Depressed Cardiac Mechanical Energetic Efficiency: A Contributor to Cardiovascular Risk in Common Metabolic Diseases—From Mechanisms to Clinical Applications

 and
and 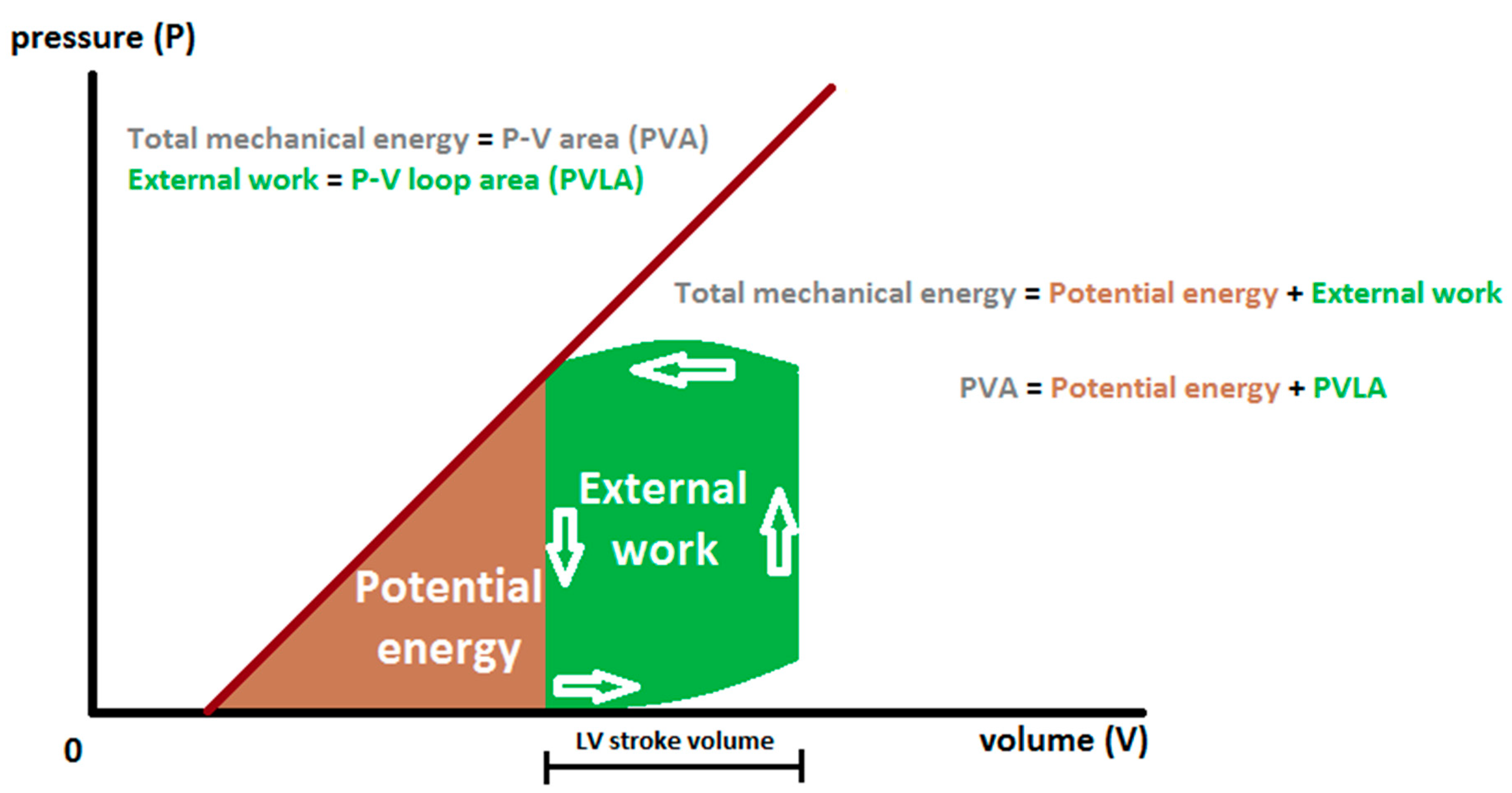{kind=link}
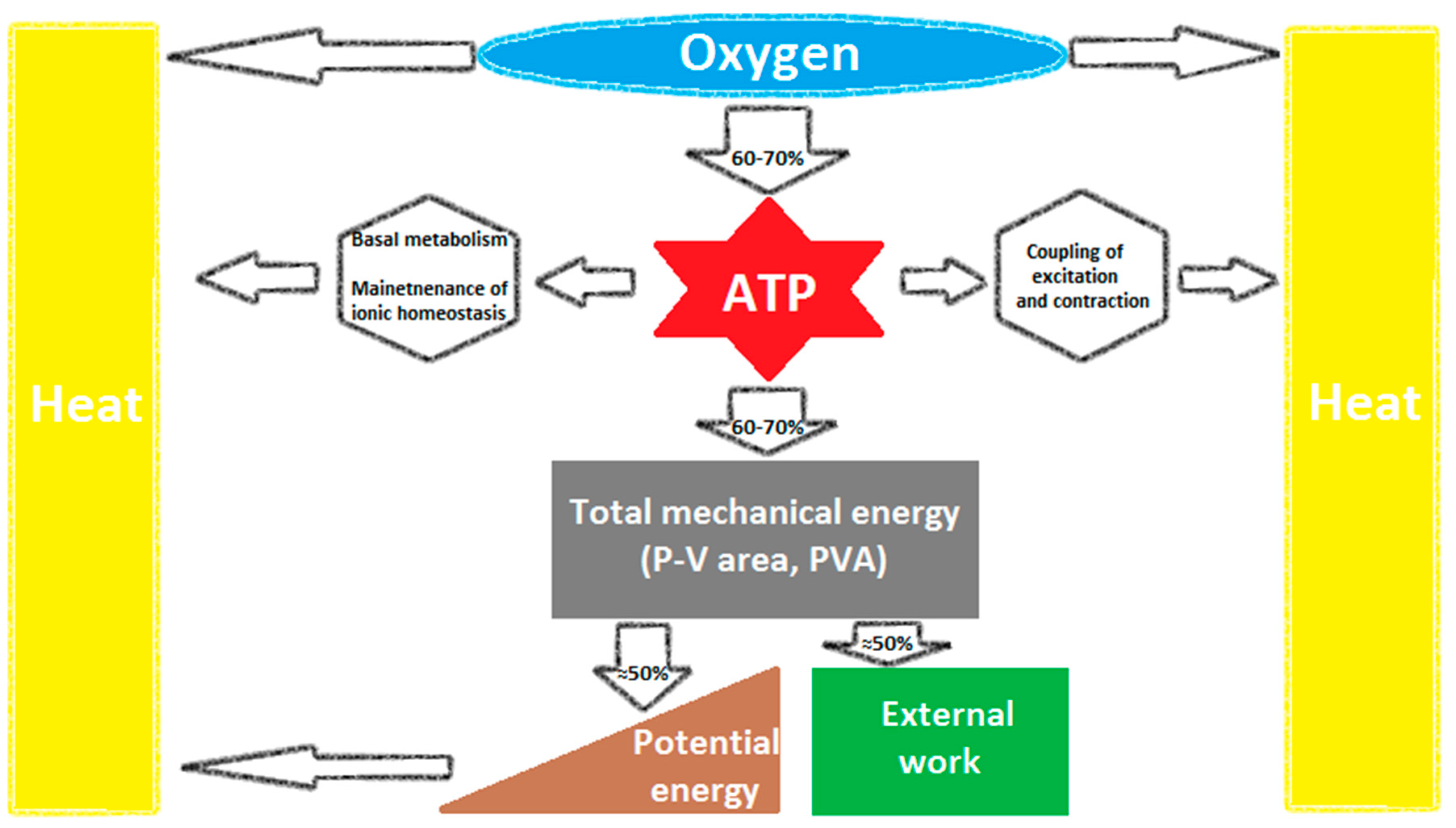{kind=link}
Abstract
:1. Introduction
2. Cardiac Mechanical Energetic Efficiency—Basic Concepts
3. Myocardial Mechano—Energetic Efficiency Index (MEEi)—A Simple Surrogate Measure of LV Mechanical Efficiency
4. Low MEEi—A Predictor of Short- and Long-Term CV Events
5. Association of Low MEEi with Metabolic Risk Factors
6. Relevance of MEEi—Pros and Cons
7. Implications for Future Research—Treatment Effects on MEE
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Starling, E.H.; Visscher, M.B. The regulation of the energy output of the heart. J. Physiol. 1927, 62, 243–261. [Google Scholar] [CrossRef]
- Bing, R.J.; Hammond, M.M. The measurement of coronary blood flow, oxygen consumption, and efficiency of the left ventricle in man. Am. Heart J. 1949, 38, 1–24. [Google Scholar] [CrossRef]
- Suga, H. Ventricular energetics. Physiol. Rev. 1990, 70, 247–277. [Google Scholar] [CrossRef]
- Kameyama, T.; Asanoi, H.; Ishizaka, S.; Yamanishi, K.; Fujita, M.; Sasayama, S. Energy conversion efficiency in human left ventricle. Circulation 1992, 85, 988–996. [Google Scholar] [CrossRef] [Green Version]
- Baxley, W.A.; Dodge, H.T.; Rackley, C.E.; Sandler, H.; Pugh, D. Left ventricular mechanical efficiency in man with heart disease. Circulation 1977, 55, 564–568. [Google Scholar] [CrossRef] [Green Version]
- Nichols, A.B.; Pearson, M.H.; Sciacca, R.R.; Cannon, P.J. Left ventricular mechanical efficiency in coronary artery disease. J. Am. Coll. Cardiol. 1986, 7, 270–279. [Google Scholar] [CrossRef] [Green Version]
- De Simone, G.; Izzo, R.; Losi, M.A.; Stabile, E.; Rozza, F.; Canciello, G.; Mancusi, C.; Trimarco, V.; De Luca, N.; Trimarco, B. Depressed myocardial energetic efficiency is associated with increased cardiovascular risk in hypertensive left ventricular hypertrophy. J. Hypertens. 2016, 34, 1846–1853. [Google Scholar] [CrossRef]
- Knaapen, P.; Germans, T.; Knuuti, J.; Paulus, W.J.; Dijkmans, P.A.; Allaart, C.P.; Lammertsma, A.A.; Visser, F.C. Myocardial energetics and efficiency: Current status of the noninvasive approach. Circulation 2007, 115, 918–927. [Google Scholar] [CrossRef] [Green Version]
- De Simone, G.; Chinali, M.; Galderisi, M.; Benincasa, M.; Girfoglio, D.; Botta, I.; D’Addeo, G.; de Divitiis, O. Myocardial mechano-energetic efficiency in hypertensive adults. J. Hypertens. 2009, 27, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Beanlands, R.S.; Bach, D.S.; Raylman, R.; Armstrong, W.F.; Wilson, V.; Montieth, M.; Moore, C.K.; Bates, E.; Schwaiger, M. Acute effects of dobutamine on myocardial oxygen consumption and cardiac efficiency measured using carbon-11 acetate kinetics in patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 1993, 22, 1389–1398. [Google Scholar] [CrossRef] [Green Version]
- Güçlü, A.; Knaapen, P.; Harms, H.J.; Vonk, A.B.; Stooker, W.; Groepenhoff, H.; Lammertsma, A.A.; van Rossum, A.C.; Germans, T.; van der Velden, J. Myocardial efficiency is an important determinant of functional improvement after aortic valve replacement in aortic valve stenosis patients: A combined PET and CMR study. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 882–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, N.H.; Sörensen, J.; Harms, H.J.; Kim, W.Y.; Nielsen, R.; Tolbod, L.P.; Frøkiær, J.; Bouchelouche, K.; Dodt, K.K.; Sihm, I.; et al. Myocardial oxygen consumption and efficiency in aortic valve stenosis patients with and without heart failure. J. Am. Heart Assoc. 2017, 6, e004810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schramm, W. The units of measurement of the ventricular stroke work: A review study. J. Clin. Monit. Comput. 2010, 24, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Losi, M.A.; Izzo, R.; Mancusi, C.; Wang, W.; Roman, M.J.; Lee, E.T.; Howard, B.V.; Devereux, R.B.; de Simone, G. Depressed myocardial energetic efficiency increases risk of incident heart failure: The Strong Heart Study. J. Clin. Med. 2019, 8, 1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancusi, C.; de Simone, G.; Best, L.G.; Wang, W.; Zhang, Y.; Roman, M.J.; Lee, E.T.; Howard, B.V.; Devereux, R.B. Myocardial mechano-energetic efficiency and insulin resistance in non-diabetic members of the Strong Heart Study cohort. Cardiovasc. Diabetol. 2019, 18, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancusi, C.; Losi, M.A.; Izzo, R.; Canciello, G.; Manzi, M.V.; Sforza, A.; De Luca, N.; Trimarco, B.; de Simone, G. Effect of diabetes and metabolic syndrome on myocardial mechano-energetic efficiency in hypertensive patients. The Campania Salute Network. J. Hum. Hypertens. 2017, 31, 395–399. [Google Scholar] [CrossRef]
- Fiorentino, T.V.; Miceli, S.; Succurro, E.; Sciacqua, A.; Andreozzi, F.; Sesti, G. Non-alcoholic fatty liver disease is associated with a decreased myocardial mechano-energetic efficiency. J. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Kato, T.; Niizuma, S.; Inuzuka, Y.; Kawashima, T.; Okuda, J.; Tamaki, Y.; Iwanaga, Y.; Narazaki, N.; Matsuda, T.; Soga, T.; et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ. Heart Fail. 2010, 3, 420–430. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Fernández-Checa, J.C. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J. Biol. Chem. 1997, 272, 11369–11377. [Google Scholar] [CrossRef] [Green Version]
- Schubert, K.M.; Scheid, M.P.; Duronio, V. Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J. Biol. Chem. 2000, 275, 13330–13335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratford, S.; DeWald, D.B.; Summers, S.A. Ceramide dissociates 3′-phosphoinositide production from pleckstrin homology domain translocation. Biochem. J. 2001, 354 Pt 2, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Law, B.A.; Liao, X.; Moore, K.S.; Southard, A.; Roddy, P.; Ji, R.; Szulc, Z.; Bielawska, A.; Schulze, P.C.; Cowart, L.A. Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J. 2018, 32, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelsson, E.; Sundström, J.; Arnlöv, J.; Zethelius, B.; Lind, L. Insulin resistance and risk of congestive heart failure. JAMA 2005, 294, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Aon, M.A.; Stanley, B.A.; Sivakumaran, V.; Kembro, J.M.; O’Rourke, B.; Paolocci, N.; Cortassa, S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: An experimental-computational study. J. Gen. Physiol. 2012, 139, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, L.; Battiprolu, P.K.; Fukushima, A.; Nguyen, K.; Milner, K.; Gupta, A.; Altamimi, T.; Byrne, N.; Mori, J.; et al. Malonyl CoA decarboxylase inhibition improves cardiac function post-myocardial infarction. JACC Basic Transl. Sci. 2019, 4, 385–400. [Google Scholar] [CrossRef]
- De Marco, M.; Gerdts, E.; Mancusi, C.; Roman, M.J.; Lønnebakken, M.T.; Lee, E.T.; Howard, B.V.; Devereux, R.B.; de Simone, G. Influence of left ventricular stroke volume on incident heart failure in a population with preserved ejection fraction (from the Strong Heart Study). Am. J. Cardiol. 2017, 119, 1047–1052. [Google Scholar] [CrossRef] [Green Version]
- Seko, Y.; Kato, T.; Shiba, M.; Morita, Y.; Yamaji, Y.; Haruna, Y.; Nakane, E.; Haruna, T.; Inoko, M. Staging cardiac damage in patients with hypertension. Hypertension 2019, 74, 1357–1365. [Google Scholar] [CrossRef]
- Généreux, P.; Pibarot, P.; Redfors, B.; Mack, M.J.; Makkar, R.R.; Jaber, W.A.; Svensson, L.G.; Kapadia, S.; Tuzcu, E.M.; Thourani, V.H.; et al. Staging classification of aortic stenosis based on the extent of cardiac damage. Eur. Heart J. 2017, 38, 3351–3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devereux, R.B.; Bang, C.N.; Roman, M.J.; Palmieri, V.; Boman, K.; Gerdts, E.; Nieminen, M.S.; Papademetriou, V.; Wachtell, K.; Hille, D.A.; et al. Left ventricular wall stress-mass-heart rate product and cardiovascular events in treated hypertensive patients: LIFE Study. Hypertension 2015, 66, 945–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bella, J.N.; Devereux, R.B.; Roman, M.J.; Palmieri, V.; Liu, J.E.; Paranicas, M.; Welty, T.K.; Lee, E.T.; Fabsitz, R.R.; Howard, B.V. Separate and joint effects of systemic hypertension and diabetes mellitus on left ventricular structure and function in American Indians (the Strong Heart Study). Am. J. Cardiol. 2001, 87, 1260–1265. [Google Scholar] [CrossRef]
- Ernande, L.; Bergerot, C.; Rietzschel, E.R.; De Buyzere, M.L.; Thibault, H.; Pignonblanc, P.G.; Croisille, P.; Ovize, M.; Groisne, L.; Moulin, P.; et al. Diastolic dysfunction in patients with type 2 diabetes mellitus: Is it really the first marker of diabetic cardiomyopathy? J. Am. Soc. Echocardiogr. 2011, 24, 1268–1275. [Google Scholar] [CrossRef]
- Giorda, C.B.; Cioffi, G.; de Simone, G.; Di Lenarda, A.; Faggiano, P.; Latini, R.; Lucci, D.; Maggioni, A.P.; Tarantini, L.; Velussi, M.; et al. (on behalf of the DYDA Investigators). Predictors of early-stage left ventricular dysfunction in type 2 diabetes: Results of DYDA study. Eur. J. Cardiovasc. Prev. Rehabil. 2011, 18, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, G.; Senni, M.; Tarantini, L.; Faggiano, P.; Rossi, A.; Stefenelli, C.; Russo, T.E.; Alessandro, S.; Furlanello, F.; de Simone, G. Analysis of circumferential and longitudinal left ventricular systolic function in patients with non-ischemic chronic heart failure and preserved ejection fraction (from the CARRY-IN-HFpEF study). Am. J. Cardiol. 2012, 109, 383–389. [Google Scholar] [CrossRef]
- Faden, G.; Faganello, G.; De Feo, S.; Berlinghieri, N.; Tarantini, L.; Di Lenarda, A.; Faggiano, P.; Cioffi, G. The increasing detection of asymptomatic left ventricular dysfunction in patients with type 2 diabetes mellitus without overt cardiac disease: Data from the SHORTWAVE study. Diabetes Res. Clin. Pract. 2013, 101, 309–316. [Google Scholar] [CrossRef]
- Cioffi, G.; Rossi, A.; Targher, G.; Zoppini, G.; de Simone, G.; Devereux, R.B.; Bonora, E.; Vassanelli, C. Usefulness of subclinical left ventricular midwall dysfunction to predict cardiovascular mortality in patients with type 2 diabetes mellitus. Am. J. Cardiol. 2014, 113, 1409–1414. [Google Scholar] [CrossRef]
- Galderisi, M. Diastolic dysfunction and diabetic cardiomyopathy: Evaluation by Doppler echocardiography. J. Am. Coll. Cardiol. 2006, 48, 1548–1551. [Google Scholar] [CrossRef] [Green Version]
- Seferović, P.M.; Paulus, W.J. Clinical diabetic cardiomyopathy: A two-faced disease with restrictive and dilated phenotypes. Eur. Heart J. 2015, 36, 1718–1727. [Google Scholar] [CrossRef]
- Shah, S.J.; Lam, C.S.P.; Svedlund, S.; Saraste, A.; Hage, C.; Tan, R.S.; Beussink-Nelson, L.; Ljung Faxén, U.; Fermer, M.L.; Broberg, M.A.; et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur. Heart J. 2018, 39, 3439–3450. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Dal Canto, E. Distinct myocardial targets for diabetes therapy in heart failure with preserved or reduced ejection fraction. JACC Heart Fail. 2018, 6, 1–7. [Google Scholar] [CrossRef]
- Brainin, P.; Frestad, D.; Prescott, E. The prognostic value of coronary endothelial and microvascular dysfunction in subjects with normal or non-obstructive coronary artery disease: A systematic review and meta-analysis. Int. J. Cardiol. 2018, 254, 1–9. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Palmieri, V.; Koren, M.J.; Mensah, G.A.; Roman, M.J.; Devereux, R.B. Prognostic implications of the compensatory nature of left ventricular mass in arterial hypertension. J. Hypertens. 2001, 19, 119–125. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Gottdiener, J.S.; Chinali, M.; Maurer, M.S. Left ventricular mass predicts heart failure not related to previous myocardial infarction: The Cardiovascular Health Study. Eur. Heart J. 2008, 29, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, G.; Rossi, A.; Zoppini, G.; Targher, G.; de Simone, G.; Devereux, R.B.; Vassanelli, C.; Bonora, E. Inappropriate left ventricular mass independently predicts cardiovascular mortality in patients with type 2 diabetes. Int. J. Cardiol. 2013, 168, 4953–4956. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Fryer, L.G.; Parbu-Patel, A.; Carling, D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 2002, 277, 25226–25232. [Google Scholar] [CrossRef] [Green Version]
- Spiering, M.J. The mystery of metformin. J. Biol. Chem. 2019, 294, 6689–6691. [Google Scholar] [CrossRef] [Green Version]
- Larsen, A.H.; Jessen, N.; Nørrelund, H.; Tolbod, L.P.; Harms, H.J.; Feddersen, S.; Nielsen, F.; Brøsen, K.; Hansson, N.H.; Frøkiaer, J.; et al. A randomised, double-blind, placebo-controlled trial of metformin on myocardial efficiency in insulin-resistant chronic heart failure patients without diabetes. Eur. J. Heart Fail. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV protection in the EMPA-REG OUTCOME trial: A “thrifty substrate” hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R. Evolving concepts of myocardial energy metabolism: More than just fats and carbohydrates. Circ. Res. 2016, 119, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.L.; Zhang, L.; Wagg, C.; Al Batran, R.; Gopal, K.; Levasseur, J.; Leone, T.; Dyck, J.R.B.; Ussher, J.R.; Muoio, D.M.; et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 2019, 115, 1606–1616. [Google Scholar] [CrossRef]
- Ho, K.L.; Karwi, Q.G.; Wagg, C.; Zhang, L.; Vo, K.; Altamimi, T.; Uddin, G.M.; Ussher, J.R.; Lopaschuk, G.D. Ketones can become the major fuel source for the heart but do not increase cardiac efficiency. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Verma, S.; Rawat, S.; Ho, K.L.; Wagg, C.S.; Zhang, L.; Teoh, H.; Dyck, J.E.; Uddin, G.M.; Oudit, G.Y.; Mayoux, E.; et al. Empagliflozin increases cardiac energy production in diabetes: Novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC Basic Transl. Sci. 2018, 3, 575–587. [Google Scholar] [CrossRef]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na +/H + exchanger, lowering of cytosolic Na + and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Huang, C.; Kirshenbaum, L.; Karmazyn, M. NHE-1 inhibition improves impaired mitochondrial permeability transition and respiratory function during postinfarction remodelling in the rat. J. Mol. Cell. Cardiol. 2005, 38, 135–143. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; San Antonio, R.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef]
- De Simone, G.; Chinali, M.; Mureddu, G.F.; Cacciatore, G.; Lucci, D.; Latini, R.; Masson, S.; Vanasia, M.; Maggioni, A.P.; Boccanelli, A. AREA-in-CHF Investigators. Effect of canrenone on left ventricular mechanics in patients with mild systolic heart failure and metabolic syndrome: The AREA-in-CHF study. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 783–791. [Google Scholar] [CrossRef]
- Marino, P.N.; Binda, G.; Calzaducca, E.; Panizza, A.; Ferrari, I.; Bellacosa, I.; Ambrosio, G. Transcatheter aortic valve replacement acutely improves left ventricular mechanical efficiency in severe aortic stenosis: Effects of different phenotypes. Clin. Res. Cardiol. 2020, 109, 819–831. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juszczyk, A.; Jankowska, K.; Zawiślak, B.; Surdacki, A.; Chyrchel, B. Depressed Cardiac Mechanical Energetic Efficiency: A Contributor to Cardiovascular Risk in Common Metabolic Diseases—From Mechanisms to Clinical Applications. J. Clin. Med. 2020, 9, 2681. https://doi.org/10.3390/jcm9092681
Juszczyk A, Jankowska K, Zawiślak B, Surdacki A, Chyrchel B. Depressed Cardiac Mechanical Energetic Efficiency: A Contributor to Cardiovascular Risk in Common Metabolic Diseases—From Mechanisms to Clinical Applications. Journal of Clinical Medicine. 2020; 9(9):2681. https://doi.org/10.3390/jcm9092681
Chicago/Turabian StyleJuszczyk, Albert, Karolina Jankowska, Barbara Zawiślak, Andrzej Surdacki, and Bernadeta Chyrchel. 2020. "Depressed Cardiac Mechanical Energetic Efficiency: A Contributor to Cardiovascular Risk in Common Metabolic Diseases—From Mechanisms to Clinical Applications" Journal of Clinical Medicine 9, no. 9: 2681. https://doi.org/10.3390/jcm9092681
APA StyleJuszczyk, A., Jankowska, K., Zawiślak, B., Surdacki, A., & Chyrchel, B. (2020). Depressed Cardiac Mechanical Energetic Efficiency: A Contributor to Cardiovascular Risk in Common Metabolic Diseases—From Mechanisms to Clinical Applications. Journal of Clinical Medicine, 9(9), 2681. https://doi.org/10.3390/jcm9092681