Targeting β2-Adrenergic Receptors Shows Therapeutical Benefits in Clear Cell Renal Cell Carcinoma from Von Hippel–Lindau Disease

 , , and
, , and 
Abstract
:1. Introduction
2. Results
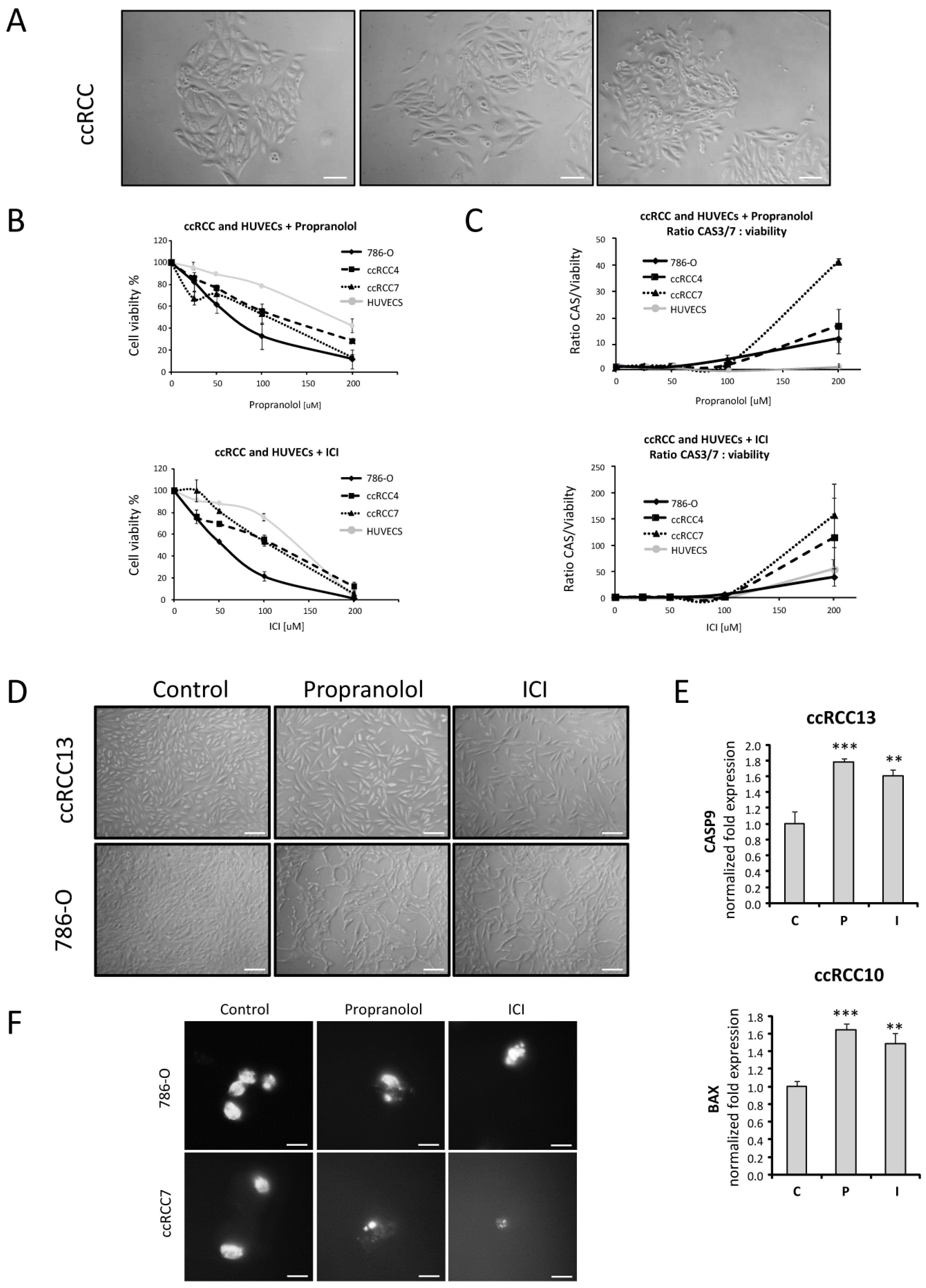
2.1. Isolation and Cultivation of Primary ccRCC Tumor Cultures from Surgical Specimens
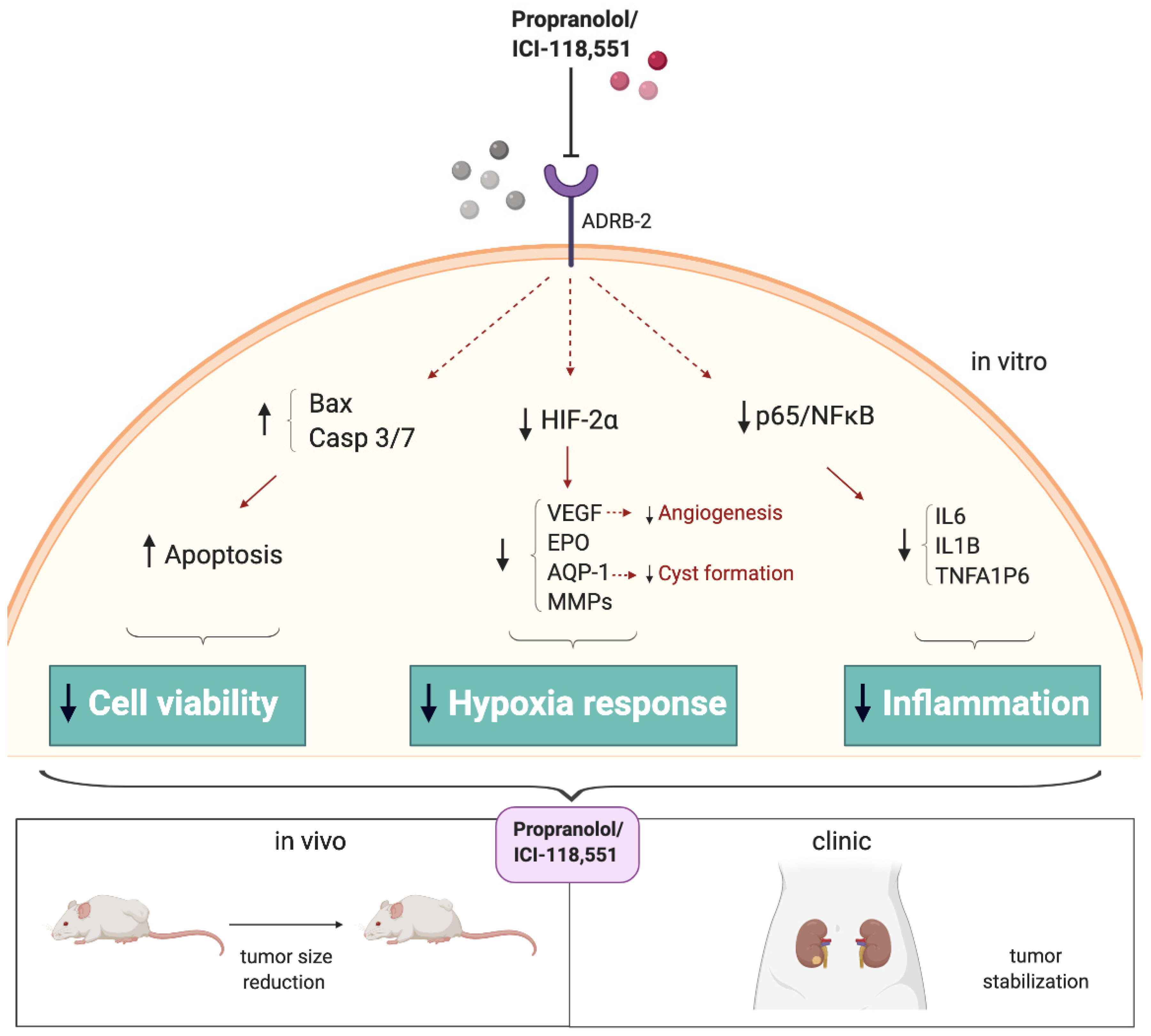
2.2. Cell Viability of ccRCC Primary Tumors and RCC 786-O Cell Line Is Impaired after ADRB Blockers Treatment by Triggering Apoptosis
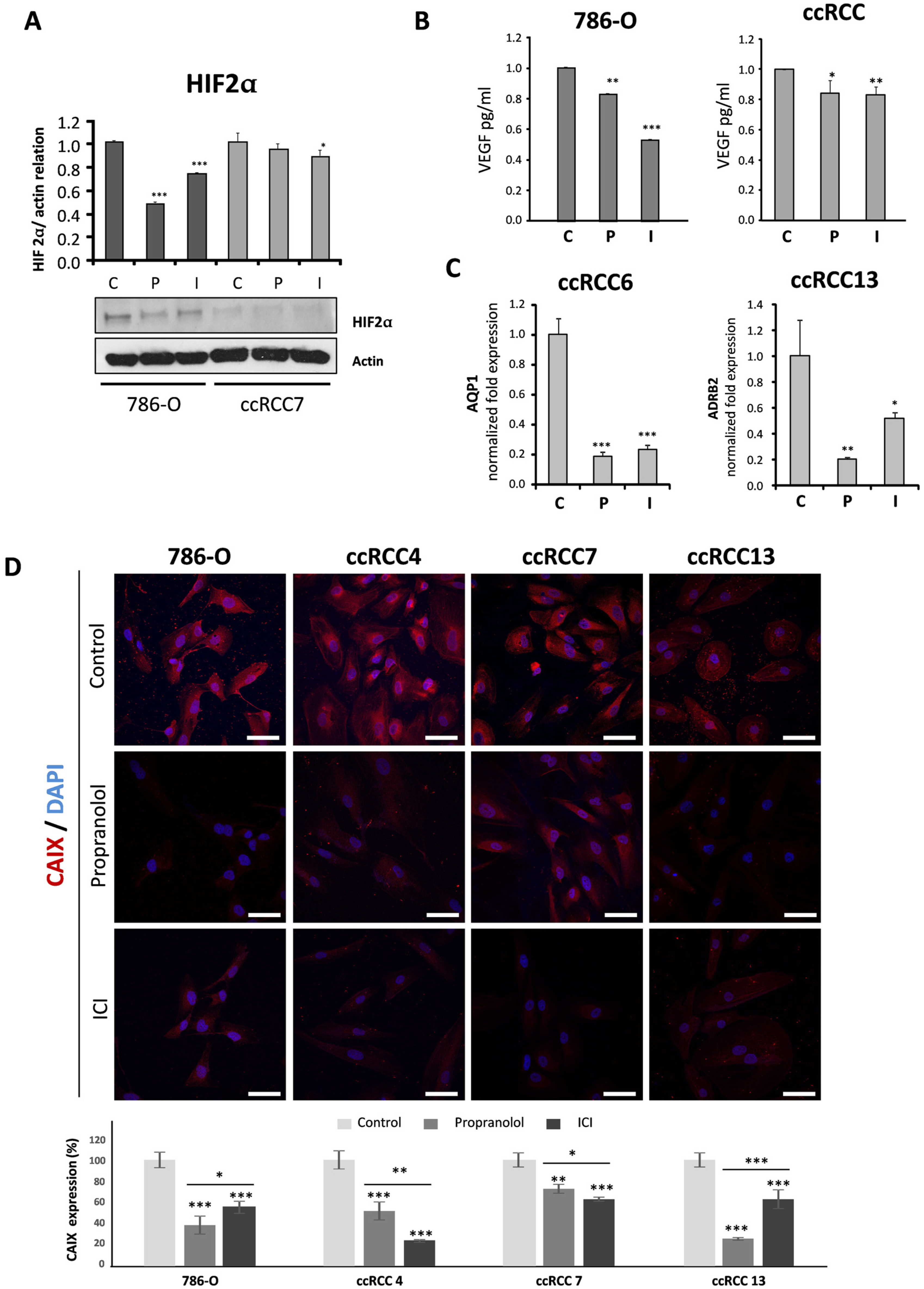
2.3. Propranolol and ICI-118,551 Decrease HIF, VEGF, and CAIX Expression
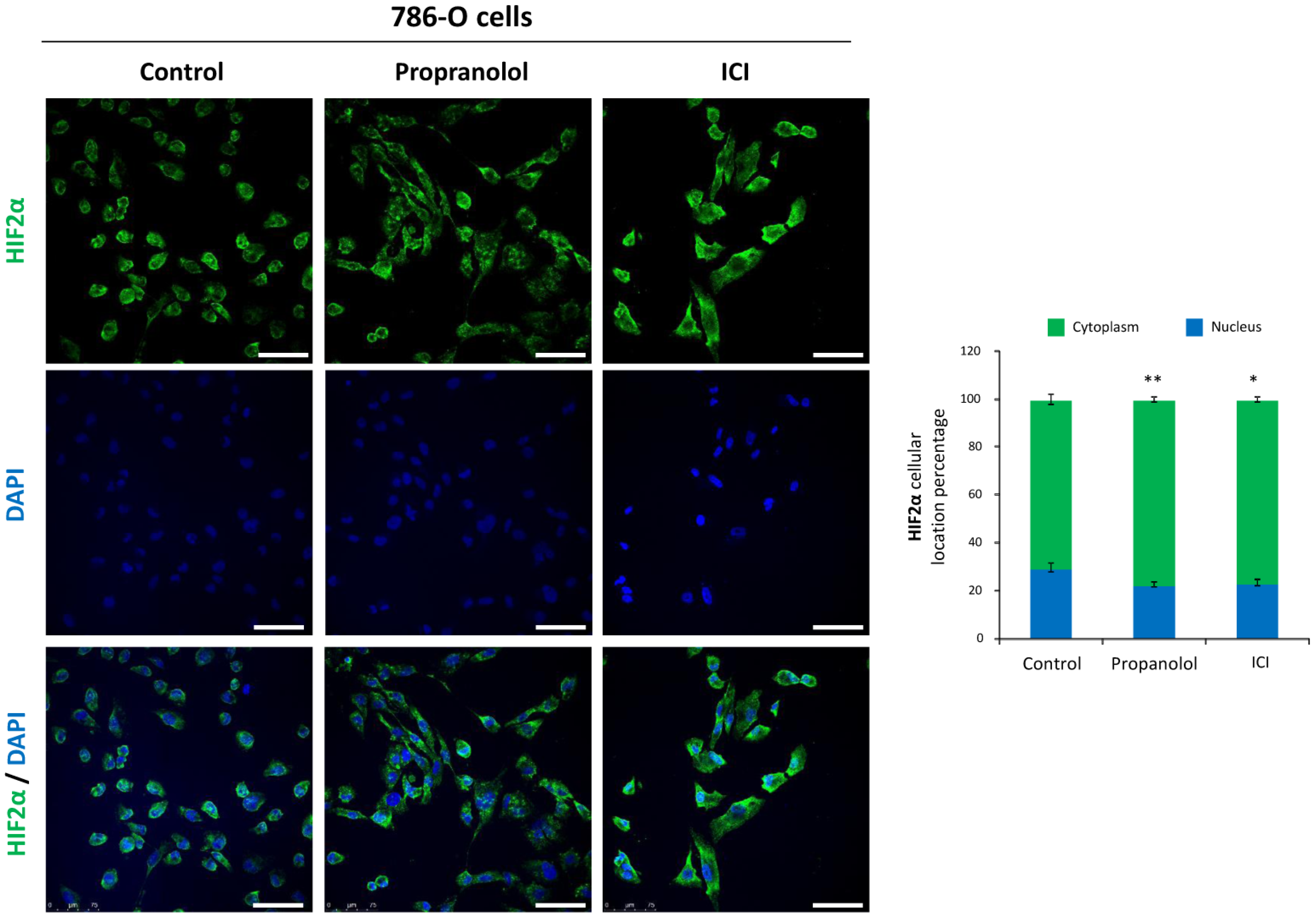
2.4. HIF-2α Cell Distribution Is Altered by the ADRB Antagonists
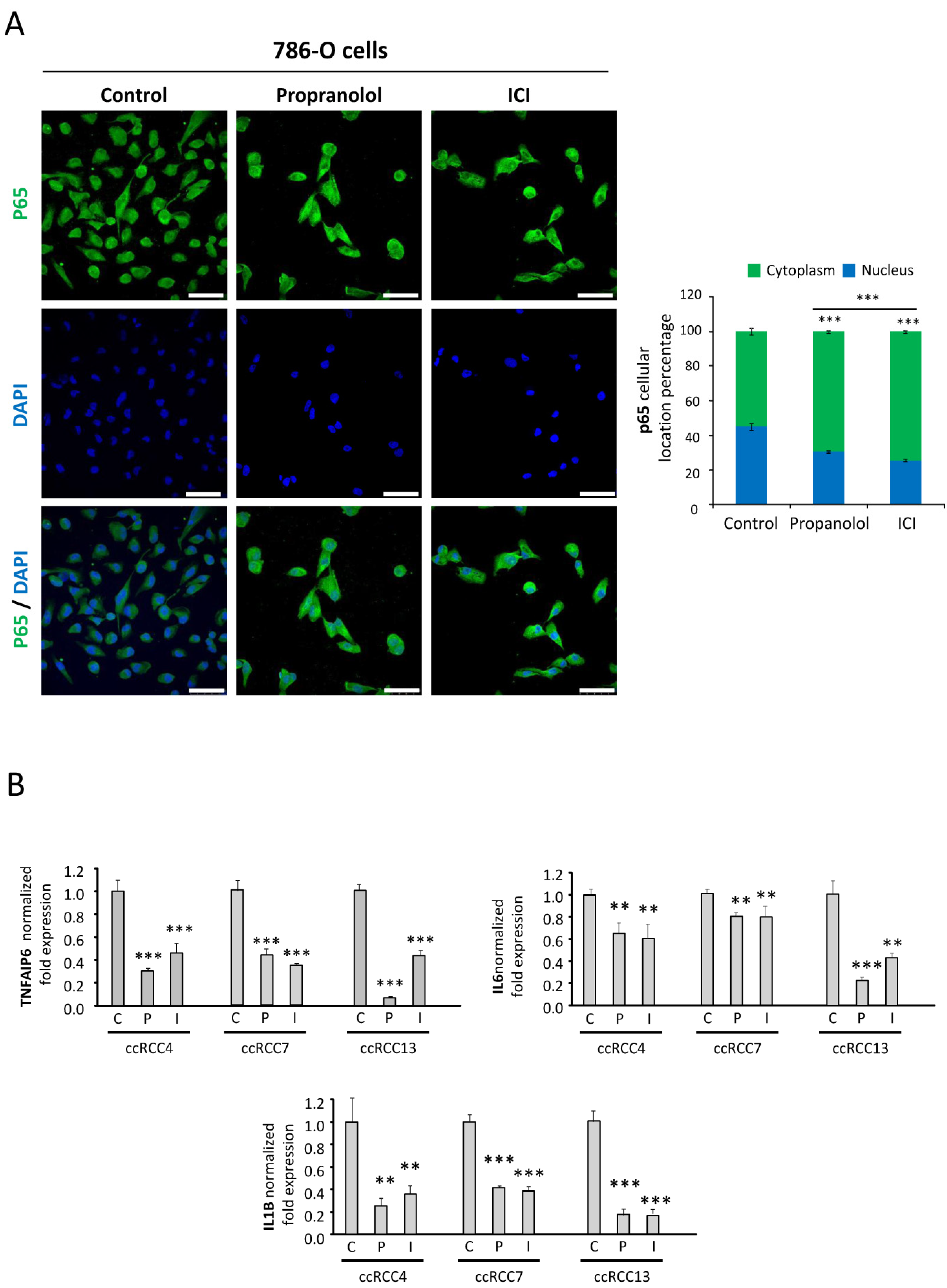
2.5. NFκB-p65 Pathway Is Targeted by the ADRB Blockers
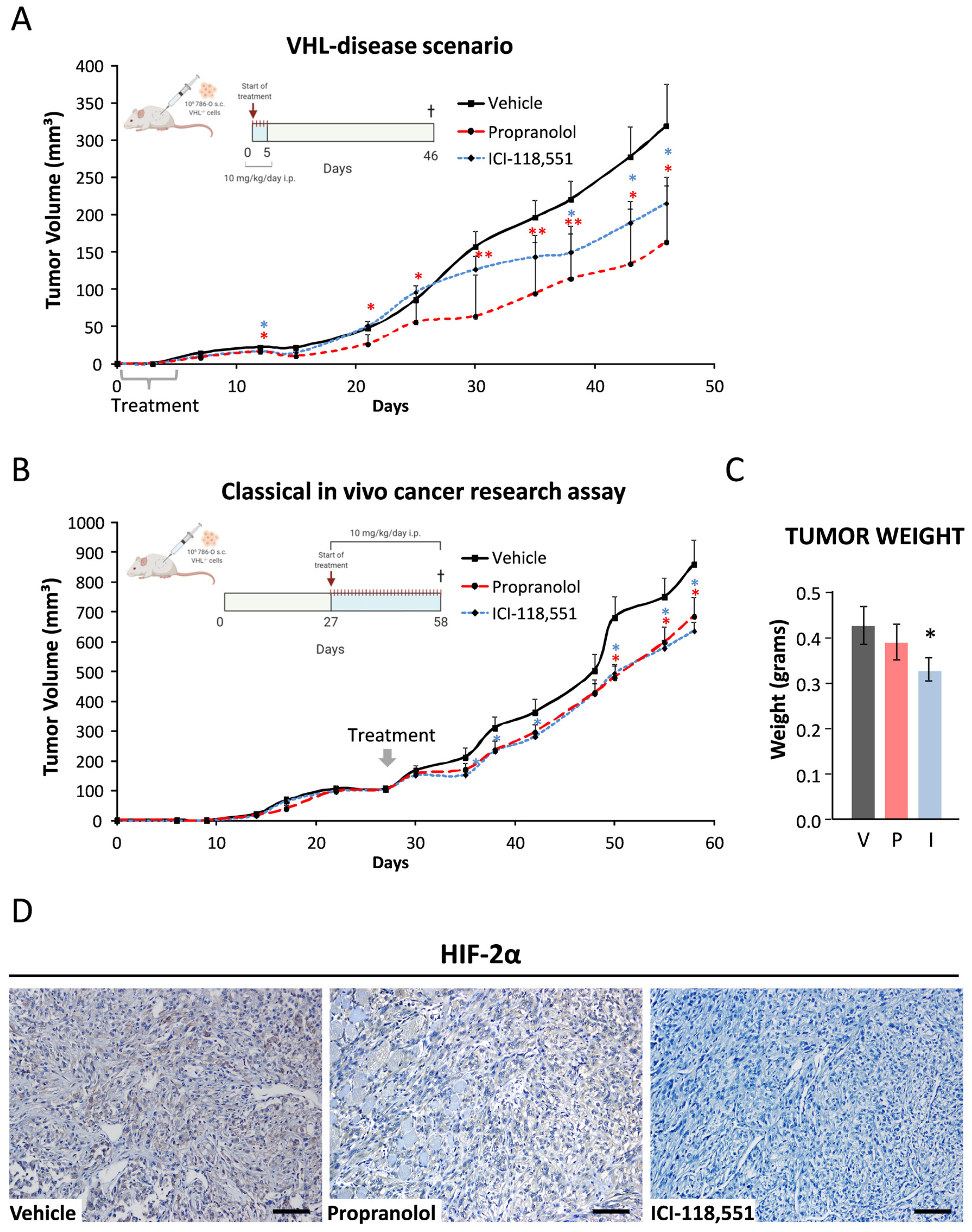
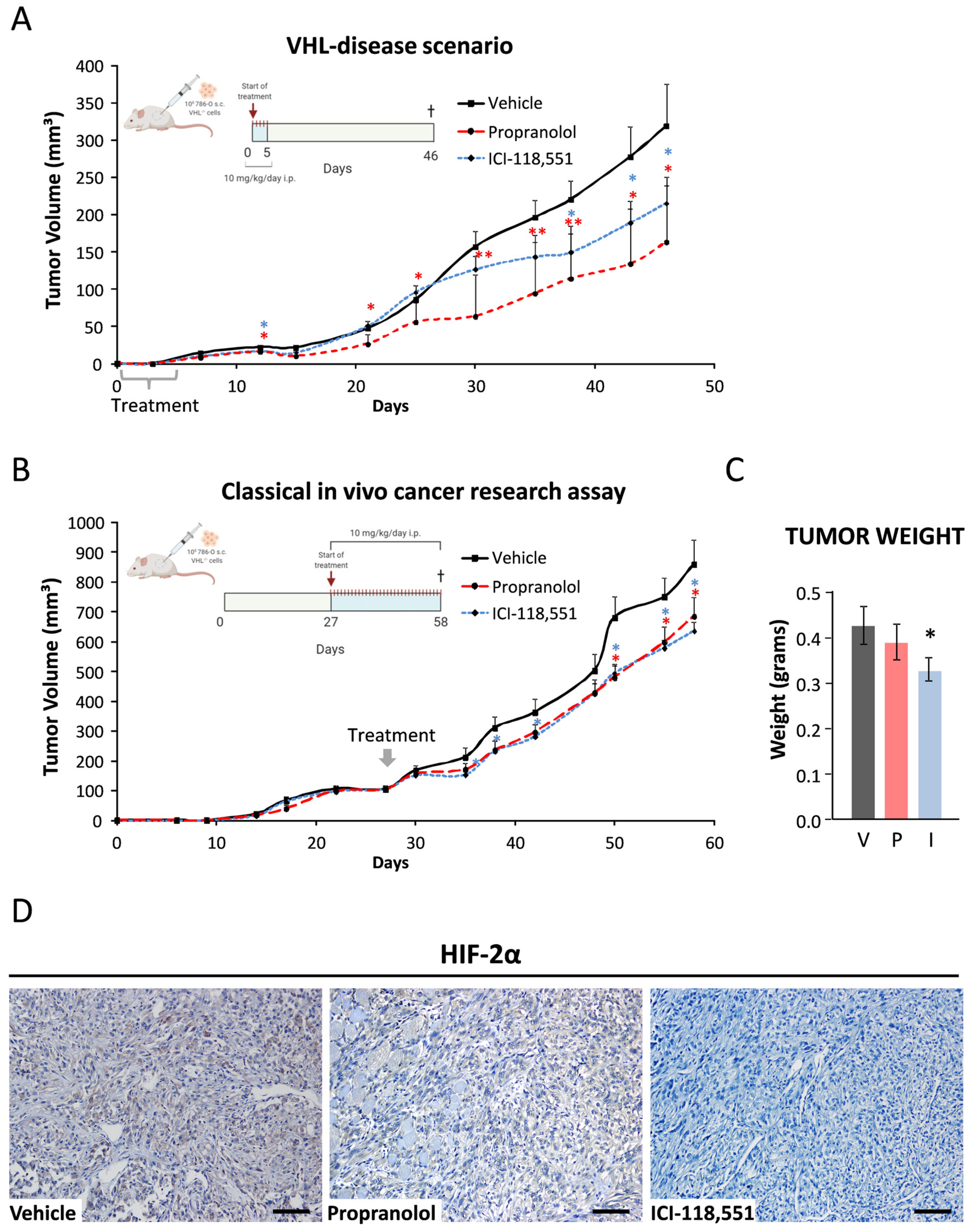
2.6. ICI-118,551 and Propranolol Treatment Decreases the Growth Rate of ccRCC VHL−/− 786-O Tumor Xenograft in NSG Mice
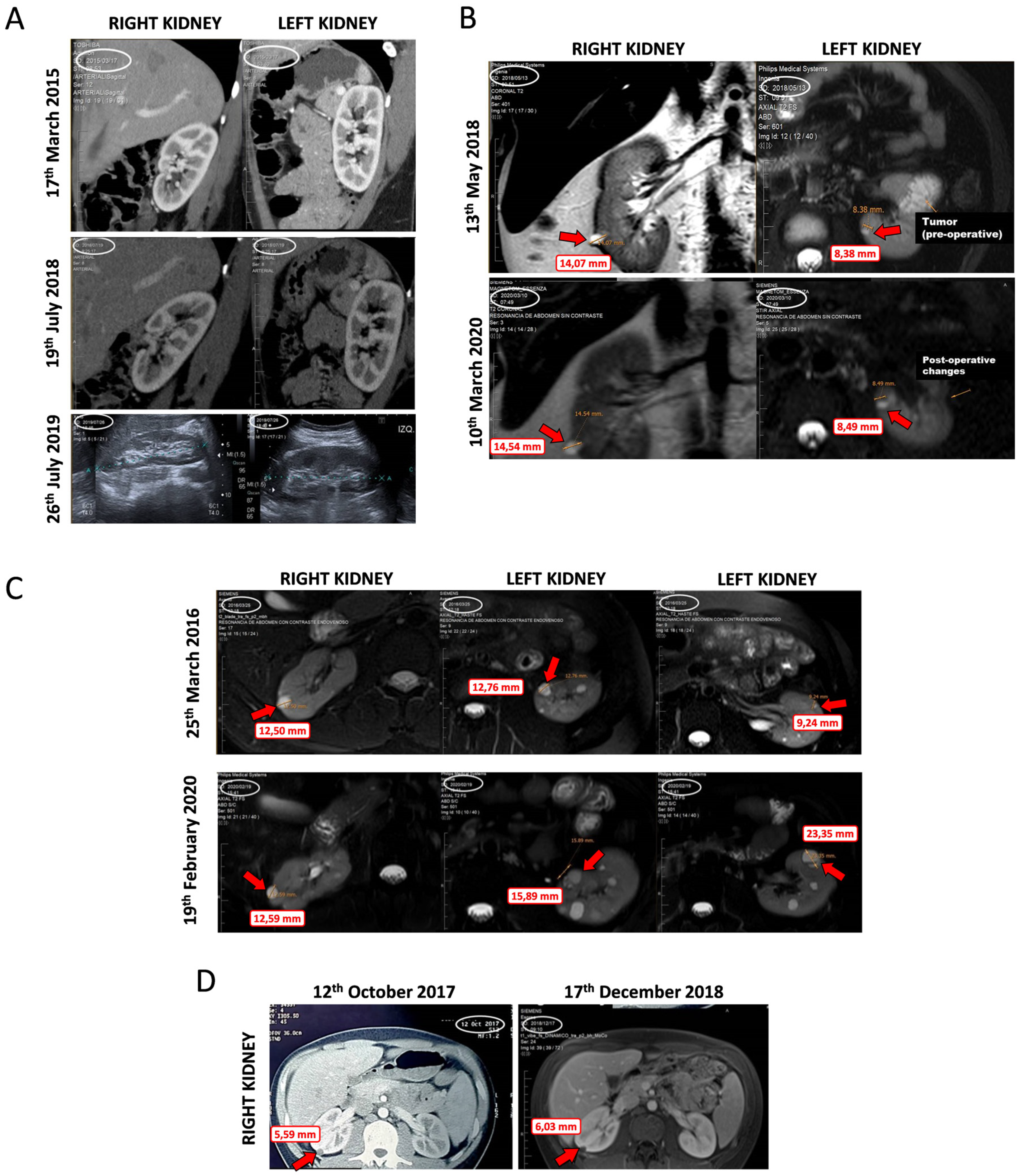
2.7. Data from VHL Patients Treated with Propranolol as Off-Label Prescription
3. Discussion
4. Materials and Methods
4.1. Ethics Approval and Consent to Participate
4.2. Cell Culture and Treatments
4.3. Cell Viability Assay
4.4. Caspase Activation Assay
4.5. Real-Time Quantitative PCR
4.6. Western Blot Analysis for HIF-2α
4.7. VEGF Determination
4.8. Immunofluorescence Microscopy
4.9. In Vivo Tumor Xenografts
4.10. Immunohistochemistry
4.11. Clinical Data from Patients
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Shuin, T.; Yamasaki, I.; Tamura, K.; Okuda, H.; Furihata, M.; Ashida, S. Von Hippel-Lindau disease: Molecular pathological basis, clinical criteria, genetic testing, clinical features of tumors and treatment. Jpn. J. Clin. Oncol. 2006, 36, 337–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, E.R.; Neumann, H.P.H.; Richard, S. Von Hippel-Lindau disease: A clinical and scientific review. Eur. J. Hum. Genet. 2011, 19, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Bader, H.L.; Hsu, T. Systemic VHL gene functions and the VHL disease. FEBS. Lett. 2012, 586, 1562–1569. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, M.; Foulkes, W.D. Pheochromocytoma and paraganglioma syndromes: Genetics and management update. Curr. Oncol. 2014, 21, e8–e17. [Google Scholar] [CrossRef]
- Richard, S.; Gardie, B.; Couvé, S.; Gad, S. Von Hippel-Lindau: How a rare disease illuminates cancer biology. Semin. Cancer. Biol. 2013, 23, 26–37. [Google Scholar] [CrossRef]
- Vortmeyer, A.O.; Falke, E.A.; Gläsker, S.; Li, J.; Oldfield, E.H. Nervous system involvement in von Hippel-Lindau disease: Pathology and mechanisms. Acta. Neuropathol. 2013, 125, 333–350. [Google Scholar] [CrossRef]
- Neumann, H.P.H.; Bender, B.U.; Berger, D.P.; Laubenberger, J.; Schultze-Seemann, W.; Wetterauer, U.; Ferstl, F.J.; Herbst, E.W.; Schwarzkopf, G.; Hes, F.J.; et al. Prevalence, morphology and biology of renal cell carcinoma in von Hippel-Lindau disease compared to sporadic renal cell carcinoma. J. Urol. 1998, 160, 1248–1254. [Google Scholar] [CrossRef]
- Bausch, B.; Jilg, C.; Gläsker, S.; Vortmeyer, A.; Lützen, N.; Anton, A.; Eng, C.; Neumann, H.P.H. Renal cancer in von Hippel-Lindau disease and related syndromes. Nat. Rev. Nephrol. 2013, 9, 529–538. [Google Scholar] [CrossRef]
- Montani, M.; Heinimann, K.; Von Teichman, A.; Rudolph, T.; Perren, A.; Moch, H. VHL-gene deletion in single renal tubular epithelial cells and renal tubular cysts: Further evidence for a cyst-dependent progression pathway of clear cell renal carcinoma in von hippel-lindau disease. Am. J. Surg. Pathol. 2010, 34, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Joosten, S.C.; Smits, K.M.; Aarts, M.J.; Melotte, V.; Koch, A.; Tjan-Heijnen, V.C.; Van Engeland, M. Epigenetics in renal cell cancer: Mechanisms and clinical applications. Nat. Rev. Urol. 2018, 15, 430–451. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Brunet, J.P.; Di Napoli, A.; Mertz, K.D.; Seeley, A.; Pires, M.M.; Linhart, D.; Worrell, R.A.; Moch, H.; Rubin, M.A.; et al. Patterns of gene expression and copy-number alterations in von-Hippel Lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009, 69, 4674–4681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capitanio, J.F.; Mazza, E.; Motta, M.; Mortini, P.; Reni, M. Corrigendum to “Mechanisms, indications and results of salvage systemic therapy for sporadic and von Hippel-Lindau related hemangioblastomas of the central nervous system” [Crit. Rev. Oncol. Hematol. 86 (2013) 69–84]. Crit. Rev. Oncol. Hematol. 2014, 90, 91. [Google Scholar] [CrossRef]
- Jonasch, E.; McCutcheon, I.E.; Gombos, D.S.; Ahrar, K.; Perrier, N.D.; Liu, D.; Robichaux, C.C.; Villarreal, M.F.; Weldon, J.A.; Woodson, A.H.; et al. Pazopanib in patients with von Hippel-Lindau disease: A single-arm, single-centre, phase 2 trial. Lancet Oncol. 2018, 19, 1351–1359. [Google Scholar] [CrossRef]
- Albiñana, V.; De Las-Heras, K.V.G.; Serrano-Heras, G.; Segura, T.; Perona-Moratalla, A.B.; Mota-Pérez, M.; De Campos, J.M.; Botella, L.M. Propranolol reduces viability and induces apoptosis in hemangioblastoma cells from von Hippel-Lindau patients. Orphanet. J. Rare Dis. 2015, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cifola, I.; Bianchi, C.; Mangano, E.; Bombelli, S.; Frascati, F.; Fasoli, E.; Ferrero, S.; Di Stefano, V.; Zipeto, M.A.; Magni, F.; et al. Renal cell carcinoma primary cultures maintain genomic and phenotypic profile of parental tumor tissues. BMC Cancer 2011, 11, 244. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, A.M.; Albiñana, V.; Gallardo-Vara, E.; Recio-Poveda, L.; de Rojas-P, I.; de Las-Heras, K.V.G.; Aguirre, D.T.; Botella, L.M. The β2-adrenergic receptor antagonist ICI-118,551 blocks the constitutively activated HIF signalling in hemangioblastomas from von Hippel-Lindau disease. Sci. Rep. 2019, 9, 10062. [Google Scholar] [CrossRef]
- Albiñana, V.; Escribano, R.M.J.; Soler, I.; Padial, L.R.; Recio-Poveda, L.; De Las-Heras, K.V.G.; Botella, L.M. Repurposing propranolol as a drug for the treatment of retinal haemangioblastomas in von Hippel-Lindau disease. Orphanet. J. Rare. Dis. 2017, 12, 122. [Google Scholar] [CrossRef] [Green Version]
- González-Rodríguez, B.; De Las-Heras, K.V.G.; Aguirre, D.T.; Rodríguez-Padial, L.; Albiñana, V.; Recio-Poveda, L.; Cuesta, A.M.; Botella, L.M.; Jiménez-Escribano, R.M. Evaluation of the safety and effectiveness of oral propranolol in patients with von Hippel-Lindau disease and retinal hemangioblastomas: Phase III clinical trial. BMJ. Open. Ophthalmol. 2019, 4, e000203. [Google Scholar] [CrossRef] [Green Version]
- Treatment of Von Hippel-Lindau (VHL)-Related Hemangioblastoma With PTK787/ZK 222584. Available online: https://clinicaltrials.gov/ct2/show/NCT00052013 (accessed on 31 May 2020).
- Eyetech Study Group. Preclinical and phase 1A clinical evaluation of an anti-VEGF pegylated aptamer (EYE001) for the treatment of exudative age-related macular degeneration. Retina 2002, 22, 143–152. [Google Scholar] [CrossRef] [PubMed]
- 17AAG to Treat Kidney Tumors in Von Hippel-Lindau Disease. Available online: https://clinicaltrials.gov/ct2/show/results/NCT00088374 (accessed on 31 May 2020).
- Wong, W.T.; Liang, K.J.; Hammel, K.; Coleman, H.R.; Chew, E.Y. Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel-Lindau disease. Ophthalmology 2008, 115, 1957–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonasch, E.; McCutcheon, I.E.; Waguespack, S.G.; Wen, S.; Davis, D.W.; Smith, L.A.; Tannir, N.M.; Gombos, D.S.; Fuller, G.N.; Matin, S.F. Pilot trial of sunitinib therapy in patients with von Hippel-Lindau disease. Ann. Oncol. 2011, 22, 2661–2666. [Google Scholar] [CrossRef] [PubMed]
- Evaluation of Sunitinib Malate in Patients With Von Hippel-Lindau Syndrome (VHL) Who Have VHL Lesions to Follow. Available online: https://clinicaltrials.gov/ct2/show/results/NCT00330564 (accessed on 31 May 2020).
- Phase II Study of Vandetanib in Individuals With Kidney Cancer. Available online: https://clinicaltrials.gov/ct2/show/results/NCT00566995 (accessed on 31 May 2020).
- Knickelbein, J.E.; Jacobs-El, N.; Wong, W.T.; Wiley, H.E.; Cukras, C.A.; Meyerle, C.B.; Chew, E.Y. Systemic Sunitinib Malate Treatment for Advanced Juxtapapillary Retinal Hemangioblastomas Associated with von Hippel-Lindau Disease. Ophthalmol. Retin. 2017, 1, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunitinib Malate to Treat Advanced Eye Disease in Patients With Von Hippel-Lindau Syndrome (VHL3). Available online: https://clinicaltrials.gov/ct2/show/results/NCT00673816 (accessed on 31 May 2020).
- Oudard, S.; Elaidi, R.; Brizard, M.; Le Rest, C.; Caillet, V.; Deveaux, S.; Benoit, G.; Corréas, J.M.; Benoudiba, F.; David, P.; et al. Sunitinib for the treatment of benign and malignant neoplasms from von Hippel-Lindau disease: A single-arm, prospective phase II clinical study from the PREDIR group. Oncotarget 2016, 7, 85306–85317. [Google Scholar] [CrossRef] [Green Version]
- Welsh, S.; Fife, K.; Matakidou, A.; Mullin, J.; Machin, A.; Qian, W.; Ingleson, V.; Dalchau, K.M.; Whittaker, P.; Warren, A.; et al. A phase II clinical study evaluating the efficacy and safety of neoadjuvant and adjuvant sunitinib in previously untreated patients with metastatic renal cell carcinoma (mRCC) (NeoSun). J. Clin. Oncol. 2017, 35, e16087. [Google Scholar] [CrossRef]
- Pilié, P.; Hasanov, E.; Matin, S.F.; Woodson, A.H.H.; Marcott, V.D.; Bird, S.; Slack, R.S.; Fuller, G.N.; McCutcheon, I.E.; Jonasch, E. Pilot study of dovitinib in patients with von Hippel-Lindau disease. Oncotarget 2018, 9, 23390–23395. [Google Scholar] [CrossRef] [Green Version]
- TKI 258 in Von Hippel-Lindau Syndrome (VHL). Available online: https://clinicaltrials.gov/ct2/show/results/NCT01266070 (accessed on 31 May 2020).
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.; Maughan, B.J.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Phase II study of the oral HIF-2α inhibitor MK-6482 for Von Hippel-Lindau disease–associated renal cell carcinoma. J. Clin. Oncol. 2020, 38, 5003. [Google Scholar] [CrossRef]
- Lobo, N.C.; Gedye, C.; Apostoli, A.J.; Brown, K.R.; Paterson, J.; Stickle, N.; Robinette, M.; Fleshner, N.; Hamilton, R.J.; Kulkarni, G.; et al. Efficient generation of patient-matched malignant and normal primary cell cultures from clear cell renal cell carcinoma patients: Clinically relevant models for research and personalized medicine. BMC Cancer 2016, 16, 485. [Google Scholar] [CrossRef] [Green Version]
- Shepard, M.J.; Bugarini, A.; Edwards, N.A.; Lu, J.; Zhang, Q.; Wu, T.; Zhuang, Z.; Chittiboina, P. Repurposing propranolol as an antitumor agent in von Hippel-Lindau disease. J. Neurosurg. 2019, 131, 1106–1114. [Google Scholar] [CrossRef] [Green Version]
- Yin, T.; Yu, S.; Xiao, L.; Zhang, J.; Liu, C.; Lu, Y.; Liu, C. Correlation between the expression of aquaporin 1 and hypoxia-inducible factor 1 in breast cancer tissues. J. Huazhong Univ. Sci. Technol. 2008, 28, 346–348. [Google Scholar] [CrossRef] [PubMed]
- Abreu-Rodríguez, I.; Silva, R.S.; Martins, A.P.; Soveral, G.; Toledo-Aral, J.J.; López-Barneo, J.; Echevarría, M. Functional and transcriptional induction of aquaporin-1 gene by hypoxia; analysis of promoter and role of HIF-1α. PLoS ONE 2011, 6, e28385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, P.; Pal, S.; Dutta, V.; Boruah, D.; Chandran, V.M.; Bhatoe, H.S. Correlation of expression pattern of aquaporin-1 in primary central nervous system tumors with tumor type, grade, proliferation, microvessel density, contrast-enhancement and perilesional edema. J. Cancer Res. Ther. 2012, 8, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, C.; Feldman, M.J.; Wang, H.; Pang, Y.; Maggio, D.M.; Zhu, D.; Nesvick, C.L.; Dmitriev, P.; Bullova, P.; et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 2017, 8, 56110–56125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J. Clin. Investig. 2001, 107, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Malec, V.; Coulson, J.M.; Urbé, S.; Clague, M.J. Combined Analyses of the VHL and Hypoxia Signaling Axes in an Isogenic Pairing of Renal Clear Cell Carcinoma Cells. J. Proteome Res. 2015, 14, 5263–5272. [Google Scholar] [CrossRef]
- Chang, P.Y.; Huang, W.Y.; Lin, C.L.; Huang, T.C.; Wu, Y.Y.; Chen, J.H.; Kao, C.H. Propranolol reduces cancer risk. Medicine 2015, 94, e1097. [Google Scholar] [CrossRef]
- Wilding, A.; Ingham, S.L.; Lalloo, F.; Clancy, T.; Huson, S.M.; Moran, A.; Evans, D.G. Life expectancy in hereditary cancer predisposing diseases: An observational study. J. Med. Genet. 2012, 49, 264–269. [Google Scholar] [CrossRef]
- Van Velthoven, V.; Reinacher, P.C.; Klisch, J.; Neumann, H.P.H.; Gläsker, S.; Bristol, R.E.; Spetzler, R.F.; Bricolo, A.; Benzel, E.C.; Lefranc, F.; et al. Treatment of Intramedullary Hemangioblastomas, with Special Attention to Von Hippel-Lindau Disease. Neurosurgery 2003, 53, 1306–1314. [Google Scholar] [CrossRef]
- Giles, R.H.; Gläsker, S. The first prospective trial for von Hippel-Lindau disease: Pazopanib. Lancet Oncol. 2018, 19, 1267–1269. [Google Scholar] [CrossRef]
- Gläsker, V.; Klingler, J.H. RareConnect.org Webinar: Hemangioblastomas of the Central Nervous System in Patients with VHL. Available online: https://youtu.be/F0wSl9AQxrg (accessed on 26 November 2018).
- Huang, Q.; Tan, Q.; Mao, K.; Yang, G.; Ma, G.; Luo, P.; Wang, S.; Mei, P.; Wu, F.; Xu, J.; et al. The role of adrenergic receptors in lung cancer. Am. J. Cancer. Res. 2018, 8, 2227–2237. [Google Scholar] [PubMed]
- Xiao, M.B.; Jin, D.D.; Jiao, Y.J.; Ni, W.K.; Liu, J.X.; Qu, L.S.; Lu, C.H.; Ni, R.Z.; Jiang, F.; Chen, W.C. β2-AR regulates the expression of AKR1B1 in human pancreatic cancer cells and promotes their proliferation via the ERK1/2 pathway. Mol. Biol. Rep. 2018, 45, 1863–1871. [Google Scholar] [CrossRef]
- Cui, B.; Luo, Y.; Tian, P.; Peng, F.; Lu, J.; Yang, Y.; Su, Q.; Liu, B.; Yu, J.; Luo, X.; et al. Stress-induced epinephrine enhances lactate dehydrogenase A and promotes breast cancer stem-like cells. J. Clin. Investig. 2019, 129, 1030–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Giorgi, V.; Grazzini, M.; Benemei, S.; Marchionni, N.; Botteri, E.; Pennacchioli, E.; Geppetti, P.; Gandini, S. Propranolol for Off-label Treatment of Patients With Melanoma: Results From a Cohort Study. JAMA Oncol. 2018, 4, e172908. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, E.; Street, J.; Pouchy, C.; Carre, M.; Gifford, A.J.; Murray, J.; Norris, M.D.; Trahair, T.; Andre, N.; Kavallaris, M. β-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma. Br. J. Cancer 2013, 108, 2485–2494. [Google Scholar] [CrossRef] [Green Version]
- Phadke, S.; Clamon, G. Beta blockade as adjunctive breast cancer therapy: A review. Crit. Rev. Oncol. Hematol. 2019, 138, 173–177. [Google Scholar] [CrossRef]
- Léauté-Labrèze, C.; De La Roque, E.D.; Hubiche, T.; Boralevi, F.; Thambo, J.B.; Taïeb, A. Propranolol for severe hemangiomas of infancy. N. Engl. J. Med. 2008, 358, 2649–2651. [Google Scholar] [CrossRef]
- Lamy, S.; Lachambre, M.P.; Lord-Dufour, S.; Béliveau, R. Propranolol suppresses angiogenesis in vitro: Inhibition of proliferation, migration, and differentiation of endothelial cells. Vasc. Pharmacol. 2010, 53, 200–208. [Google Scholar] [CrossRef]
- Storch, C.H.; Hoeger, P.H. Propranolol for infantile haemangiomas: Insights into the molecular mechanisms of action. Br. J. Dermatol. 2010, 163, 269–274. [Google Scholar] [CrossRef]
- Wong, A.; Hardy, K.L.; Kitajewski, A.M.; Shawber, C.J.; Kitajewski, J.K.; Wu, J.K. Propranolol accelerates adipogenesis in hemangioma stem cells and causes apoptosis of hemangioma endothelial cells. Plast. Reconstr. Surg. 2012, 130, 1012–1021. [Google Scholar] [CrossRef]
- Munabi, N.C.O.; England, R.W.; Edwards, A.K.; Kitajewski, A.A.; Tan, Q.K.; Weinstein, A.; Kung, J.E.; Wilcox, M.; Kitajewski, J.K.; Shawber, C.J.; et al. Propranolol Targets Hemangioma Stem Cells via cAMP and Mitogen-Activated Protein Kinase Regulation. Stem Cells Transl. Med. 2016, 5, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Xia, G.; Qiu, F.; Wu, C.; Denmon, A.P.; Zi, X. Physapubescin selectively induces apoptosis in VHL-null renal cell carcinoma cells through down-regulation of HIF-2α and inhibits tumor growth. Sci. Rep. 2016, 6, 32582. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofano, C.; Minervini, A.; Menicagli, M.; Salinitri, G.; Bertacca, G.; Pefanis, G.; Masieri, L.; Lessi, F.; Collecchi, P.; Minervini, R.; et al. Nuclear expression of hypoxia-inducible factor-1α in clear cell renal cell carcinoma is involved in tumor progression. Am. J. Surg. Pathol. 2007, 31, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Uniacke, J.; Holterman, C.E.; Lachance, G.; Franovic, A.; Jacob, M.D.; Fabian, M.R.; Payette, J.; Holcik, M.; Pause, A.; Lee, S. An oxygen-regulated switch in the protein synthesis machinery. Nature 2012, 486, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Minamishima, Y.A.; Yan, Q.; Schlisio, S.; Ebert, B.L.; Zhang, X.; Zhang, L.; Kim, W.Y.; Olumi, A.F.; Kaelin, W.G., Jr. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappaB agonist Card9 by CK2. Mol. Cell 2007, 28, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1αa by NF-κB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Castellanos, M.A.; Gómez de las Heras, K.V.; Díaz-Redondo, T.; González-Flores, E.; Albiñana, V.; Botella, L.M. Case Report: Propranolol increases the therapeutic response to temozolomide in a patient with metastatic paraganglioma. F1000Research 2017, 6, 2087. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Xu, Q.; Zuo, Y.; Liu, L.; Liu, S.; Chen, L.; Wang, K.; Lei, Y.; Zhao, X.; Li, Y. Isoprenaline/β2-AR activates Plexin-A1/VEGFR2 signals via VEGF secretion in gastric cancer cells to promote tumor angiogenesis. BMC Cancer 2017, 17, 875. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial Registration | Phase | Title | Intervention | Number of Patients | Status Start Date | Outcome | Link to Results |
|---|---|---|---|---|---|---|---|
| NCT00052013 | 2 | Treatment of Von Hippel-Lindau (VHL)-Related Hemangioblastoma With PTK787/ZK 222584 | PTK787 ZK 222584 (vatalanib) | 11 | C Feb 2003 | 100% discontinued (adverse events). | [21] |
| NCT00056199 | 1 | EYE001 to Treat Retinal Tumors in Patients With Von Hippel-Lindau Syndrome | EYE001(anti-VEGF aptamer) | 5 | C Mar 2003 | 80% showed stabilization or improvement of vision. | [22] |
| NCT00088374 | 2 | 17AAG to Treat Kidney Tumors in Von Hippel-Lindau Disease | 1 DMAG | 9 | C Jul 2004 | Study did not meet accrual. | [23] |
| 18FDG | |||||||
| [15-O] H2O | |||||||
| EPL diluent | |||||||
| NCT00089765 | 1 | Ranibizumab Injections to Treat Retinal Tumors in Patients With Von Hippel-Lindau Syndrome | Ranibizumab | 5 | C Aug 2004 | Not clear therapeutic effect. “Minimal beneficial effects on most VHL-related Retinal Capillary HB” | [24] |
| NCT00330564 | 2 | Evaluation of Sunitinib Malate in Patients With Von Hippel-Lindau Syndrome (VHL) Who Have VHL Lesions to Follow | SU011248 (Sunitinib) | 15 | T May 2006 | Early termination due to slow accrual. 33% showed partial response in RCC but not in HB. | [25,26] |
| NCT00470977 | 1/2 | Treatment of Exudative and Vasogenic Chorioretinal Diseases Including Variants of AMD and Other CNV Related Maculopathy | Ranibizumab injection (0.5 mg) | 18 | C May 2007 | - | - |
| EudraCT 2007-002132-29 | 2 | A Phase II Trial of Sorafenib (a tyrosine kinase inhibitor) given orally twice daily in renal cancer patients with vHL syndrome | Sorafenib | 25 | T Jan 2008 | - | - |
| NCT00566995 | 2 | Phase II Study of Vandetanib in Individuals With Kidney Cancer | ZACTIMA (Vandetanib) (ZD6474) | 37 | C Feb 2008 | 80% completed the study. None of them showed a complete response and 8% showed a partial response. | [27] |
| NCT00673816 | 1/2 | Sunitinib Malate to Treat Advanced Eye Disease in Patients With Von Hippel-Lindau Syndrome | Sunitinib Malate | 2 | T May 2008 | Study did not meet accrual plus adverse events. | [28,29] |
| NCT01015300 | 1 | Bevacizumab (Avastin) in Unresectable/Recurrent Hemangioblastoma From VonHippel-Lindau Disease | Avastin | 1 | T Dec 2009 | Study did not meet accrual. | - |
| NCT01168440 EudraCT 2009-013052-76 | 2 | A single-arm, phase II study of SU11248 (sunitinib) in patients with von Hippel-Lindau (VHL) disease | Sunitinib | 5 | C Feb 2010 | Disease progression (20%), unacceptable toxicity (60%), and lack of clinical improvement. after 7 cycles | [30] |
| EudraCT 2005-004502-82 | 2 | A Phase II Study of Neoadjuvant Sunitinib in Metastatic Renal Cell Carcinoma | Sunitinib | 16 | T Oct 2010 | Study did not meet accrual. 58% showed complete or partial response. | [31] |
| NCT01436227 | 2 | Pazopanib in Von Hippel-Lindau (VHL) Syndrome | Pazopanib | 32 | O Jan 2012 | 80% discontinued (progressive disease, loss of quality of life, or intolerance). 42% showed partial response and 55% a complete or partial response in ccRCC. | [15] |
| NCT01266070 | 2 | TKI 258 in Von Hippel-Lindau Syndrome (VHL) | Dovitinib | 6 | T Nov 2012 | 33% discontinued plus the trial met toxicity stopping rule. | [32,33] |
| EudraCT 2014-003671-30 | 3 | Therapeutic effect of propranolol in a series of patients with von Hippel-Lindau disease and retinal hemangioblastomas in short, medium and long term treatment. | Propranolol | 7 | C Oct 2014 | 28% showed partial response and 72% a stable disease. No serious adverse effects were recorded. | [19,20] |
| NCT02108002 | 1 | Effect of Vorinostat on Nervous System Hemangioblastomas in Von Hippel-Lindau Disease (Missense Mutation Only) | Vorinostat | 7 | C Apr 2014 | - | - |
| NCT02859441 | 1/2 | A Phase I/II Trial for Intravitreous Treatment of Severe Ocular Von Hippel-Lindau Disease Using a Combination of the PDGF Antagonist E10030 and the VEGF Antagonist Ranibizumab | Ranibizumab & E10030 (anti-PDGF pegylated aptamer) | 3 | C Aug 2016 | - | - |
| NCT03108066 | 2 | PT2385 for the Treatment of Von Hippel-Lindau Disease-Associated Clear Cell Renal Cell Carcinoma | PT2385 Tablets (HIF-2α inhibitor) | 4 | O May 2017 | - | - |
| NCT03401788 | 2 | A Phase 2 Study of PT2977 for the Treatment of Von Hippel Lindau Disease-Associated Renal Cell Carcinoma | PT2977 (HIF-2α inhibitor) | 61 | C Mar 2018 | 27.9% showed partial response. | [34] |
| Case | Gender, Age | Lesions Prior to Propranolol Treatment | Propranolol Treatment Initiation (months) | Propranolol Treatment Doses (mg/kg/day) | Response to Propranolol Treatment |
|---|---|---|---|---|---|
| 1 | Female, 32 | Bilateral retinal HB | January 2017 (36 months) | Started at 0.66 and increased to 1.5 | No lesions detected along the treatment |
| 2 | Male, 24 | Medulla HB extracted and partial nephrectomy due to ccRCC left renal lesions | May 2018 (22 months) | Started at 0.66 and increased to 1.2 | Stable bilateral cystic lesions after starting treatment |
| 3 | Male, 28 | Left eye retinal HB, multiple tumoral and cystic lesions | August 2016 (47 months) | Started at 0.5 and increased to 1.4 | Stable bilateral lesions but one after starting treatment |
| 4 | Male, 22 | Right eye retinal HB and extracted cerebellum HB | April 2018 (16 months) | 1.8 | Stable 3 bilateral cystic lesions after starting treatment |
| Gene | Sequence |
|---|---|
| 18S Fwd 18S Rev | 5′-CTCAACACGGGAAACCTCAC-3′ 5′-CGCTCCACCAACTAAGAACG-3′ |
| ADRB-1 Fwd ADRB-1 Rev | 5′-GTGGAAGATGGGTGGGTTAG-3′ 5′-GAGCCACGATGATCGATTTTA-3′ |
| ADRB-2 Fwd ADRB-2 Rev | 5′-CCATGTCCAGAACCTTAGCC-3′ 5′-GATCTGCGGAGTCCATGC -3′ |
| AQP-1 Fwd AQP-1 Rev | 5′-GGAGGGTCCCGATGATCT-3′ 5′-CCTCCCTGACTGGGAACTC-3′ |
| BAX Fwd BAX Rev | 5′-CACTCCCGCCACAAAGAT-3′ 5′-CAAGACCAGGGTGGTTGG-3′ |
| CAIX Fwd CAIX Rev | 5′-TGCCGTCAATTAAGCATAAGG-3′ 5′-GTCCAGTAATCTGGGCAGGTA′-3 |
| CASP9 Fwd CASP9 Rev | 5′-CCCAAGCTCTTTTTCATCCA-3′ 5′-TTACTGCCAGGGGACTCGT-3′ |
| IL-6 Fwd IL-6 Rev | 5′-CAGGAGCCCAGCTATGAACT-3′ 5′-GAAGGCAGCAGGCAACAC-3′ |
| IL-1β Fwd IL-1β Rev | 5′-CTGTCCTGCGTGTTGAAAGA-3′ 5′-TTGGGTAATTTTTGGGATCTACA-3′ |
| TNFAIP6 Fwd TNFAIP6 Rev | 5′-GGCCATCTCGCAACTTACA-3′ 5′-GCAGCACAGACATGAAATCC-3′ |
| MDR Fwd MDR Rev | 5′-TTGAAATGAAATGTTGTCTGG-3′ 5′-CAAAGAAACAACGGTTCGG-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albiñana, V.; Gallardo-Vara, E.; de Rojas-P, I.; Recio-Poveda, L.; Aguado, T.; Canto-Cano, A.; Aguirre, D.T.; Serra, M.M.; González-Peramato, P.; Martínez-Piñeiro, L.; et al. Targeting β2-Adrenergic Receptors Shows Therapeutical Benefits in Clear Cell Renal Cell Carcinoma from Von Hippel–Lindau Disease. J. Clin. Med. 2020, 9, 2740. https://doi.org/10.3390/jcm9092740
Albiñana V, Gallardo-Vara E, de Rojas-P I, Recio-Poveda L, Aguado T, Canto-Cano A, Aguirre DT, Serra MM, González-Peramato P, Martínez-Piñeiro L, et al. Targeting β2-Adrenergic Receptors Shows Therapeutical Benefits in Clear Cell Renal Cell Carcinoma from Von Hippel–Lindau Disease. Journal of Clinical Medicine. 2020; 9(9):2740. https://doi.org/10.3390/jcm9092740
Chicago/Turabian StyleAlbiñana, Virginia, Eunate Gallardo-Vara, Isabel de Rojas-P, Lucia Recio-Poveda, Tania Aguado, Ana Canto-Cano, Daniel T. Aguirre, Marcelo M. Serra, Pilar González-Peramato, Luis Martínez-Piñeiro, and et al. 2020. "Targeting β2-Adrenergic Receptors Shows Therapeutical Benefits in Clear Cell Renal Cell Carcinoma from Von Hippel–Lindau Disease" Journal of Clinical Medicine 9, no. 9: 2740. https://doi.org/10.3390/jcm9092740
APA StyleAlbiñana, V., Gallardo-Vara, E., de Rojas-P, I., Recio-Poveda, L., Aguado, T., Canto-Cano, A., Aguirre, D. T., Serra, M. M., González-Peramato, P., Martínez-Piñeiro, L., Cuesta, A. M., & Botella, L. M. (2020). Targeting β2-Adrenergic Receptors Shows Therapeutical Benefits in Clear Cell Renal Cell Carcinoma from Von Hippel–Lindau Disease. Journal of Clinical Medicine, 9(9), 2740. https://doi.org/10.3390/jcm9092740