
Transcriptome Analysis Reveals Candidate Genes Regulating the Skin and Hair Diversity of Xinji Fine-Wool Sheep and Tan Sheep

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Skin Tissue Sample Information
2.2. RNA Extraction, Library Construction, and RNA Sequencing
2.3. Sequencing Data Mapping and Transcriptome Assembly
2.4. Analysis of Differentially Expressed Genes
2.5. Clustering and Correlation Analysis
2.6. Functional Enrichment and Protein–Protein Interaction Analyses of DEGs
2.7. Relative Gene Expression Analysis by Quantitative Real-Time PCR (qPCR)
2.8. qPCR Statistical Analysis
3. Results
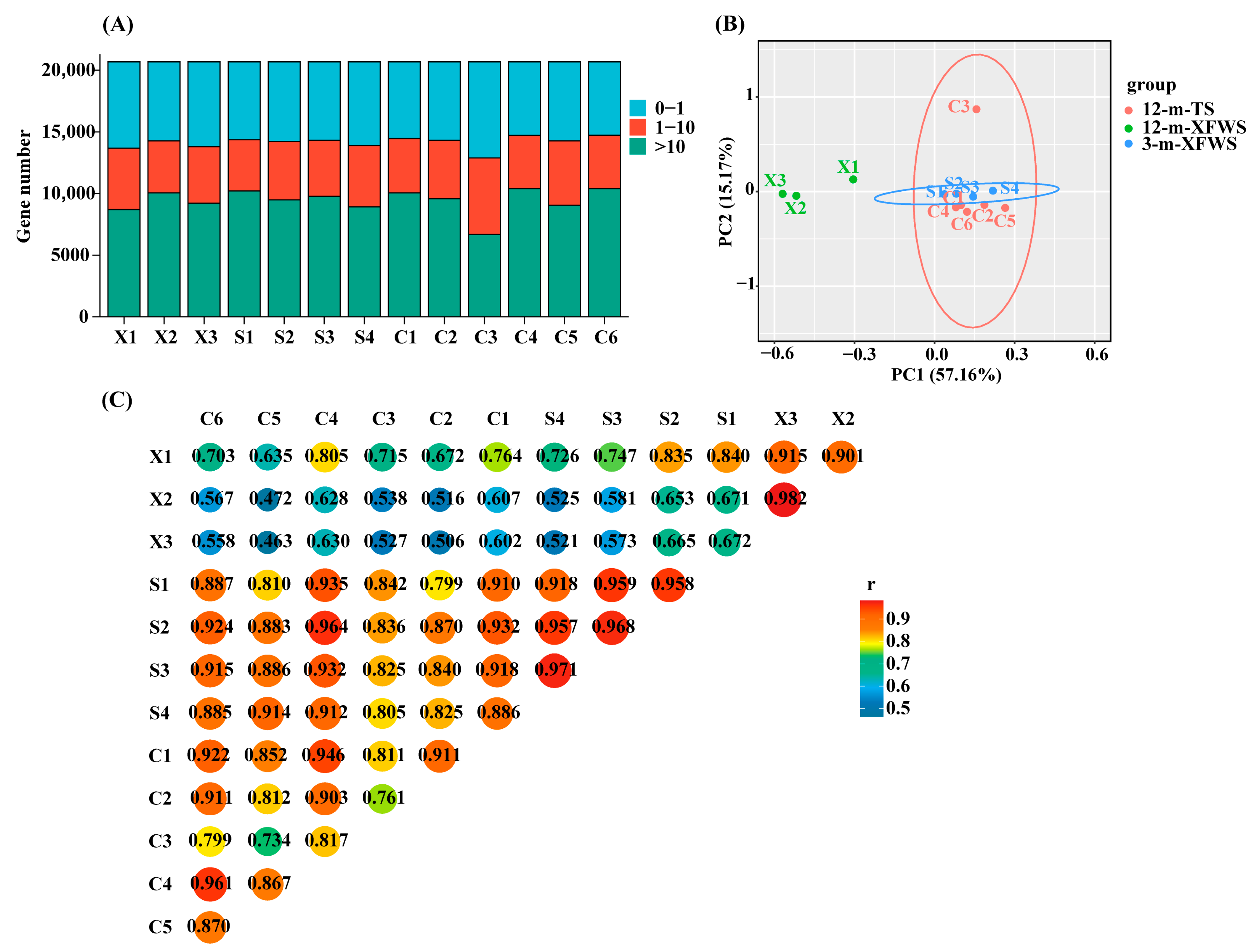
3.1. Transcriptome Sequencing and Alignment
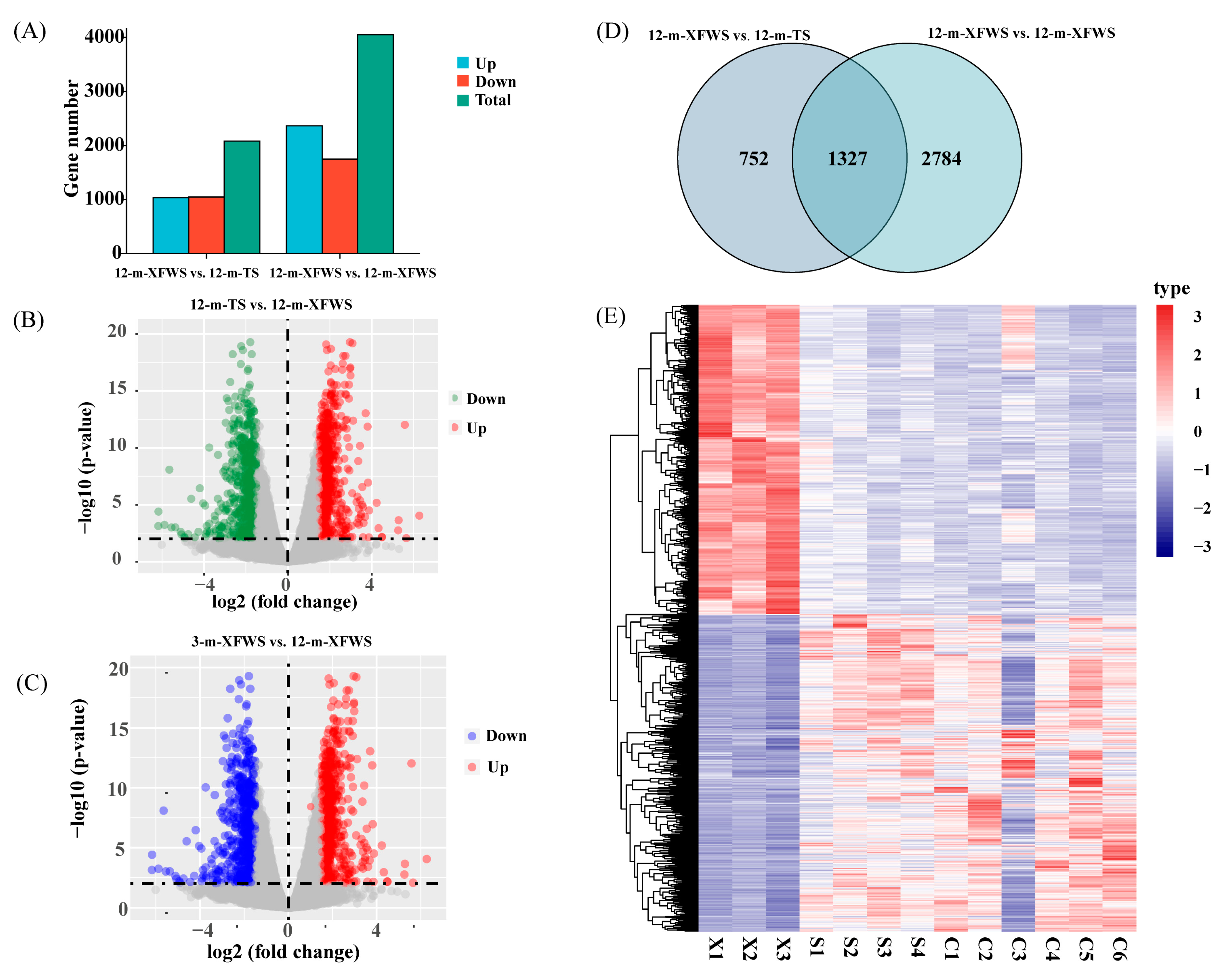
3.2. Analysis of the Differentially Expressed Genes
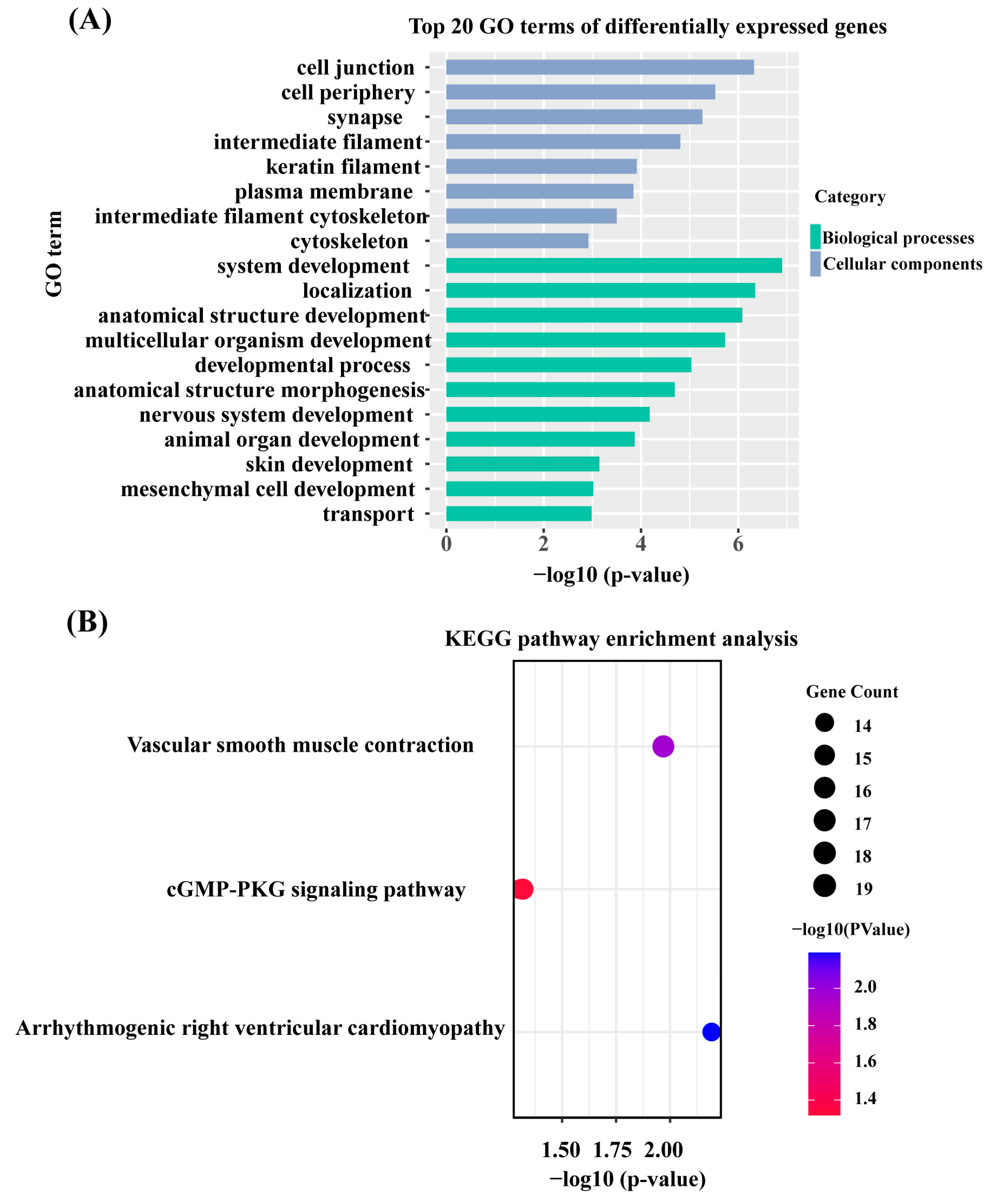
3.3. GO Enrichment and KEGG Pathway Analysis
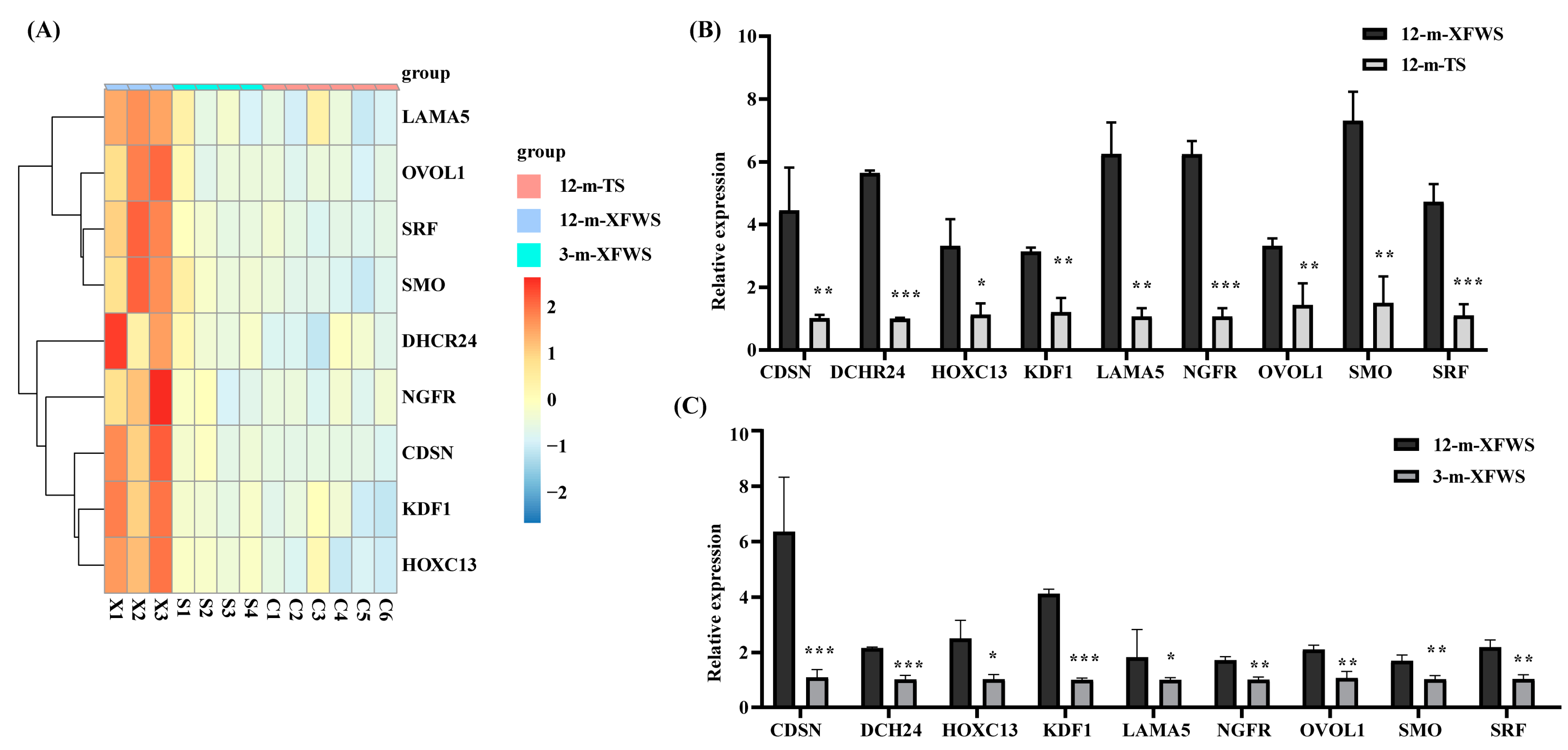
3.4. Identification of Candidate Genes for Hair Follicle Development
3.5. Analysis and Annotation of Transcription Factors
4. Discussion
4.1. RNA-Seq Analysis and Shoulder-Skin-Related DEGs in Different Breeds and Ages
4.2. DEGs Correlated with Hair Follicle Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schneider, M.R.; Schmidt-Ullrich, R.; Paus, R. The Hair Follicle as a Dynamic Miniorgan. Curr. Biol. 2009, 19, R132–R142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, S.J. The Tylotrich (Hair) follicle of the american opossum. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1968, 160, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Nixon, A.J.; Broad, L.; Saywell, D.P.; Pearson, A.J. Transforming growth factor-alpha immunoreactivity during induced hair follicle growth cycles in sheep and ferrets. J. Histochem. Cytochem. 1996, 44, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Grymowicz, M.; Rudnicka, E.; Podfigurna, A.; Napierala, P.; Smolarczyk, R.; Smolarczyk, K.; Meczekalski, B. Hormonal Effects on Hair Follicles. Int. J. Mol. Sci. 2020, 21, 5342. [Google Scholar] [CrossRef]
- Rogers, G.E. Biology of the wool follicle: An excursion into a unique tissue interaction system waiting to be re-discovered. Exp. Dermatol. 2006, 15, 931–949. [Google Scholar] [CrossRef]
- Wang, X.; Ma, Q.; Zhao, Z. Research progress of regulator genes of Tan sheep furcolor. Biot. Resour. 2019, 41, 143–148. [Google Scholar]
- Wu, C.; Wang, C. Polymorphism analysis of five reproductive hormone receptor genes in Xinji fine wool sheep. Sci. Technol. 2019, 55, 63–69. [Google Scholar]
- Moore, G.; Jackson, N.; Isaacs, K.; Brown, G. Pattern and Morphogenesis in Skin. J. Theor. Biol. 1998, 191, 87–94. [Google Scholar] [CrossRef]
- Bandmann, H.J.; Bosse, K. Histology and anatomy of the hair follicle in the course of the hair cycle. Arch. Klin. Exp. Dermatol. 1966, 227, 390–409. [Google Scholar] [CrossRef]
- Hollis, D.E.; Chapman, R.E.; Panaretto, B.A.; Moore, G. Morphological Changes in the Skin and Wool Fibres of Merino Sheep Infused with Mouse Epidermal Growth Factor. Aust. J. Biol. Sci. 1983, 36, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, J.M.; Frenkel, M.J.; Reis, P.J. Changes in the matrix proteins of wool and mouse hair following the administra-tion of depilatory compounds. Aust. J. Biol. Sci. 1980, 33, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Tisserant, E.; Da Silva, C.; Kohler, A.; Morin, E.; Wincker, P.; Martin, F. Deep RNA sequencing improved the structural anno-tation of the Tuber melanosporum transcriptome. New Phytol. 2011, 189, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, G.C.; Arnaud, M.B.; Inglis, D.O.; Skrzypek, M.S.; Binkley, G.; Simison, M.; Miyasato, S.R.; Binkley, J.; Orvis, J.; Shah, P.; et al. The Aspergillus Genome Database: Multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucleic Acids Res. 2014, 42, D705–D710. [Google Scholar] [CrossRef] [Green Version]
- La, Y.; He, X.; Zhang, L.; Di, R.; Wang, X.; Gan, S.; Zhang, X.; Zhang, J.; Hu, W.; Chu, M. Comprehensive Analysis of Differentially Expressed Profiles of mRNA, lncRNA, and circRNA in the Uterus of Seasonal Reproduction Sheep. Genes 2020, 11, 301. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; You, S.; Yao, Y.; Liu, Z.-J.; Hazi, W.; Li, C.-Y.; Zhang, X.-Y.; Hou, X.-X.; Wei, J.-C.; Li, X.-Y.; et al. Expression profiles of circular RNAs in sheep skeletal muscle. Asian-Australas. J. Anim. Sci. 2018, 31, 1550–1557. [Google Scholar] [CrossRef]
- Li, Y.; Kong, L.; Deng, M.; Lian, Z.; Han, Y.; Sun, B.; Guo, Y.; Liu, G.; Liu, D. Heat Stress-Responsive Transcriptome Analysis in the Liver Tissue of Hu Sheep. Genes 2019, 10, 395. [Google Scholar] [CrossRef] [Green Version]
- Mou, C.; Thomason, H.A.; Willan, P.M.; Clowes, C.; Harris, W.E.; Drew, C.F.; Dixon, J.; Dixon, M.J.; Headon, D.J. Enhanced ectodys-plasin-A receptor (EDAR) signaling alters multiple fiber characteristics to produce the East Asian hair form. Hum. Mutat. 2008, 29, 1405–1411. [Google Scholar] [CrossRef] [Green Version]
- Kamberov, Y.G.; Wang, S.; Tan, J.; Gerbault, P.; Wark, A.; Tan, L.; Yang, Y.; Li, S.; Tang, K.; Chen, H.; et al. Modeling Recent Human Evolution in Mice by Expression of a Selected EDAR Variant. Cell 2013, 152, 691–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Y.; Li, S.; Zheng, X.; Chen, W.; Li, X.; Liu, Z.; Hu, Y.; Qiao, H.; Qi, Q.; Pei, Q.; et al. Transcriptome Reveals Long Non-coding RNAs and mRNAs Involved in Primary Wool Follicle Induction in Carpet Sheep Fetal Skin. Front. Physiol. 2018, 9, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, W.K.W.; Freem, L.; Zhao, D.; Painter, K.J.; Woolley, T.E.; Gaffney, E.A.; McGrew, M.J.; Tzika, A.; Milinkovitch, M.C.; Schneider, P.; et al. Feather arrays are patterned by interacting signalling and cell density waves. PLoS Biol. 2019, 17, e3000132. [Google Scholar] [CrossRef] [Green Version]
- Drew, C.F.; Lin, C.M.; Jiang, T.X.; Blunt, G.; Mou, C.; Chuong, C.M.; Headon, D.J. The Edar subfamily in feather placode formation. Dev. Biol. 2007, 305, 232–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, C.; Pitel, F.; Gourichon, D.; Vignoles, F.; Tzika, A.; Tato, P.; Yu, L.; Burt, D.W.; Bed’Hom, B.; Tixier-Boichard, M.; et al. Cryptic Patterning of Avian Skin Confers a Developmental Facility for Loss of Neck Feathering. PLoS Biol. 2011, 9, e1001028. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Guo, T.; Yuan, C.; Liu, J.; Guo, J.; Feng, R.; Niu, C.; Sun, X.; Yang, B. Integrated Analysis of the Roles of Long Noncoding RNA and Coding RNA Expression in Sheep (Ovis aries) Skin during Initiation of Secondary Hair Follicle. PLoS ONE 2016, 11, e0156890. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.Y.; Yang, H.; Shi, G.Q.; Shen, M.; Yang, J.Q.; Yang, Y.L.; Liu, X.J. Expression profile analysis of microRNAs during hair folli-cle development in the sheep foetus. Biosci. Biotechnol. Biochem. 2019, 83, 1045–1061. [Google Scholar] [CrossRef]
- Zhao, R.; Li, J.; Liu, N.; Li, H.; Liu, L.; Yang, F.; Li, L.; Wang, Y.; He, J. Transcriptomic Analysis Reveals the Involvement of lncRNA-miRNA-mRNA Networks in Hair Follicle Induction in Aohan Fine Wool Sheep Skin. Front. Genet. 2020, 11, 590. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript as-sembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differen-tiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive func-tional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Reimand, J.; Arak, T.; Adler, P.; Kolberg, L.; Reisberg, S.; Peterson, H.; Vilo, J. g: Profiler-a web server for functional interpreta-tion of gene lists (2016 update). Nucleic Acids Res. 2016, 44, W83–W89. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Potter, C.S.; Pruett, N.D.; Kern, M.J.; Baybo, M.A.; Godwin, A.R.; Potter, K.A.; Peterson, R.L.; Sundberg, J.P.; Awgulewitsch, A. The Nude Mutant Gene Foxn1 Is a HOXC13 Regulatory Target during Hair Follicle and Nail Differentiation. J. Investig. Dermatol. 2011, 131, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.-W.; Chu, Y.-K.; Yang, H.; Yan, X.-H.; Rong, E.-G.; Li, H.; Wang, N. Functional Analysis of Sheep POU2F3 Isoforms. Biochem. Genet. 2019, 58, 335–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wu, K.; Wang, L.; Wang, Z.; Han, W.; Chen, D.; Wei, Y.; Su, R.; Wang, R.; Liu, Z.; et al. Comparative study on seasonal hair follicle cycling by analysis of the transcriptomes from cashmere and milk goats. Genome 2020, 112, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dong, H. Study on the development law of hair follicle of Gansu alpine fine wool sheep and its hybrid sheep. Chin. Herbiv. Anim. Sci. 2019, 39, 67–69. [Google Scholar]
- Dreesen, O.; Chojnowski, A.; Ong, P.F.; Zhao, T.Y.; Common, J.E.; Lunny, D.; Lane, E.B.; Lee, S.J.; Vardy, L.A.; Stewart, C.L.; et al. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J. Cell Biol. 2013, 200, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Taimen, P.; Pfleghaar, K.; Shimi, T.; Möller, D.; Ben-Harush, K.; Erdos, M.R.; Adam, S.A.; Herrmann, H.; Medalia, O.; Collins, F.S.; et al. A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc. Natl. Acad. Sci. USA 2009, 106, 20788–20793. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Taketomi, Y.; Isogai, Y.; Miki, Y.; Sato, H.; Masuda, S.; Nishito, Y.; Morioka, K.; Ishimoto, Y.; Suzuki, N.; et al. Hair fol-licular expression and function of group X secreted phospholipase A2 in mouse skin. J. Biol. Chem. 2011, 286, 11616–11631. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Hindes, A.; Burns, C.J.; Koppel, A.C.; Kiss, A.; Yin, Y.; Ma, L.; Blumenberg, M.; Khnykin, D.; Jahnsen, F.L.; et al. Serum Response Factor Controls Transcriptional Network Regulating Epidermal Function and Hair Follicle Morphogenesis. J. Investig. Dermatol. 2013, 133, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, X.; Chen, W.; Sun, W.; Hussain, Z.; Wang, S.; Wang, J. Analysis of lncRNAs Expression Profiles in Hair Follicle of Hu Sheep Lambskin. Animal 2020, 10, 1035. [Google Scholar] [CrossRef]
- Li, S.; Chen, W.; Zheng, X.; Liu, Z.; Yang, G.; Hu, X.; Mou, C. Comparative investigation of coarse and fine wool sheep skin indicates the early regulators for skin and wool diversity. Gene 2020, 758, 144968. [Google Scholar] [CrossRef]
- Leclerc, E.A.; Huchenq, A.; Mattiuzzo, N.R.; Metzger, D.; Chambon, P.; Ghyselinck, N.B.; Serre, G.; Jonca, N.; Guerrin, M. Cor-neodesmosin gene ablation induces lethal skin-barrier disruption and hair-follicle degeneration related to desmo-some dysfunction. J. Cell Sci. 2009, 122, 2699–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Tsuji, G.; Ohno, F.; Uchi, H.; Nakahara, T.; Hashimoto-Hachiya, A.; Yoshida, Y.; Yamamoto, O.; Oda, Y.; Furue, M. Activation of the OVOL1-OVOL2 Axis in the Hair Bulb and in Pilomatricoma. Am. J. Pathol. 2016, 186, 1036–1043. [Google Scholar] [CrossRef] [Green Version]
- Gilanchi, S.; Esmaeilzade, B.; Eidi, A.; Barati, M.; Mehrabi, S.; Ghoroghi, F.M.; Nobakht, M. Neuronal differentiation of rat hair follicle stem cells: The involvement of the neuroprotective factor Seladin-1 (DHCR24). Iran. Biomed. J. 2014, 18, 136–142. [Google Scholar] [PubMed]
- Veraitch, O.; Mabuchi, Y.; Matsuzaki, Y.; Sasaki, T.; Okuno, H.; Tsukashima, A.; Amagai, M.; Okano, H.; Ohyama, M. Induction of hair follicle dermal papilla cell properties in human induced pluripotent stem cell-derived multipotent LNGFR(+)THY-1(+) mesenchymal cells. Sci. Rep. 2017, 7, 42777. [Google Scholar] [CrossRef]
- Yang, X.; Cui, Y.; Yue, J.; He, H.; Yu, C.; Liu, P.; Liu, J.; Ren, X.; Meng, Y. The histological characteristics, age-related thickness change of skin, and expression of the HSPs in the skin during hair cycle in yak (Bos grunniens). PLoS ONE 2017, 12, e0176451. [Google Scholar] [CrossRef]
- Qi, Y.; Fu, S.; He, X.; Wang, B.; Da, L.; Te, R.; Yuejun, M.; Suzhen, S.; Zhang, W.; Liu, Y. Preliminary comparison of skin transcriptome from sheep with different wool fibre diameters. Anim. Prod. Sci. 2021, 61, 708. [Google Scholar] [CrossRef]
- Zhuang, Z.H.; Zhong, Y.; Chen, Y.H.; Zhang, Z.W. Research progress on the roles of Krüppel-like factors in muscle tissues. Hereditas 2018, 40, 733–748. [Google Scholar]
- Vanhoutteghem, A.; Bouche, C.; Maciejewski-Duval, A.; Hervé, F.; Djian, P. Basonuclins and disco: Orthologous zinc finger proteins essential for development in vertebrates and arthropods. Biochimie 2011, 93, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Runko, A.P.; Sagerström, C.G. A novel subfamily of zinc finger genes involved in embryonic development. J. Cell Biochem. 2004, 93, 887–895. [Google Scholar] [CrossRef]
- Tsuji, G.; Ito, T.; Chiba, T.; Mitoma, C.; Nakahara, T.; Uchi, H.; Furue, M. The role of the OVOL1-OVOL2 axis in normal and dis-eased human skin. J. Dermatol. Sci. 2018, 90, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, M.W.; Jiang, T.-X.; Plikus, M.V.; Guerrero-Juarez, C.F.; Lin, C.-H.; Schafer, C.; Maxson, R.; Widelitz, R.B.; Chuong, C.-M.; Lin, C.-H. Msx2 Supports Epidermal Competency during Wound-Induced Hair Follicle Neogenesis. J. Investig. Dermatol. 2018, 138, 2041–2050. [Google Scholar] [CrossRef] [Green Version]
- Depew, M.J.; Liu, J.K.; Long, J.E.; Presley, R.; Meneses, J.J.; Pedersen, R.A.; Rubenstein, J. Dlx5 regulates regional development of the branchial arches and sensory capsules. Development 1999, 126, 3831–3846. [Google Scholar] [CrossRef]
- Zhao, B.; Luo, H.; He, J.; Huang, X.; Chen, S.; Fu, X.; Zeng, W.; Tian, Y.; Liu, S.; Li, C.J.; et al. Comprehensive transcriptome and methylome analysis delineates the biological basis of hair follicle development and wool-related traits in Me-rino sheep. BMC Biol. 2021, 19, 197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sun, F.; Jin, H.; Dalrymple, B.P.; Cao, Y.; Wei, T.; Vuocolo, T.; Zhang, M.; Piao, Q.; Ingham, A.B. A comparison of transcriptomic patterns measured in the skin of Chinese fine and coarse wool sheep breeds. Sci. Rep. 2017, 7, 14301. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, T.; Liang, B.; Zhao, Y.; Han, J.; Pu, Y.; Wang, C.; Ma, Y.; Jiang, L. Transcriptome Analysis Reveals Candidate Genes Regulating the Skin and Hair Diversity of Xinji Fine-Wool Sheep and Tan Sheep. Agriculture 2022, 12, 15. https://doi.org/10.3390/agriculture12010015
Bai T, Liang B, Zhao Y, Han J, Pu Y, Wang C, Ma Y, Jiang L. Transcriptome Analysis Reveals Candidate Genes Regulating the Skin and Hair Diversity of Xinji Fine-Wool Sheep and Tan Sheep. Agriculture. 2022; 12(1):15. https://doi.org/10.3390/agriculture12010015
Chicago/Turabian StyleBai, Tianyou, Benmeng Liang, Yuhetian Zhao, Jiangang Han, Yabin Pu, Chunxin Wang, Yuehui Ma, and Lin Jiang. 2022. "Transcriptome Analysis Reveals Candidate Genes Regulating the Skin and Hair Diversity of Xinji Fine-Wool Sheep and Tan Sheep" Agriculture 12, no. 1: 15. https://doi.org/10.3390/agriculture12010015
APA StyleBai, T., Liang, B., Zhao, Y., Han, J., Pu, Y., Wang, C., Ma, Y., & Jiang, L. (2022). Transcriptome Analysis Reveals Candidate Genes Regulating the Skin and Hair Diversity of Xinji Fine-Wool Sheep and Tan Sheep. Agriculture, 12(1), 15. https://doi.org/10.3390/agriculture12010015