Selective Apheresis of C-Reactive Protein for Treatment of Indications with Elevated CRP Concentrations

 , and
, and 
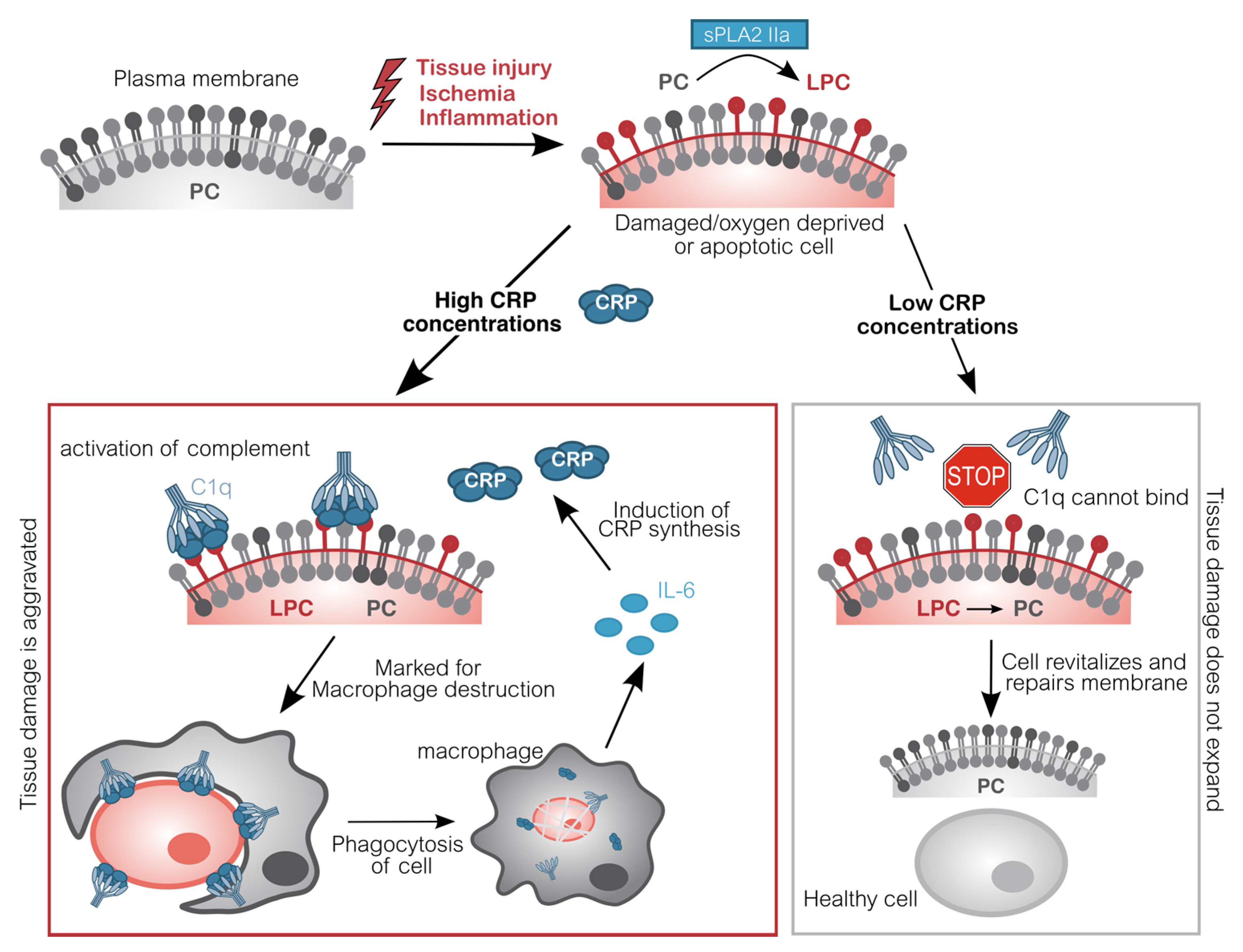
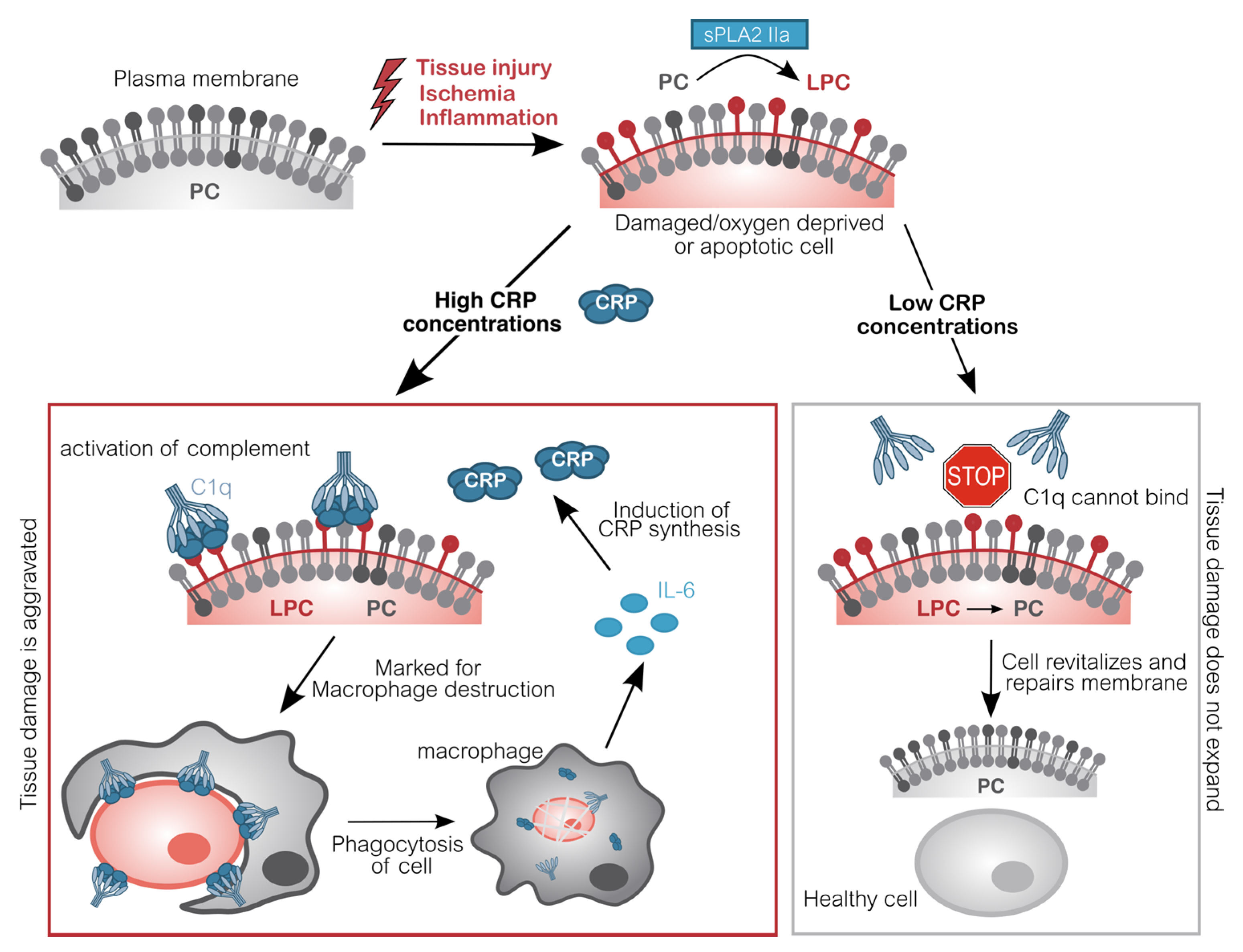{kind=link}
{kind=link}
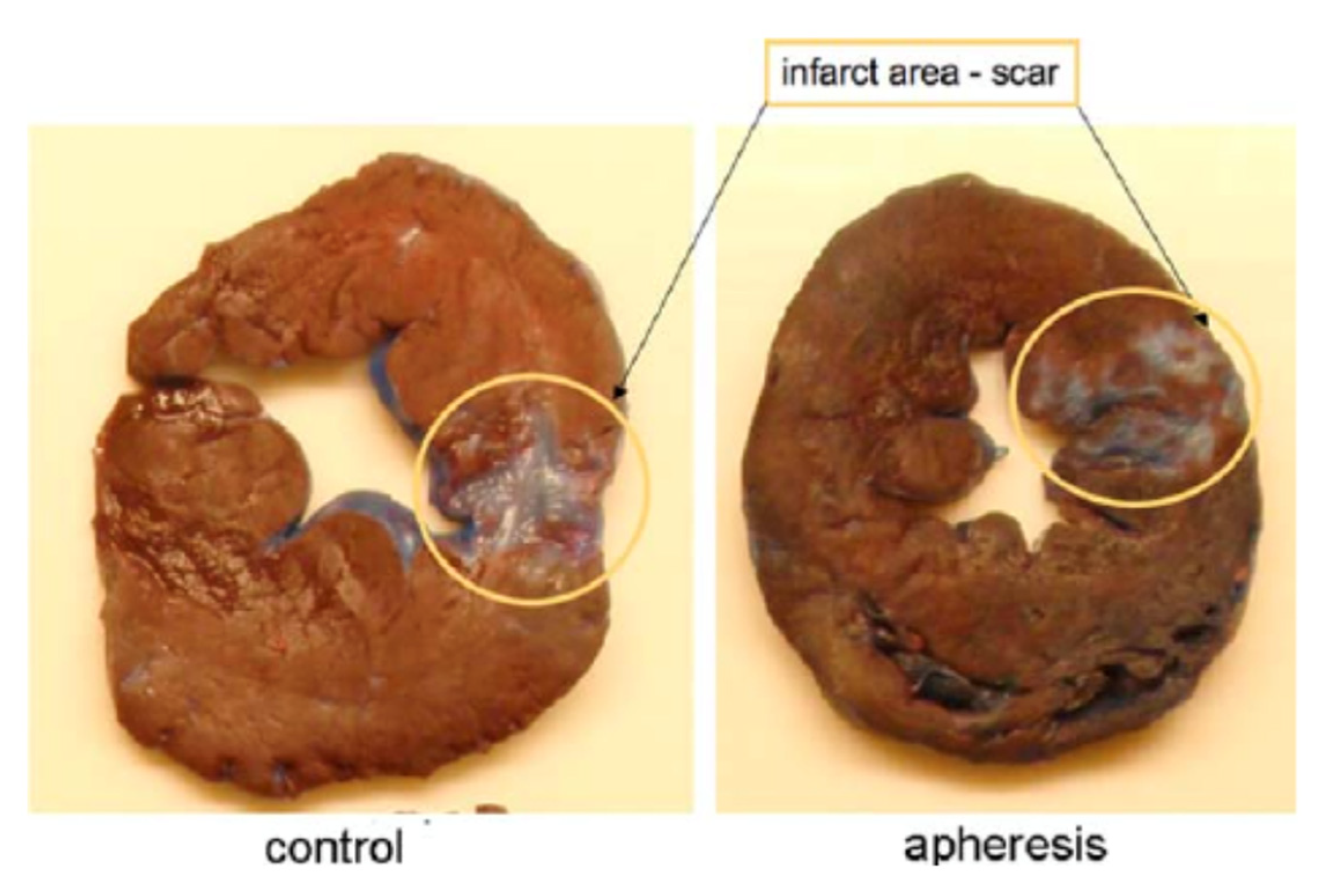{kind=link}
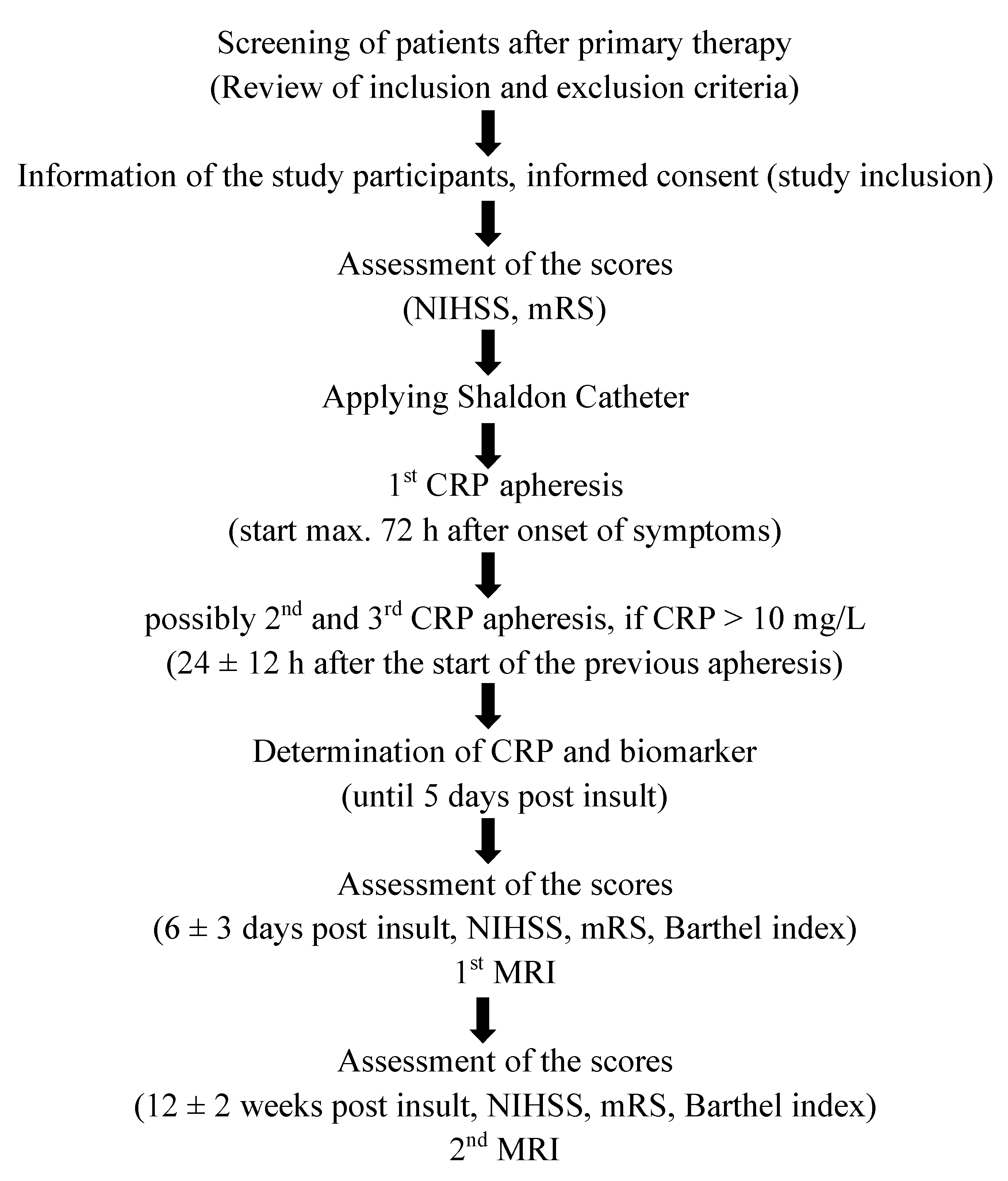{kind=link}
Abstract
:1. General Introduction
2. Role of CRP
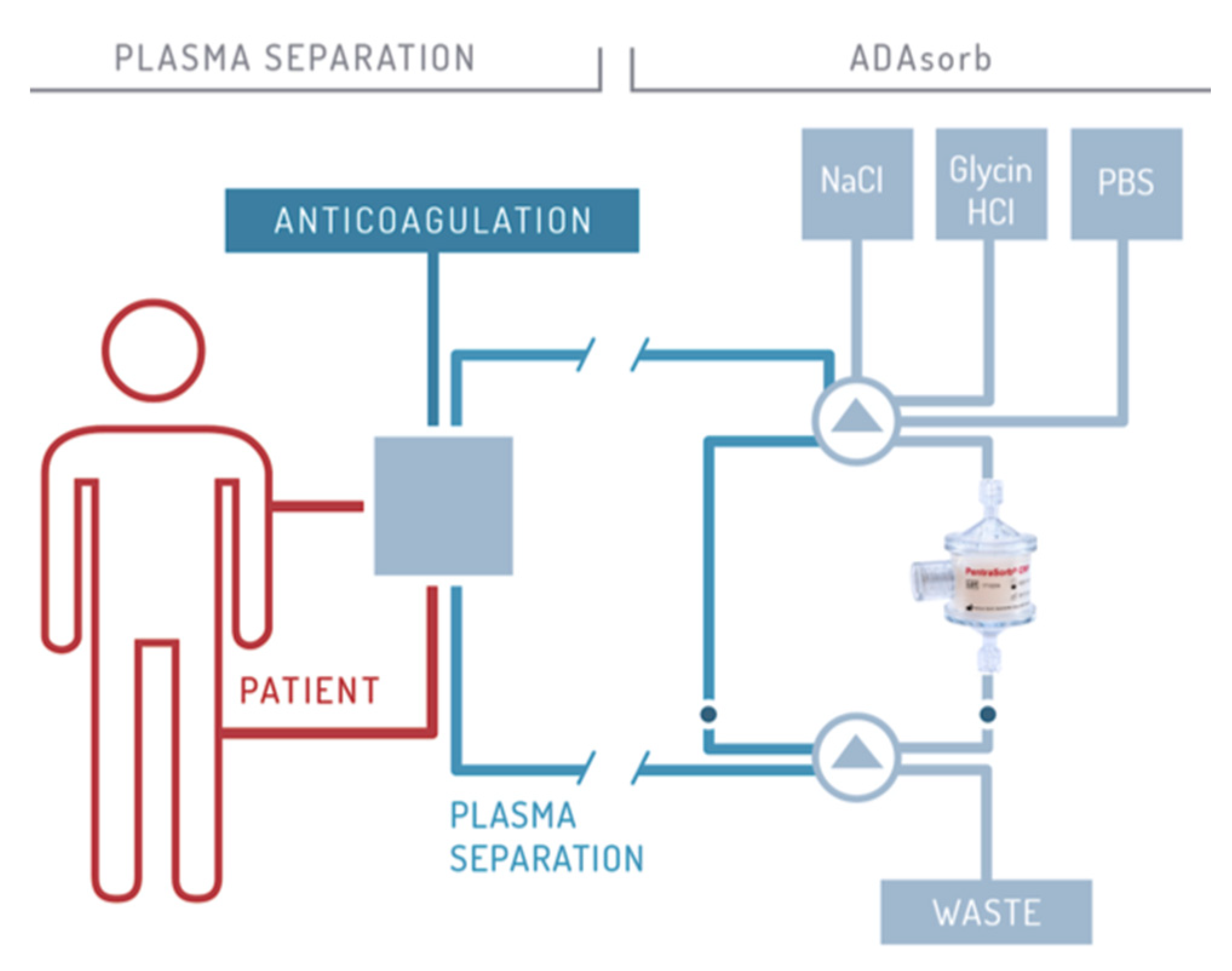
3. CRP Apheresis
4. CRP Apheresis after Ischemic Tissue Damage
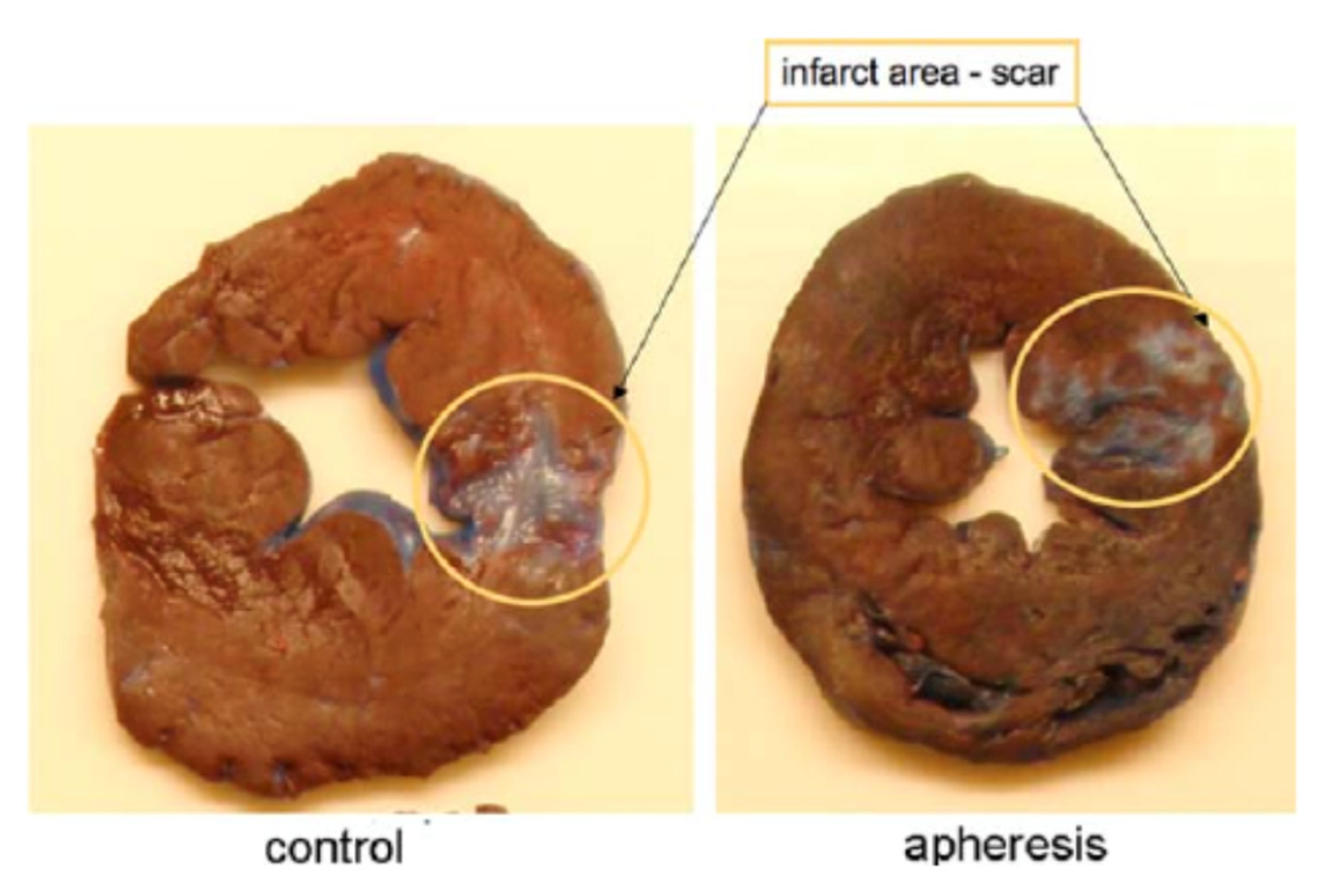
4.1. Myocardial Infarction
4.2. Ischemic Stroke
5. CRP Apheresis in Other Indications
6. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, U.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef] [Green Version]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in Wound Repair: Molecular and Cellular Mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.-B.; Hernández-Reséndiz, S.; Crespo-Avilan, G.E.; Mukhametshina, R.T.; Kwek, X.-Y.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 2018, 186, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Anzai, T. Inflammatory Mechanisms of Cardiovascular Remodeling. Circ. J. 2018, 82, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Neher, M.D.; Weckbach, S.; Flierl, M.A.; Huber-Lang, M.; Stahel, P.F. Molecular mechanisms of inflammation and tissue injury after major trauma-is complement the “bad guy”? J. Biomed. Sci. 2011, 18, 90. [Google Scholar] [CrossRef] [Green Version]
- Day, J.; Taylor, K. The systemic inflammatory response syndrome and cardiopulmonary bypass. Int. J. Surg. 2005, 3, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillett, W.S.; Francis, T. Serological Reactions in Pneumonia with a Non-Protein Somatic Fraction of Pneumococcus. J. Exp. Med. 1930, 52, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Kunze, R. C-Reactive Protein: From Biomarker to Trigger of Cell Death? Ther. Apher. Dial. 2019, 23, 494–496. [Google Scholar] [CrossRef]
- Mortensen, R.F. C-Reactive Protein, Inflammation, and Innate Immunity. Immunol. Res. 2001, 24, 163–176. [Google Scholar] [CrossRef]
- Du Clos, T.W. Pentraxins: Structure, Function, and Role in Inflammation. ISRN Inflamm. 2013, 2013, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Toniatti, C.; Arcone, R.; Majello, B.; Ganter, U.; Arpaia, G.; Ciliberto, G. Regulation of the human C-reactive protein gene, a major marker of inflammation and cancer. Mol. Biol. Med. 1990, 7, 199–212. [Google Scholar] [PubMed]
- Zhang, D.; Sun, M.; Samols, D.; Kushner, I. STAT3 Participates in Transcriptional Activation of the C-reactive Protein Gene by Interleukin-6. J. Biol. Chem. 1996, 271, 9503–9509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, F.; Torzewski, J.; Kamenz, J.; Veit, K.; Hombach, V.; Dedio, J.; Ivashchenko, Y. Interleukin-1β stimulates acute phase response and C-reactive protein synthesis by inducing an NFκB- and C/EBPβ-dependent autocrine interleukin-6 loop. Mol. Immunol. 2008, 45, 2678–2689. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, B.; Bader, A.; Poli, V.; Rüther, U. Interleukin-6 is necessary, but not sufficient, for induction of the humanC-reactive protein gene in vivo. Biochem. J. 1997, 325, 617–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushner, I.; Broder, M.L.; Karp, D. Control of the Acute Phase Response. J. Clin. Investig. 1978, 61, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, K.; Harmoinen, A.; Hermens, W.; Simoons, M.L.; Van De Werf, F.; Verstraete, M. Serum C-reactive protein and infarct size in myocardial infarct patients with a closed versus an open infarct-related coronary artery after thrombolytic therapy. Eur. Hear. J. 1993, 14, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Dimitrijević, O.; Stojcevski, B.D.; Ignjatović, S.; Singh, N.M. Serial Measurements of C-Reactive Protein After Acute Myocardial Infarction in Predicting One-Year Outcome. Int. Hear. J. 2006, 47, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, A.S.; Martinsson, A.; Wretlind, B.; Ahnve, S. IL-6 levels in acute and post myocardial infarction: Their relation to CRP levels, infarction size, left ventricular systolic function, and heart failure. Eur. J. Intern. Med. 2004, 15, 523–528. [Google Scholar] [CrossRef]
- Szalai, A.J.; Briles, D.E.; Volanakis, J.E. Role of complement in C-reactive-protein-mediated protection of mice from Streptococcus pneumoniae. Infect. Immun. 1996, 64, 4850–4853. [Google Scholar] [CrossRef] [Green Version]
- Mold, C.; Rodic-Polic, B.; Du Clos, T.W. Protection from Streptococcus pneumoniae Infection by C-Reactive Protein and Natural Antibody Requires Complement But Not Fcγ Receptors. J. Immunol. 2002, 168, 6375–6381. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.-K.; Binder, C.J.; Torzewski, M.; Witztum, J.L. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: Phosphorylcholine of oxidized phospholipids. Proc. Natl. Acad. Sci. USA 2002, 99, 13043–13048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.P.; Mold, C.; Du Clos, T.W. Sublytic complement attack exposes C-reactive protein binding sites on cell membranes. J. Immunol. 1994, 152, 2995–3005. [Google Scholar] [PubMed]
- Sparkes, B.L.; Woods, K.; Roth, M.; Welti, R.; Fleming, S.D. Phospholipase A2 alters membrane lipid composition during ischemia/reperfusion (39.55). J. Immunol. 2009, 182, 39.55. [Google Scholar]
- Yagami, T.; Yamamoto, Y.; Koma, H. The Role of Secretory Phospholipase A2 in the Central Nervous System and Neurological Diseases. Mol. Neurobiol. 2013, 49, 863–876. [Google Scholar] [CrossRef]
- Murakami, M.; Taketomi, Y.; Sato, H.; Yamamoto, K. Secreted phospholipase A2 revisited. J. Biochem. 2011, 150, 233–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujioka, D.; Kawabata, K.-I.; Ishimoto, Y.; Suzuki, N.; Hanasaki, K.; Sato, R.; Hasebe, H.; Kobayashi, T.; Saito, Y.; Kanazawa, M.; et al. Abstract 1435: Reduction in Myocardial Ischemia-reperfusion Injury in Group X Secretory Phospholipase A2-deficient Mice. Circulation 2006, 114, II_275. [Google Scholar] [CrossRef]
- Yano, T.; Fujioka, D.; Saito, Y.; Kobayashi, T.; Nakamura, T.; Obata, J.-E.; Kawabata, K.; Watanabe, K.; Watanabe, Y.; Mishina, H.; et al. Group V secretory phospholipase A2 plays a pathogenic role in myocardial ischaemia–reperfusion injury. Cardiovasc. Res. 2010, 90, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Nijmeijer, R.; Lagrand, W.K.; Baidoshvili, A.; Lubbers, Y.T.P.; Hermens, W.T.; Meijer, C.J.L.M.; Visser, C.A.; Hack, C.E.; Niessen, H.W.M. Secretory type II phospholipase A(2) binds to ischemic myocardium during myocardial infarction in humans. Cardiovasc. Res. 2002, 53, 138–146. [Google Scholar] [CrossRef]
- Nijmeijer, R.; Willemsen, M.; Meijer, C.J.L.M.; Visser, C.A.; Verheijen, R.H.; Gottlieb, R.A.; Hack, C.E.; Niessen, H.W.M. Type II secretory phospholipase A2 binds to ischemic flip-flopped cardiomyocytes and subsequently induces cell death. Am. J. Physiol. Circ. Physiol. 2003, 285, H2218–H2224. [Google Scholar] [CrossRef]
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Goda, T.; Miyahara, Y. Calcium-independent binding of human C-reactive protein to lysophosphatidylcholine in supported planar phospholipid monolayers. Acta Biomater. 2017, 48, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Kushner, I.; Kaplan, M.H. Studies of acute phase protein: I. An Immunohistochemical method for the localization of cx-reactive protein in rabbits. Association with necrosis in local inflammatory lesions. J. Exp. Med. 1961, 114, 961–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narkates, A.J.; Volanakis, J.E. C-reactive protein binding specificities: Artificial and natural phospholipid bilayers. Ann. N. Y. Acad. Sci. 1982, 389, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Kushner, I.; Rakita, L.; Kaplan, M.H. Studies of acute-phase protein. II. Localization of Cx-reactive protein in heart in induced myocardial infarction in rabbits. J. Clin. Investig. 1963, 42, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, B.; Führnrohr, B.; Müller, R.; Sheriff, A. CRP and the disposal of dying cells: Consequences for systemic lupus erythematosus and rheumatoid arthritis. Autoimmunity 2007, 40, 295–298. [Google Scholar] [CrossRef]
- Gaboriaud, C.; Juanhuix, J.; Gruez, A.; Lacroix, M.; Darnault, C.; Pignol, D.; Verger, D.; Fontecilla-Camps, J.C.; Arlaud, G.J. The Crystal Structure of the Globular Head of Complement Protein C1q Provides a Basis for Its Versatile Recognition Properties. J. Biol. Chem. 2003, 278, 46974–46982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, R.; Kemp, J.A.; Sabin, C.; Pepys, M.B. Human C-Reactive Protein Increases Cerebral Infarct Size after Middle Cerebral Artery Occlusion in Adult Rats. Br. J. Pharmacol. 2004, 24, 1214–1218. [Google Scholar] [CrossRef] [Green Version]
- Hack, C.; Wolbink, G.-J.; Schalkwijk, C.; Speijer, H.; Hermens, W.T.; Bosch, H.V.D. A role for secretory phospholipase A2 and C-reactive protein in the removal of injured cells. Immunol. Today 1997, 18, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Griselli, M.; Herbert, J.; Hutchinson, W.; Taylor, K.; Sohail, M.; Krausz, T.; Pepys, M.B. C-Reactive Protein and Complement Are Important Mediators of Tissue Damage in Acute Myocardial Infarction. J. Exp. Med. 1999, 190, 1733–1740. [Google Scholar] [CrossRef] [Green Version]
- Nijmeijer, R.; Lagrand, W.K.; Lubbers, Y.T.P.; Visser, C.A.; Meijer, C.J.L.M.; Niessen, H.W.M.; Hack, C.E. C-Reactive Protein Activates Complement in Infarcted Human Myocardium. Am. J. Pathol. 2003, 163, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Sheriff, A.; Schindler, R.; Vogt, B.; Abdel-Aty, H.; Unger, J.K.; Bock, C.; Gebauer, F.; Slagman, A.; Jerichow, T.; Mans, D.; et al. Selective apheresis of C-reactive protein: A new therapeutic option in myocardial infarction? J. Clin. Apher. 2014, 30, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Slagman, A.; Bock, C.; Abdel-Aty, H.; Vogt, B.; Gebauer, F.; Janelt, G.; Wohlgemuth, F.; Morgenstern, R.; Yapici, G.; Puppe, A.; et al. Specific Removal of C-Reactive Protein by Apheresis in a Porcine Cardiac Infarction Model. Blood Purif. 2011, 31, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepys, M.B.; Hirschfield, G.M.; Tennent, G.A.; Gallimore, J.R.; Kahan, M.C.; Bellotti, V.; Hawkins, P.N.; Myers, R.M.; Smith, M.D.; Polara, A.; et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature 2006, 440, 1217–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ries, W.; Heigl, F.; Garlichs, C.; Sheriff, A.; Torzewski, J. Die CRP-Apherese: Eine neue Therapiemöglichkeit bei Inflammation; Nephro-News, Medicom VerlagsgmbH Bruck: Mur, Austria, 2019; pp. 23–27. [Google Scholar]
- Ries, W.; Heigl, F.; Garlichs, C.; Sheriff, A.; Torzewski, J. Selective C-Reactive Protein-Apheresis in Patients. Ther. Apher. Dial. 2019, 23, 570–574. [Google Scholar] [CrossRef]
- Braig, D.; Nero, T.L.; Koch, H.-G.; Kaiser, B.; Wang, X.; Thiele, J.R.; Morton, C.J.; Zeller, J.; Kiefer, J.; Potempa, L.A.; et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat. Commun. 2017, 8, 14188. [Google Scholar] [CrossRef] [Green Version]
- Thiele, J.R.; Habersberger, J.; Braig, D.; Schmidt, Y.; Goerendt, K.; Maurer, V.; Bannasch, H.; Scheichl, A.; Woollard, K.J.; Von Dobschütz, E.; et al. Dissociation of Pentameric to Monomeric C-Reactive Protein Localizes and Aggravates Inflammation. Circulation 2014, 130, 35–50. [Google Scholar] [CrossRef] [Green Version]
- McFadyen, J.D.; Kiefer, J.; Braig, D.; Loseff-Silver, J.; Potempa, L.A.; Eisenhardt, S.U.; Peter, K. Dissociation of C-Reactive Protein Localizes and Amplifies Inflammation: Evidence for a Direct Biological Role of C-Reactive Protein and Its Conformational Changes. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, H.-Y.; Li, W.; Shen, Z.-Y.; Wang, Y.-D.; Ji, S.-R.; Wu, Y. An ELISA Assay for Quantifying Monomeric C-Reactive Protein in Plasma. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Thiele, J.R.; Zeller, J.; Bannasch, H.; Stark, G.B.; Peter, K.; Eisenhardt, S.U. Targeting C-Reactive Protein in Inflammatory Disease by Preventing Conformational Changes. Mediat. Inflamm. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Caprio, V.; Badimon, L.; Di Napoli, M.; Fang, W.-H.; Ferris, G.R.; Guo, B.; Iemma, R.S.; Liu, D.; Zeinolabediny, Y.; Slevin, M. pCRP-mCRP Dissociation Mechanisms as Potential Targets for the Development of Small-Molecule Anti-Inflammatory Chemotherapeutics. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef]
- Prasad, K. C-Reactive Protein (CRP)-Lowering Agents. Cardiovasc. Drug Rev. 2006, 24, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Mattecka, S.; Brunner, P.; Hähnel, B.; Kunze, R.; Vogt, B.; Sheriff, A. PentraSorb C-Reactive Protein: Characterization of the Selective C-Reactive Protein Adsorber Resin. Ther. Apher. Dial. 2019, 23, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boljevic, D.; Nikolic, A.; Rusovic, S.; Lakcevic, J.; Bojic, M.; Balint, B. A Promising Innovative Treatment for ST-Elevation Myocardial Infarction: The Use of C-Reactive Protein Selective Apheresis: Case Report. Blood Purif. 2020, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ries, W.; Sheriff, A.; Heigl, F.; Zimmermann, O.; Garlichs, C.D.; Torzewski, J. “First in Man”: Case Report of Selective C-Reactive Protein Apheresis in a Patient with Acute ST Segment Elevation Myocardial Infarction. Case Rep. Cardiol. 2018, 2018, 1–4. [Google Scholar] [CrossRef]
- Torzewski, J.; Heigl, F.; Zimmermann, O.; Wagner, F.; Schumann, C.; Hettich, R.; Bock, C.; Kayser, S.; Sheriff, A. First-in-Man: Case Report of Selective C-Reactive Protein Apheresis in a Patient with SARS-CoV-2 Infection. Am. J. Case Rep. 2020, 21, e925020. [Google Scholar]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- Heusch, G.; Gersh, B.J. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: A continual challenge. Eur. Hear. J. 2016, 38, 774–784. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Boil. 2012, 298, 229–317. [Google Scholar] [CrossRef] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Pegues, M.A.; McCrory, M.A.; Zarjou, A.; Szalai, A.J. C-reactive protein exacerbates renal ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2013, 304, F1358–F1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valtchanova-Matchouganska, A.; Gondwe, M.; Nadar, A. The role of C-reactive protein in ischemia/reperfusion injury and preconditioning in a rat model of myocardial infarction. Life Sci. 2004, 75, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Stone, G.W.; Selker, H.P.; Thiele, H.; Patel, M.R.; Udelson, J.E.; Ohman, E.; Maehara, A.; Eitel, I.; Granger, C.B.; Jenkins, P.L.; et al. Relationship between Infarct Size and Outcomes Following Primary PCI. J. Am. Coll. Cardiol. 2016, 67, 1674–1683. [Google Scholar] [CrossRef]
- De Waha, S.; Patel, M.R.; Granger, C.B.; Ohman, E.M.; Maehara, A.; Eitel, I.; Ben-Yehuda, O.; Jenkins, P.; Thiele, H.; Stone, G.W. Relationship between microvascular obstruction and adverse events following primary percutaneous coronary intervention for ST-segment elevation myocardial infarction: An individual patient data pooled analysis from seven randomized trials. Eur. Hear. J. 2017, 38, 3502–3510. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Regulation of the Inflammatory Response in Cardiac Repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Danesh, J.; Wheeler, J.G.; Hirschfield, G.M.; Eda, S.; Eiriksdottir, G.; Rumley, A.; Lowe, G.D.O.; Pepys, M.B.; Gudnason, V. C-Reactive Protein and Other Circulating Markers of Inflammation in the Prediction of Coronary Heart Disease. N. Engl. J. Med. 2004, 350, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Koenig, W.; Sund, M.; Fröhlich, M.; Fischer, H.G.; Löwel, H.; Döring, A.; Hutchinson, W.L.; Pepys, M.B. C-Reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: Results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation 1999, 99, 237–242. [Google Scholar] [PubMed] [Green Version]
- Verma, S.; Szmitko, P.E.; Ridker, P.M. C-reactive protein comes of age. Nat. Clin. Pract. Neurol. 2005, 2, 29–36. [Google Scholar] [CrossRef]
- Beranek, J.T. C-reactive protein and complement in myocardial infarction and postinfarction heart failure. Eur. Hear. J. 1997, 18, 1834–1835. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Qi, X.; Li, Q.; Jia, W.; Wei, L.; Huang, A.; Liu, K.; Li, Z. Increased complements and high-sensitivity C-reactive protein predict heart failure in acute myocardial infarction. Biomed. Rep. 2016, 5, 761–765. [Google Scholar] [CrossRef]
- Mani, P.; Puri, R.; Schwartz, G.G.; Nissen, S.E.; Shao, M.; Kastelein, J.J.P.; Menon, V.; Lincoff, A.M.; Nicholls, S.J. Association of Initial and Serial C-Reactive Protein Levels With Adverse Cardiovascular Events and Death After Acute Coronary Syndrome. JAMA Cardiol. 2019, 4, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suleiman, M.; Khatib, R.; Agmon, Y.; Mahamid, R.; Boulos, M.; Kapeliovich, M.; Levy, Y.; Beyar, R.; Markiewicz, W.; Hammerman, H.; et al. Early Inflammation and Risk of Long-Term Development of Heart Failure and Mortality in Survivors of Acute Myocardial Infarction. J. Am. Coll. Cardiol. 2006, 47, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, C.; Sheriff, A.; Zimmermann, S.; Schaefauer, L.; Schlundt, C.; Raaz, D.; Garlichs, C.D.; Achenbach, S. C-reactive protein levels predict systolic heart failure and outcome in patients with first ST-elevation myocardial infarction treated with coronary angioplasty. Arch. Med. Sci. 2017, 5, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Reindl, M.; Reinstadler, S.J.; Feistritzer, H.-J.; Klug, G.; Tiller, C.; Mair, J.; Mayr, A.; Jaschke, W.; Metzler, B. Relation of inflammatory markers with myocardial and microvascular injury in patients with reperfused ST-elevation myocardial infarction. Eur. Hear. J. Acute Cardiovasc. Care 2016, 6, 640–649. [Google Scholar] [CrossRef]
- Mevorach, D.; Mascarenhas, J.O.; Gershov, D.; Elkon, K.B. Complement-dependent Clearance of Apoptotic Cells by Human Macrophages. J. Exp. Med. 1998, 188, 2313–2320. [Google Scholar] [CrossRef]
- Lagrand, W.K.; Niessen, H.W.M.; Wolbink, G.-J.; Jaspars, L.H.; Visser, C.A.; Verheugt, F.W.; Meijer, C.J.; Hack, C.E. C-Reactive Protein Colocalizes With Complement in Human Hearts During Acute Myocardial Infarction. Circulation 1997, 95, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.D.; Hennan, J.K.; Marks, R.M.; Lucchesi, B.R. C-Reactive-Protein-Associated Increase in Myocardial Infarct Size after Ischemia/Reperfusion. J. Pharmacol. Exp. Ther. 2002, 303, 1007–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krijnen, P.A.; Meischl, C.; Nijmeijer, R.; Visser, C.A.; Hack, C.E.; Niessen, H.W. Inhibition of sPLA2-IIA, C-reactive protein or complement: New therapy for patients with acute myocardial infarction? Cardiovasc. Hematol. Disord. Drug Targets 2006, 6, 113–123. [Google Scholar] [CrossRef]
- Heinecke, J.W. Chemical knockout of C-reactive protein in cardiovascular disease. Nat. Methods 2006, 2, 300–301. [Google Scholar] [CrossRef]
- Kitsis, R.N.; Jialal, I. Limiting Myocardial Damage during Acute Myocardial Infarction by Inhibiting C-Reactive Protein. N. Engl. J. Med. 2006, 355, 513–515. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Catanese, L.; Tarsia, J.; Fisher, M. Acute Ischemic Stroke Therapy Overview. Circ. Res. 2017, 120, 541–558. [Google Scholar] [CrossRef] [PubMed]
- Anrather, J.; Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Muir, K.W.; Tyrrell, P.; Sattar, N.; Warburton, E. Inflammation and ischaemic stroke. Curr. Opin. Neurol. 2007, 20, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Montaner, J.; Fernández-Cadenas, I.; Molina, C.A.; Ribo, M.; Huertas, R.; Rosell, A.; Penalba, A.; Ortega, L.; Chacoón, P.; Álvarez-Sabín, J. Poststroke C-Reactive Protein Is a Powerful Prognostic Tool Among Candidates for Thrombolysis. Stroke 2006, 37, 1205–1210. [Google Scholar] [CrossRef] [Green Version]
- Winbeck, K.; Poppert, H.; Etgen, T.; Conrad, B.; Sander, D. Prognostic relevance of early serial C-reactive protein measurements after first ischemic stroke. Stroke 2002, 33, 2459–2464. [Google Scholar] [CrossRef]
- Arenillas, J.F.; Álvarez-Sabín, J.; Molina, C.A.; Chacoón, P.; Montaner, J.; Rovira, A.; Ibarra, B.; Quintana, M. C-Reactive Protein Predicts Further Ischemic Events in First-Ever Transient Ischemic Attack or Stroke Patients with Intracranial Large-Artery Occlusive Disease. Stroke 2003, 34, 2463–2468. [Google Scholar] [CrossRef] [Green Version]
- Di Napoli, M.; Papa, F.; Bocola, V. Prognostic Influence of Increased C-Reactive Protein and Fibrinogen Levels in Ischemic Stroke. Stroke 2001, 32, 133–138. [Google Scholar] [CrossRef]
- Elkind, M.S.V.; Tai, W.; Coates, K.; Paik, M.C.; Sacco, R.L. High-Sensitivity C-Reactive Protein, Lipoprotein-Associated Phospholipase A2, and Outcome After Ischemic Stroke. Arch. Intern. Med. 2006, 166, 2073–2080. [Google Scholar] [CrossRef]
- Woodward, M.; Lowe, G.D.; Campbell, D.J.; Colman, S.; Rumley, A.; Chalmers, J.P.; Neal, B.C.; Patel, A.; Jenkins, A.J.; E Kemp, B.; et al. Associations of Inflammatory and Hemostatic Variables with the Risk of Recurrent Stroke. Stroke 2005, 36, 2143–2147. [Google Scholar] [CrossRef]
- Muir, K.W.; Weir, C.J.; Alwan, W.; Squire, I.B.; Lees, K.R. C-reactive protein and outcome after ischemic stroke. Stroke 1999, 30, 981–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringleb, P.A.; Bousser, M.-G.; Ford, G.; Bath, P.; Brainin, M.; Caso, V.; Cervera, Á.; Chamorro, A.; Cordonnier, C.; Csiba, L.; et al. Ischaemic Stroke and Transient Ischaemic Attack. In European Handbook of Neurological Management; Wiley: Hoboken, NJ, USA, 2010; pp. 101–158. [Google Scholar]
- Kayser, S.; Kunze, R.; Sheriff, A. Selective C-reactive protein apheresis for Covid-19 patients suffering from organ damage. Ther. Apher. Dial. 2020. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, M.; Schwaninger, M.; Cappelli, R.; Ceccarelli, E.; Di Gianfilippo, G.; Donati, C.; Emsley, H.; Forconi, S.; Hopkins, S.J.; Masotti, L.; et al. Evaluation of C-Reactive Protein Measurement for Assessing the Risk and Prognosis in Ischemic Stroke. Stroke 2005, 36, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.A.E.; Visseren, F.L.; Grobbee, D.E. Screening for C-reactive protein in CVD prediction. Nat. Rev. Cardiol. 2012, 10, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, C.R.; Librizzi, L.; Closhen, D.; Pflanzner, T.; Lessmann, V.; Pietrzik, C.U.; De Curtis, M.; Luhmann, H.J. Mechanisms of C-Reactive Protein-Induced Blood–Brain Barrier Disruption. Stroke 2009, 40, 1458–1466. [Google Scholar] [CrossRef] [Green Version]
- Elwood, E.; Lim, Z.; Naveed, H.; Galea, I. The effect of systemic inflammation on human brain barrier function. Brain Behav. Immun. 2017, 62, 35–40. [Google Scholar] [CrossRef]
- Lasek-Bal, A.; Jedrzejowska-Szypulka, H.; Student, S.; Warsz-Wianecka, A.; Zareba, K.; Puz, P.; Bal, W.; Pawletko, K.; Lewin-Kowalik, J. The importance of selected markers of inflammation and blood-brain barrier damage for short-term ischemic stroke prognosis. J. Psysiol. Pharmacol. 2019, 70, 209–217. [Google Scholar]
- Khandkar, C.; Vaidya, K.; Patel, S. Colchicine for Stroke Prevention: A Systematic Review and Meta-analysis. Clin. Ther. 2019, 41, 582–590.e3. [Google Scholar] [CrossRef]
- Dove, A. CD18 trials disappoint again. Nat. Biotechnol. 2000, 18, 817–818. [Google Scholar] [CrossRef]
- Pawluk, H.; Woźniak, A.; Grześk, G.; Kołodziejska, R.; Kozakiewicz, M.; Kopkowska, E.; Grzechowiak, E.; Kozera, G. The Role of Selected Pro-Inflammatory Cytokines in Pathogenesis of Ischemic Stroke. Clin. Interv. Aging 2020, 15, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Mizuma, A.; Yenari, M.A. Anti-Inflammatory Targets for the Treatment of Reperfusion Injury in Stroke. Front. Neurol. 2017, 8, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drieu, A.; Levard, D.; Vivien, D.; Rubio, M. Anti-inflammatory treatments for stroke: From bench to bedside. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418789854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.; Demello, V.; Sobel, B.E. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation 1976, 53, 204–206. [Google Scholar]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kayser, S.; Brunner, P.; Althaus, K.; Dorst, J.; Sheriff, A. Selective Apheresis of C-Reactive Protein for Treatment of Indications with Elevated CRP Concentrations. J. Clin. Med. 2020, 9, 2947. https://doi.org/10.3390/jcm9092947
Kayser S, Brunner P, Althaus K, Dorst J, Sheriff A. Selective Apheresis of C-Reactive Protein for Treatment of Indications with Elevated CRP Concentrations. Journal of Clinical Medicine. 2020; 9(9):2947. https://doi.org/10.3390/jcm9092947
Chicago/Turabian StyleKayser, Stefan, Patrizia Brunner, Katharina Althaus, Johannes Dorst, and Ahmed Sheriff. 2020. "Selective Apheresis of C-Reactive Protein for Treatment of Indications with Elevated CRP Concentrations" Journal of Clinical Medicine 9, no. 9: 2947. https://doi.org/10.3390/jcm9092947
APA StyleKayser, S., Brunner, P., Althaus, K., Dorst, J., & Sheriff, A. (2020). Selective Apheresis of C-Reactive Protein for Treatment of Indications with Elevated CRP Concentrations. Journal of Clinical Medicine, 9(9), 2947. https://doi.org/10.3390/jcm9092947