Molecular Operational Taxonomic Units (MOTUs) up to genus level from roots, crown and main stem and leaves of cowpea at 99% level of sequence similarity.
3.2. Cluster Analysis
The percentage relative abundances (Supplementary Table S1) showed that the Ascomycota was highest in the roots with 58.74% and least in the crown stem with 13.45%. The Basidiomycota had the highest diversity in the main stem with 15.65% and the least diversity in the leaves where this phylum was absent (Supplementary Table S1). Overall, fungal diversity was highest in the roots followed by the leaves, main stem and the least was the crown stem (Figure 1). Some of the MOTUs were unidentified or unassigned.
In the Ascomycota (Supplementary Table S1), the Dothideomycetes were by far the most abundant with 45.14% in the leaves, followed with 29.14% in the roots, 23.92% in the main stem and 7.79% in the crown stem. The Dothideomycetes was represented by five genera across three orders and four families. The Eurotiomycetes was highest in the roots (4.32%) and least in the leaves (0.21%). The Eurotiomycetes was represented with three genera across one order and family. The Leotiomycetes was highest in the roots with 2.55% and least in the leaves with 0.21%, represented by one genus of uncertain position (Geomyces). The Sordariomycetes (22.73%) resided mostly in the roots and was made up of 11 genera, six orders and nine families.
MOTUs in the Basidiomycota (Supplementary Table S1) resided in the Agaricomycetes with two genera, two orders and two families. Tremellomycetes relative abundance was highest in the main stem (15.42%) and lowest in roots (0.92%) and was represented by one genus, order and family, namely Cryptococcus. The Ustilaginomycetes occurred in main stems (0.23%) and in roots (0.21%). They were represented by two genera from two orders and families. The Mucoromycota was highest in the roots with 0.35% and least in the leaves with 0.10% consisting of one genus, order and species, namely Rhizopus.
In total there were 43 genera (Supplementary Table S1). Roots had the highest number of MOTUs (22 MOTUs) and relative abundances (67.64%), followed by leaves (12, 51.06%). The most abundant genera (Figure 1, Supplementary Table S1) in the roots were MOTUs assigned as Macrophomina (22.86%), Fusarium (7.38%) and Phoma (5.68%) while in the crown stem, the most abundant MOTUs were assigned as Phoma (7.79%) and Geomyces (2.04%). In the main stem, the most abundant MOTUs were Cladosporium (16.45%), Cryptococcus (15.42%) and Phoma (7.47%). Phoma (37.02%), Cladosporium (4.43%) and Epicoccum (2.32%) were prominent in the leaves. A large proportion of MOTUs could not be satisfactorily assigned at order level or higher (Figure 1).
For the plant tissues (roots, main stem, crown stem, leaves) (Supplementary Table S1), eight genera assigned by the pipeline were dominant (having relative abundances higher than 2%). However, these genera showed varying patterns of colonization, especially with regards to presence below and above ground. Macrophomina (22.86%) was only found in the roots. Aspergillus was more present in the roots (3.83%) and less so in leaves (0.21%), similarly to Colletotrichum (4.90%) but with the difference that the latter co-occurred in the crown. Cladosporium was only found in above soil parts in this study, being dominant in the main stem at 16.45% but also present in leaves at 4.43%. A MOTU assigned as Epicoccum only occurred in leaves (2.32%).
MOTUs found in all of the tissues included Phoma, which were highly dominant in the cowpea plants, with relative abundances higher than 5% in all tissues but being most prominent in leaves (37.02%). Geomyces occurred below ground in roots (2.55%) and the crown (2.04%) but to a lesser degree in the main stem (0.23%) and leaves (0.21%). Similarly, Fusarium was more prominent in the roots (7.38% and 3.62% as Haematonectria) but present in the other plant parts to a lesser degree (0.37% in the crown, 0.11% in stems, 0.10% in leaves). Interestingly, the remaining MOTU assigned as Cryptococcus with relative abundances higher than 2% were dominant in the main stem (15.42%) and then only again found in the roots (0.92%).
MOTUs that represented possible phytopathogen groups were detected in the plant parts (Figure 1, Supplementary Table S1). MOTUs assigned as Macrophomina, Cladosporium, Phoma, Epicoccum and Fusarium that occurred relatively dominantly represented genera known to include plant pathogens [51,52]. Other potential pathogenic MOTUs occurring in low relative abundances were Cochliobolus, Thecaphora and Ustilago. Some of these MOTUS are also known to include taxa known to cause diseases or mycotoxin issues of cowpea, such as Rhizopus [12], Colletotrichum [53] and Fusarium [19].
The PCoA analysis based on pairwise unweighted UniFrac distances (Figure 2) showed two principal coordinates explained at 86.4% of the variations (68.1% for PC1 and 18.3% for PC2, respectively). The analysis showed that the four plant parts orientated into two distinct groups. The first group was comprised of the above ground leaf and main stem samples, as well as the crown area just below soil level. Although these three groups did not cluster closely together, they were all positioned to the left of the plot. The second group was positioned to the right and represented the root samples.
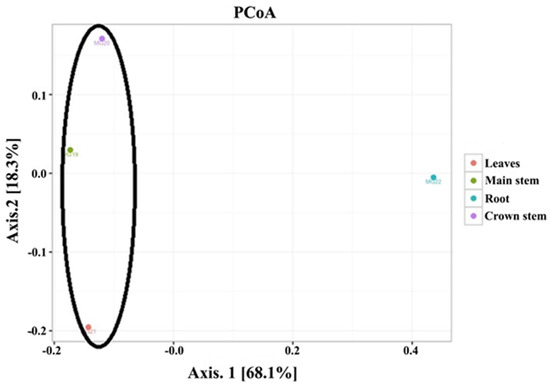
Figure 2.
Principal coordinate analysis for the four plant tissues (roots, main stem, crown stem, leaves) of cowpea.
3.3. Phylogenetic Analysis
Phylogenetic analyses were quite informative in providing a more robust indication of the possible species or species complexes that selected MOTUs could represent. Eight representatives of the MOTUs assigned as Fusarium grouped in five species complexes in the dataset of Fusarium and other closely related genera (Figure 3). These included a MOTU from roots (MG22 5414) in the F. chlamydosporum species complex (FCSC), another (MG22 137) from roots in the F. solani species complex (FSSC), two MOTUs from roots (MG22 17908) and stems (MG19 2895), respectively, in the F. oxysporum species complex (FOSC) and another set of two from roots (MG22 2563) and stems (MG19 2852), respectively, in the F. incarnatum-equiseti species complex (FIESC). A MOTU from the crown (MG20 20923) grouped in Bisifusarium, while another from roots (MG22 20923) grouped separately from sequences of any Fusarium or closely related genus (Figure 3).
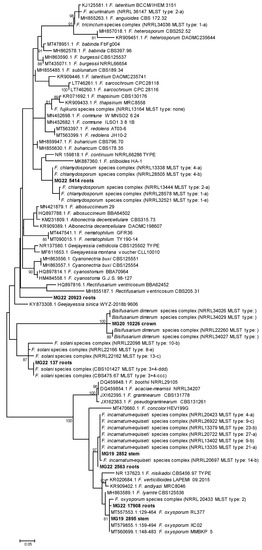
Figure 3.
Phylogram of Molecular Operational Taxonomic Units assigned as Fusarium. The phylogram was generated with Maximum Likelihood analyses of the Internal Transcribed Spacer 2 region (used evolutionary model indicated). Confidence levels ≥ 80% (1000 replicate bootstrap analysis) are indicated on the branches. Sequence reads generated in this study are indicated with MG codes.
A MOTU from the main stem that was assigned in the Ustilaginomycetes (MG19 3597) grouped with U. trichophora with a 100% Bootstrap support (Figure 4A). A MOTU from the crown (MG20 5929) grouped with sequences of R. solani, with the previously used teleomorph name of Thanetophorus cucumeris still used for some of the deposited sequences (Figure 4B). Comparisons of a crown MOTU, namely MG20 2659, showed it to possibly represent Plectosphaerella cucumerina (Figure 5), while MG22 6502 from roots grouped in Clonostachys (synonym Bionectria) but its position could not be resolved. A root MOTU (MG22 17361) grouped together with Colletotrichum coccodes and C. nigrum, separate from the known cowpea pathogens C. demiatum and C. truncatum (Figure 5).
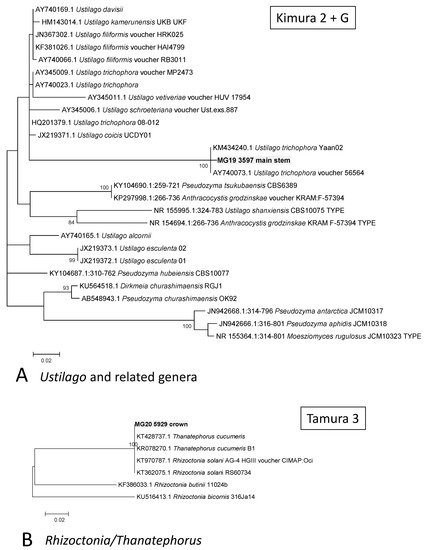
Figure 4.
Phylograms of Molecular Operational Taxonomic Units. (A) Ustilago. (B) Rhizoctonia. The phylograms were generated with Maximum Likelihood analyses of the Internal Transcribed Spacer 2 region (used evolutionary model indicated). Confidence levels ≥ 80% (1000 replicate bootstrap analysis) are indicated on the branches. Sequence reads generated in this study are indicated with MG codes.

Figure 5.
Phylograms of Molecular Operational Taxonomic Units. A Ustilago (A). B Rhizoctonia. The phylograms were generated with Maximum Likelihood analyses of the Internal Transcribed Spacer 2 region (used evolutionary model indicated). Confidence levels ≥ 80% (1000 replicate bootstrap analysis) are indicated on the branches. Sequence reads generated in this study are indicated with MG codes.
4. Discussion
Research and production of cowpea have largely been neglected in South Africa [9]. Although not as widely planted and commercialized as dry bean (Phaseolus vulgaris) or soybean (Glycine max), this crop has the potential to become an important substitute for other legumes or crops to improve food security, ensure production resilience and maintain the health of consumers. A number of prominent to rare MOTUs have been detected that are associated with cowpea at the trial site. Some of these showed possible colonization trends in specific plant tissues, while genera known to include phytopathogens were detected. This study represents the first of its kind on cowpea and is an important first step for such future studies towards crop improvement.
Results detected 43 genera from one location. Genera such as Fusarium, Cladosporium, Macrophomina, Cryptococcus and MOTUs assigned as Phoma in the Didymellaceae were among the dominant groups in this study. All of these prominent groups contain plant pathogens, except for Cryptococcus where some species have been shown to exhibit potential biocontrol [54,55,56,57,58]. Clonostachys represented another genus known to include species used for biocontrol [59,60,61,62]. Other potential pathogen genera included Plectosphaerella, Colletotrichum, Cochliobolus, Thecaphora, Ustilago and Rhizoctonia.
Previous cultivation-based studies revealed that the fungal community associated with cowpea was dominated by three genera, namely Fusarium, Cladosporium and Phoma [12]. Other fungi, such as S. rolfsii, P. ultimum, R. solani, a Rhizopus sp. and Trichoderma harzianum, were present more rarely. The prevalence of Fusarium, Cladosporium and members of the Didymellaceae such as Phoma was confirmed in this study. Cladosporium, Phoma and Fusarium were also shared among the four plant parts. NGS based studies on other crops such as rice, sugarcane, wheat and Arabidopsis thaliana found more or less the same groups to occur as those found in this study [63,64,65,66,67].
In this study the fungal community from below ground roots grouped quite separately from those of the other tissues above ground including the crown area that is at the soil–air interface. Some MOTUs were only detected in the roots, such as Macrophomina and Colletotrichum. On the other hand, the Cladosporium and Cryptococcus only occurred above ground. Others were very dominant in roots and only present in above ground tissues at low relative abundances, such as Fusarium. Phoma had an interesting occurrence where it was dominantly present in all plant tissues (>5%) but had an exceedingly high relative abundance of 37% in the leaves.
A sense of colonization patterns of tissues and variation in prominence could be obtained with the Illumina sequencing. In this study results showed that only a small number of genera were prominently associated with cowpeas. Interestingly, the absence of others was also detected, such as the commonly occurring and cosmopolitan phytopathogen genus Alternaria. The approach could thus be useful to study community structure changes from a baseline when various agronomical effects are applied and management options against plant pathogens of cowpea are tested. However, more extensive environmental sequencing studies, as well as confirmation based on isolates, are needed to confirm observed patterns.
Some of the groups detected are known to include phytopathogen species of cowpea, as well as other plants. This is despite the fact that only healthy cowpea plants were used, indicating that these fungi could be potential pathogens [68,69]. Fusarium species have previously been associated with disease symptoms on cowpea, including South Africa [12]. Species in the F. oxysporum, F. chlamydosporum, F. incarnatum-equiseti and F. solani species complexes were possibly detected in this study, which include known pathogens and mycotoxin producers [70]. Similarly, members of the Phoma and Epicoccum groups in the Didymellaceae [71,72], and Cladosporium (Cladosporiaceae) [73] include known pathogens, including on legumes [74]. Other interesting pathogens detected include smuts possibly representing Thecaphora that include pathogens of potato [75], peanut [76], rhubarbs [77] and U. trichophora, which is a pathogen of rice, and Echinochloa crus-galli [78]. Plec. cucumerina is a pathogen of horticultural crops [79] and leafy vegetable crops [80]. Colletotrichum is possibly related to C. nigrum and C. coccodes, which are both phytopathogens of peppers and tomato [54]. The important cowpea pathogens C. dematium [81] and C. truncatum [82] were not detected.
The NGS approach detected interesting MOTUs from the healthy cowpea plants, some at low relative abundances. For example, U. trichophora was detected in cowpea stem, the first report in cowpea from South Africa. However, the biological significance of this finding is unclear and in vivo assays may be required to validate it. Even though other approaches would be needed to confirm the presence of this species, phylogenetic placing of these MOTUs derived from partial ITS data was used, which also represents the first report from South Africa.
Although phylogenies generated in this study from the ITS2 region generated by Illumina sequencing are limited because they only represent a portion of the ITS region and the ITS region does not always distinguish between species of certain genera [83], valuable information could in some cases be gained. It was shown that MOTUs assigned as Fusarium or initial names consisted of more than one species complex. The identities of others could be ascertained to some degree, for example, those of R. solani and Plec. cucumerina. The presence of known pathogens of cowpea could be ruled out, even if other members of the genus were present. For example, although the identity of the Colletotrichum could not be confirmed with certainty, it was clear that it did not represent the previously reported pathogens C. truncatum and C. demiatum. This approach has also been used in a previous study to determine species identities or absences more accurately [41].
Our description of the mycobiome associated with parts of cowpea provides an interesting baseline for cowpea grown in the Potchefstroom area that can be used for further monitoring to improve risk assessments and crop improvement for this crop. Future studies with adequate samples and results for robust statistical analyses can build further on this study. Sound knowledge on the pathogens that threaten this crop is still largely lacking, even though these pathogens threaten cowpea production, which forms such an integral part of the livelihoods of many. Knowing how to improve growth and yield or how this crop improves growth of other crops and assessing the potential threat of mycotoxins will aid numerous human communities, especially those that are poor and heavily depend on the products of this crop. It is essential to gain knowledge of the cowpea core mycobiome to proceed to studies on the functions of these fungi [35,36,66,84] and how these fungal communities may change in the plants due to various biotic and abiotic changes [85]. Such an approach can be used to develop targeted control strategies that are focused on managing the most prevalent phytopathogens in a given region. Future analyses with additional biogeographical datasets of cowpea mycoflora will help to identify whether or not the core mycobiome ascribed to cowpea in this study will be similar elsewhere and what the sources are. The occurrence of important mycotoxins can also be studied. Future work on expanded biogeographical regions will help to provide such answers and build a more complete baseline on the fungi associated with cowpea.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/agriculture12030333/s1, Figure S1: Rarefaction curves for the four cowpea plant tissues (roots, main stem, crown stem, leaves); Table S1: Relative abundances of MOTUs (differentiated at 99% similarity) from the different plant parts of cowpea, excluding unassigned or unknown taxa that could not be placed in a family. Values in bold are higher than 2%, while values of 0% were omitted. Values at phylum, class, order and family level represented totals of the relative abundances of the generic names.
Author Contributions
Conceptualization, M.G.; methodology, M.G. and E.D.C.; software, E.D.C. and M.G.; validation, M.G., T.R.K., S.G. and E.D.C.; formal analysis, M.G. and E.D.C.; investigation, T.R.K.; resources, M.G.; data curation, E.D.C.; writing—original draft preparation, T.R.K.; writing—review and editing, M.G. and S.G.; supervision, M.G.; project administration, M.G.; funding acquisition, M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Agricultural Research Council Collaborative Consortium To Broaden the Food Base.
Institutional Review Board Statement
Sampling and transport of plant specimens were done in accordance with national and institutional regulations. Since studies were not done on animals or humans, no additional institutional ethics approval was needed.
Data Availability Statement
Sequencing data and sample metadata are available on management platform under BioProject ID PRJNA738463.
Acknowledgments
Staff of the Small Grain Institute (Agricultural Research Council) in Potchefstroom, South Africa, are thanked for maintenance of the trial. N. McLaren (University of the Free State) conceptualized and planted the trial.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Langyintuo, A.S.; Lowenberg-DeBoer, J.; Faye, M.; Lambert, D.; Ibro, G.; Moussa, B.; Kergna, A.; Kushwaha, S.; Musa, S.; Ntoukam, G. Cowpea supply and demand in West and Central Africa. Field Crops Res. 2003, 82, 215–231. [Google Scholar] [CrossRef]
- Singh, B.B.; Hartmann, P.; Fatokun, C.; Tamo, M.; Tarawali, S.; Ortiz, R. Recent progress on cowpea improvement. Chron. Hortic. 2003, 43, 8–12. [Google Scholar]
- Ehlers, J.D.; Hall, A.E. Cowpea (Vigna unguiculata L Walp). Field Crops Res. 1997, 53, 187–204. [Google Scholar] [CrossRef]
- Hamid, S.; Muzaffar, S.; Wani, I.; Ahmed, M.; Farooq, A.B.; Mohd, M. Physical and cooking characteristics of two cowpea cultivars grown in temperate Indian climate. J. Saudi Soc. Agric. Sci. 2016, 15, 127–134. [Google Scholar] [CrossRef]
- Menendez, C.M.; Hall, A.E.; Gepts, P. A genetic linkage map of cowpea (Vigna unguiculata) developed from a cross between two inbred, domesticated lines. Theor. Appl. Genet. 1997, 95, 1210–1217. [Google Scholar] [CrossRef]
- Quin, F.M. Introduction. In Advances in Cowpea Research; Singh, B.B., Mohan Raj, D.R., Dashiell, K.E., Jackai, L.E.N., Eds.; Sayce Publishing: Devon, UK, 1997; pp. ix–xv. [Google Scholar]
- Davis, D.W.; Oelke, E.A.; Oplinger, E.S.; Doll, J.D.; Hanson, C.V.; Putnam, D.H. Cowpea research, production and utilization. In Field Crops Manual; Bressani, R., Ed.; John Wiley and Sons: Oxford, UK, 1991. [Google Scholar]
- Ba, F.S.; Pasquet, R.S.; Gepts, P. Genetic diversity in cowpea [Vigna unguiculata (L.) Walp.] as revealed by RAPD markers. Genet. Resour. Crop Evol. 2004, 51, 539–550. [Google Scholar] [CrossRef]
- Asiwe, J.A.N. Needs assessment of cowpea production practices, constraints and utilization in South Africa. Afr. J. Biotechnol. 2009, 8, 5383–5388. [Google Scholar]
- Asiwe, J.A.N.; Belane, A.; Dakora, F.D. Evaluation of cowpea breeding lines for nitrogen fixatrion at ARC-Grain Crops Institute, Potchefstroom, South Africa. In Proceedings of the 16th International Congress on Biological Nitrogen Fixation, Big Sky, MT, USA, 14–19 June 2009. [Google Scholar]
- Okereke, G.U.; Egwu, S.E.; Nnabude, P. Effect of cowpea organic residues and fertilizer N on soil fertility, growth and yield of upland rice. In Proceedings of the Eighteenth World Congress Soil Science, Philadelphia, PA, USA, 9–15 July 2006. [Google Scholar]
- Adandonon, A.; Aveling, T.A.S.; Tamo, M. Occurrence and distribution of cowpea damping-off and stem rot and associated fungi in Benin. J. Agric. Sci. 2004, 142, 561–566. [Google Scholar] [CrossRef][Green Version]
- Singh, B.B. Advances in Cowpea Research; Sayce Publishing: Devon, UK, 1997. [Google Scholar]
- Singh, S.R.; Rachie, K.O. Cowpea Research, Production and Utilisation; John Wiley and Sons Ltd.: Chichester, UK, 1985. [Google Scholar]
- Aveling, T.A.S.; Adandonon, A. First report of pre- and postemergence damping-off of cowpea caused by Pythium ultimum in South Africa. Plant Dis. 2000, 84, 922. [Google Scholar] [CrossRef]
- Kendrick, J.B. Seed transmission of cowpea wilt. Phytopathology 1931, 21, 979–983. [Google Scholar]
- Kendrick, J.B.; Snyder, W.C. Fusarium yellows of beans. Phytopathology 1942, 32, 1010–1014. [Google Scholar]
- Pio-Ribeiro, G.; Assis-Filho, F.M.; Doenças do, C. Manual de Fitopatologia; Agronômica Ceres: São Paulo, Brazil, 1997. [Google Scholar]
- Kritzinger, Q.; Aveling, T.A.; Marasas, W.F.; Rheeder, J.P.; Van Der Westhuizen, L.; Shephard, G.S. Mycoflora and fumonisin mycotoxins associated with cowpea [Vigna unguiculata (L.) Walp] seeds. J. Agric. Food Chem. 2003, 51, 2188–2192. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.E.; Korsten, L.; Aveling, T.A.S. Infection process of Colletotrichum dematium on cowpea stems. Mycol. Res. 1999, 103, 230–234. [Google Scholar] [CrossRef]
- Allen, D.J.; Thottappilly, G.; Emechebe, A.M.; Singh, B.B. Diseases of cowpea. In In Pathology of Food and Pasture Legumes of Cowpea; Allen, D.J., Lenne, J.M., Eds.; CAB International: Wallingford, UK, 1998; pp. 267–324. [Google Scholar]
- la Grange, N.; Aveling, T.A.S. First Report of Alternaria cassiae on Cowpea. Plant Dis. 1998, 82, 1171. [Google Scholar] [CrossRef]
- Seenappa, M.; Keswani, C.L.; Kundya, T.M. Aspergillus infection and aflatoxin production in some Cowpea (Vigna unguiculata (L) Walp) lines in Tanzania. Mycopathologia 1983, 83, 103–106. [Google Scholar] [CrossRef]
- Moss, M.O. Centenary review—Mycotoxins. Mycol. Res. 1996, 100, 513–523. [Google Scholar] [CrossRef]
- Caroll, E.C.; Carroll, F.E. Studies on the incidence of coniferous needle endophytes in the Pacific Northwest. Can. J. Bot. 1978, 56, 3034–3043. [Google Scholar] [CrossRef]
- Petrini, O.; Fisher, P.J. Fungal endophytes in Salicornia perennis. Trans. Br. Mycol. Soc. 1986, 87, 647–651. [Google Scholar] [CrossRef]
- Rodriguez, R.J.; White, J.F., Jr.; Arnold, A.E.; Redman, R.S. Fungal endophytes: Diversity and functional roles. New Phytol. 2009, 182, 314–330. [Google Scholar] [CrossRef]
- Schulz, B.; Boyle, C. The endophytic continuum. Mycol. Res. 2005, 109, 661–686. [Google Scholar] [CrossRef]
- Mei, C.; Flinn, B.S. The use of beneficial microbial endophytes for plant biomass and stress tolerance improvement. Recent Pat. Biotechnol. 2010, 4, 81–95. [Google Scholar] [CrossRef]
- Khan, A.L.; Al-Harrasi, A.; Al-Rawahi, A.; Al-Farsi, Z.; Al-Mamari, A.; Waqas, M.; Asaf, S.; Elyassi, A.; Mabood, F.; Shin, J.H.; et al. Endophytic Fungi from Frankincense Tree Improves Host Growth and Produces Extracellular Enzymes and Indole Acetic Acid. PLoS ONE 2016, 11, e0158207. [Google Scholar] [CrossRef]
- David, V.; Terrat, S.; Herzine, K.; Claisse, O.; Rousseaux, S.; Tourdot-Marechal, R.; Masneuf-Pomarede, I.; Ranjard, L.; Alexandre, H. Highthroughput sequencing of amplicons for monitoring yeast biodiversity in must and during alcoholic fermentation. J. Ind. Microbiol. Biotechnol. 2014, 41, 811–821. [Google Scholar] [CrossRef]
- Cheng, W.X.; Parton, W.J.; Gonzalez-Meler, M.A.; Phillips, R.; Asao, S.; McNickle, G.G.; Brzostek, E.; Jastrow, J.D. Synthesis and modeling perspectives of rhizosphere priming. New Phytol. 2014, 201, 31–44. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, M.G.; Liu, Y.; Zhang, F.S.; Hodge, A.; Feng, G. Carbon and phosphorus exchange may enable cooperation between an arbuscular mycorrhizal fungus and a phosphate-solubilizing bacterium. New Phytol. 2016, 210, 1022–1032. [Google Scholar] [CrossRef]
- Zhu, B.; Gutknecht, J.L.M.; Herman, D.J.; Keck, D.C.; Firestone, M.K.; Cheng, W.X. Rhizosphere priming effects on soil carbon and nitrogen mineralization. Soil Biol. Biochem. 2014, 76, 183–192. [Google Scholar] [CrossRef]
- Beans, C. Core Concept: Probing the phytobiome to advance agriculture. Proc. Natl. Acad. Sci. USA 2017, 114, 8900–8902. [Google Scholar] [CrossRef]
- Leach, J.E.; Triplett, L.R.; Argueso, C.T.; Trivedi, P. Communication in the Phytobiome. Cell 2017, 169, 587–596. [Google Scholar] [CrossRef]
- Taylor, M.W.; Tsai, P.; Anfang, N.; Ross, H.A.; Goddard, M.R. Pyrosequencing reveals regional differences in fruit-associated fungal communities. Environ. Microbiol. 2014, 16, 2848–2858. [Google Scholar] [CrossRef]
- Price, L.B.; Liu, C.M.; Melendez, J.H.; Frankel, Y.M.; Engelthaler, D.; Aziz, M.; Bowers, J.; Rattray, R.; Ravel, J.; Kingsley, C.; et al. Community analysis of chronic wound bacteria using 16S rRNA gene-based pyrosequencing: Impact of diabetes and antibiotics on chronic wound microbiota. PLoS ONE 2009, 4, e6462. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Ryberg, M.; Abarenkov, K.; Sjokvist, E.; Kristiansson, E. The ITS region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiol. Lett. 2009, 296, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Kinge, T.R.; Cason, E.D.; Valverde, A.; Nyaga, M.; Gryzenhout, M. Endophytic seed mycobiome of six sorghum (Sorghum bicolor) cultivars from commercial seedlots using an Illumina sequencing approach. Mycosphere 2019, 10, 739–756. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Koljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence-Limits on Phylogenies—An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- González, V.; Tello, M.L. The endophytic mycota associated with Vitis vinifera in central Spain. Fungal Divers. 2011, 47, 29–42. [Google Scholar] [CrossRef]
- Kim, D.-H.; Kim, S.-H.; Kwon, S.-W.; Lee, J.-K.; Hong, S.-B. Mycoflora of Soybeans Used for Meju Fermentation. Mycobiology 2013, 41, 100–107. [Google Scholar] [CrossRef][Green Version]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef]
- Liu, F.; Cai, L.; Crous, P.W.; Damm, U. Circumscription of the anthracnose pathogens Colletotrichum lindemuthianum and C. nigrum. Mycologia 2013, 105, 844–860. [Google Scholar] [CrossRef]
- Qin, G.Z.; Tian, S.P. Enhancement of Biocontrol Activity of Cryptococcus laurentii by Silicon and the Possible Mechanisms Involved. Phytopathology 2005, 95, 69–75. [Google Scholar] [CrossRef]
- Li, J.K.; Li, H.; Ji, S.F.; Chen, T.; Tian, S.P.; Qin, G.Z. Enhancement of biocontrol efficacy of Cryptococcus laurentii by cinnamic acid against Penicillium italicum in citrus fruit. Postharvest Biol. Technol. 2019, 149, 42–49. [Google Scholar] [CrossRef]
- Rong, X.; McSpadden Gardener, B.B. Draft Genome Sequence of Cryptococcus flavescens Strain OH182.9_3C, a Biocontrol Agent against Fusarium Head Blight of Wheat. Genome Announc. 2013, 1, e00762-13. [Google Scholar] [CrossRef]
- Freimoser, F.M.; Rueda-Mejia, M.P.; Tilocca, B.; Migheli, Q. Biocontrol yeasts: Mechanisms and applications. World J. Microbiol. Biotechnol. 2019, 35, 154. [Google Scholar] [CrossRef]
- Jensen, D.F.; Knudsen, I.M.B.; Lubeck, M.; Mamarabadi, M.; Hockenhull, J.; Jensen, B. Development of a biocontrol agent for plant disease control with special emphasis on the near commercial fungal antagonist Clonostachys rosea strain ‘IK726’. Australas. Plant Pathol. 2007, 36, 95–101. [Google Scholar] [CrossRef]
- Cota, L.V.; Maffia, L.A.; Mizubuti, E.S.; Macedo, P.E.; Antunes, R.F. Biological control of strawberry grey mould by Clonostachys rosea under field conditions. Phytopathology 2008, 98, S41–S42. [Google Scholar]
- Xue, A.G.; Voldeng, H.D.; Savard, M.E.; Fedak, G.; Tian, X.; Hsiang, T. Biological control of fusarium head blight of wheat with Clonostachys rosea strain ACM941. Can. J. Plant Pathol. 2009, 31, 169–179. [Google Scholar]
- Sun, Z.B.; Li, S.D.; Ren, Q.; Xu, J.L.; Lu, X.; Sun, M.H. Biology and applications of Clonostachys rosea. J. Appl. Microbiol. 2020, 129, 486–495. [Google Scholar] [CrossRef]
- Chen, J.; Xu, D.L.; Chao, L.M.; Liu, H.J.; Bao, Y.Y. Microbial assemblages associated with the rhizosphere and endosphere of an herbage, Leymus chinensis. Microb. Biotechnol. 2020, 13, 1390–1402. [Google Scholar] [CrossRef]
- de Souza, R.S.C.; Okura, V.K.; Armanhi, J.S.L.; Jorrín, B.; Lozano, N.; da Silva, M.J.; González-Guerrero, M.; de Araújo, L.M.; Verza, N.C.; Bagheri, H.C.; et al. Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Sci. Rep. 2016, 6, 28774. [Google Scholar] [CrossRef]
- Eyre, A.W.; Wang, M.Y.; Oh, Y.; Dean, R.A. Identification and Characterization of the Core Rice Seed Microbiome. Phytobiomes J. 2019, 3, 148–157. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef]
- Simonin, M.; Dasilva, C.; Terzi, V.; Ngonkeu, E.L.M.; Diouf, D.; Kane, A.; Béna, G.; Moulin, L. Influence of plant genotype and soil on the wheat rhizosphere microbiome: Evidences for a core microbiome across eight African and European soils. FEMS Microbiol. Ecol. 2020, 96, fiaa067. [Google Scholar] [CrossRef]
- Stergiopoulos, I.; Gordon, T.R. Cryptic fungal infections: The hidden agenda of plant pathogens. Front. Plant Sci. 2014, 5, 506. [Google Scholar] [CrossRef]
- Herrera-León, S.; Ramiro, R.; Arroyo, M.; Díez, R.; Usera, M.A.; Echeita, M.A. Blind comparison of traditional serotyping with three multiplex PCRs for the identification of Salmonella serotypes. Res. Microbiol. 2007, 158, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.F.; Summerell, B.A. The Fusarium Laboratory Manual, 1st ed.; Blackwell Publishing Ltd.: Ames, IA, USA, 2006. [Google Scholar]
- Jayasiri, S.C.; Hyde, K.D.; Jones, E.B.G.; Jeewon, R.; Ariyawansa, H.A.; Bhat, J.D.; Camporesi, E.; Kang, J.C. Taxonomy and multigene phylogenetic evaluation of novel species in Boeremia and Epicoccum with new records of Ascochyta and Didymella (Didymellaceae). Mycosphere 2017, 8, 1080–1101. [Google Scholar] [CrossRef]
- Walter, H.; Muggia, L.; Fritscher, M.; Holler, A.; Horvat, D.; Guttenberger, H.; Simon, U.K. Multiple taxa in the Phoma-complex associate with black elder (Sambucus nigra L.). Fungal Biol. 2016, 120, 43–50. [Google Scholar] [CrossRef]
- Bensch, K.; Braun, U.; Groenewald, J.Z.; Crous, P.W. The genus Cladosporium. Stud. Mycol. 2012, 1–379. [Google Scholar] [CrossRef]
- Lodama, K.E. Fumonisin Production by and Biological Control of Fusarium Species Associated with Cowpea Seed; University of Pretoria: Pretoria, South Africa, 2011. [Google Scholar]
- Andrade, O.; Munoz, G.; Galdames, R.; Duran, P.; Honorato, R. Characterization, in vitro culture, and molecular analysis of Thecaphora solani, the causal agent of potato smut. Phytopathology 2004, 94, 875–882. [Google Scholar] [CrossRef]
- Conforto, C.; Cazon, I.; Fernandez, F.D.; Marinelli, A.; Oddino, C.; Rago, A.M. Molecular sequence data of Thecaphora frezii affecting peanut crops in Argentina. Eur. J. Plant Pathol. 2013, 137, 663–666. [Google Scholar] [CrossRef]
- Vasighzadeh, A.; Zafari, D.; Selcuk, F.; Huseyin, E.; Kursat, M.; Lutz, M.; Piatek, M. Discovery of Thecaphora schwarzmaniana on Rheum ribes in Iran and Turkey: Implications for the diversity and phylogeny of leaf smuts on rhubarbs. Mycol. Prog. 2014, 13, 881–892. [Google Scholar] [CrossRef]
- Deng, Q.D.; Yong, M.L.; Li, D.Y.; Lai, C.H.; Chen, H.M.; Fan, J.; Hu, D.W. Survey and examination of the potential alternative hosts of Villosiclava virens, the pathogen of rice false smut, in China. J. Integr. Agric. 2015, 14, 1332–1337. [Google Scholar] [CrossRef]
- Carlucci, A.; Raimondo, M.L.; Santos, J.; Phillips, A.J.L. Plectosphaerella species associated with root and collar rots of horticultural crops in southern Italy. Persoonia 2012, 28, 34–48. [Google Scholar] [CrossRef]
- Gullino, M.L.; Gilardi, G.; Garibaldi, A. Seed-Borne Fungal Pathogens of Leafy Vegetable Crops. In Global Perspectives on the Health of Seeds and Plant Propagation Material; Gullino, M., Munkvold, G., Eds.; Springer: Dordrecht, The Netherlands, 2014; Volume 6. [Google Scholar]
- Smith, J.E.; Aveling, T.A.S. Colletotrichum dematium: Causal agent of a new cowpea stem disease in South Africa. Plant Dis. 1997, 81, 832. [Google Scholar] [CrossRef]
- Latunde-Dada, A.O.; O’Connell, R.J.; Nash, C.; Lucas, J.A. Stomata penetration of cow pea (Vigna unguiculata) leaves by Colletotrichum species causing latent anthracnose. Plant Pathol. 1996, 48, 777–785. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Lemanceau, P.; Blouin, M.; Muller, D.; Moenne-Loccoz, Y. Let the core microbiota be functional. Trends Plant Sci. 2017, 22, 583–595. [Google Scholar] [CrossRef]
- Pancher, M.; Ceol, M.; Corneo, P.E.; Longa, C.M.O.; Yousaf, S.; Pertot, I.; Campisano, A. Fungal endophytic communities in grapevines (Vitis vinifera L.) respond to crop management. Appl. Environ. Microbiol. 2012, 78, 4308–4317. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).