Implications of a Change of Paradigm in Alpha1 Antitrypsin Deficiency Augmentation Therapy: From Biochemical to Clinical Efficacy


Abstract
1. Introduction
2. Initial Assumptions
2.1. Biochemical Efficacy
2.2. Protective Threshold Level
2.3. Dose
2.4. Route of Administration
2.5. Origin of AAT
2.6. Final Statement
3. Clinical Efficacy: A Change of Paradigm
4. Controversies over Indication
4.1. Age Limits
4.2. Mutations
4.3. Lung Function Limits
4.4. Indication in Liver Disease
5. Controversies in the Administration
5.1. Infusion Frequency
5.2. Correct Dose
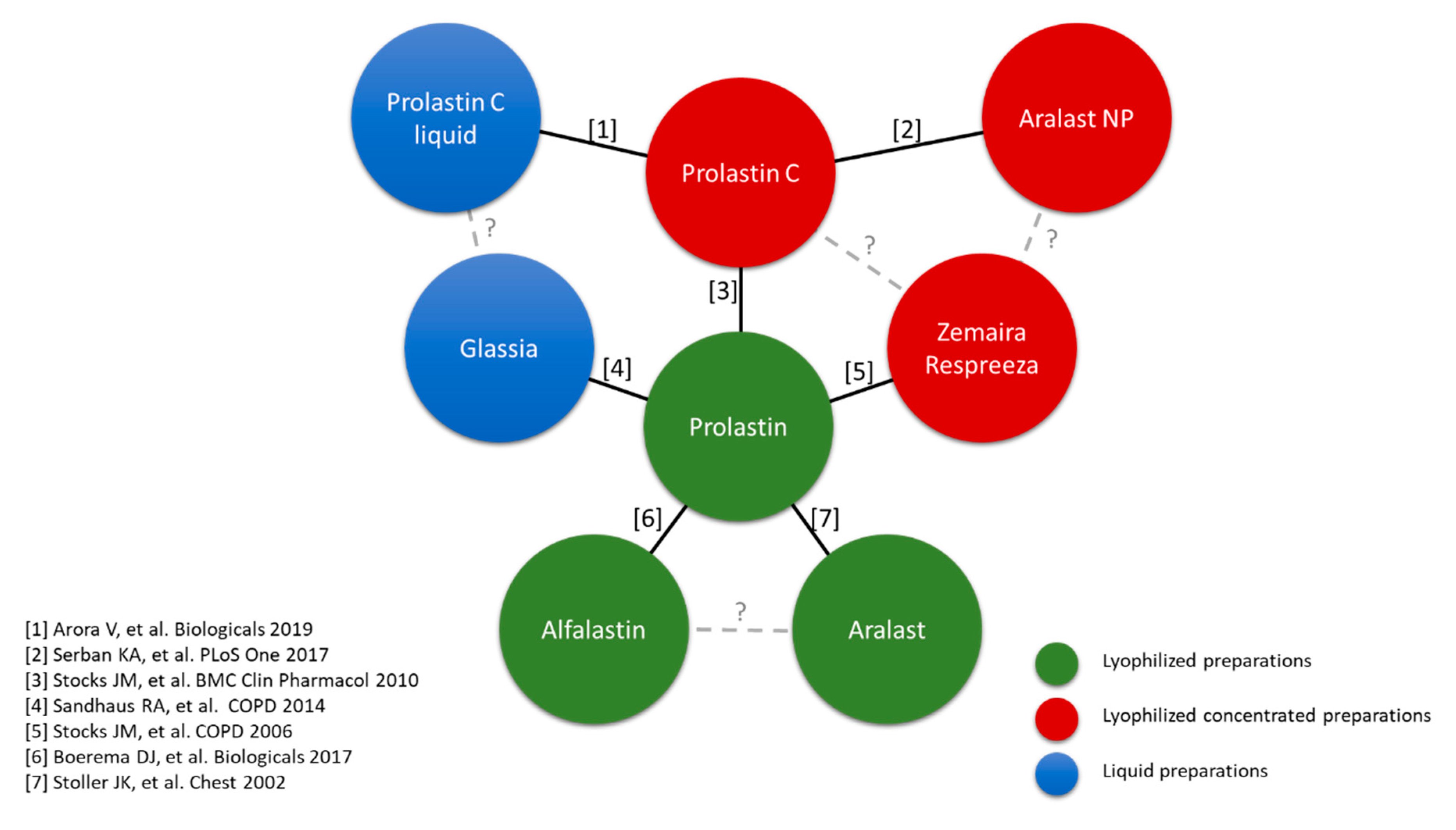
5.3. Differences between Preparations
6. Controversies in the Follow-Up
6.1. Monitoring Control in the First Few Months
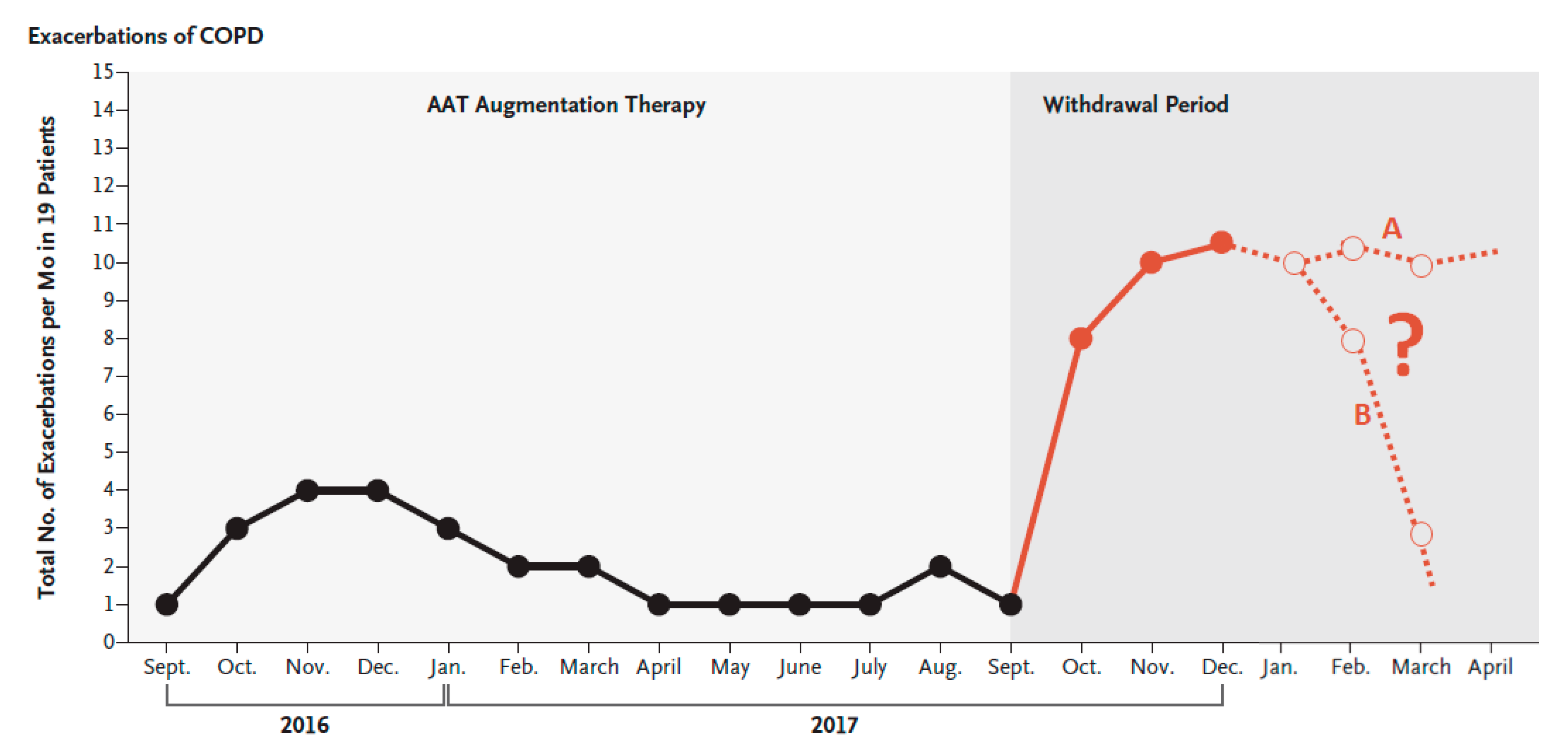
6.2. Discontinuation of Augmentation Therapy
6.3. Pulmonary Transplantation
7. The Future: A Second Change of Paradigm Coming
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha1-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, A.; Piitulainen, E.; Parr, D.G.; Deng, C.; Wencker, M.; Shaker, S.B.; Stockley, R.A. Exploring the role of CT densitometry: A randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 2009, 33, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, A.; Dijkman, J.H.; Madsen, F.; Stoel, B.; Hutchison, D.C.; Ulrik, C.S.; Skovgaard, L.T.; Kok-Jensen, A.; Rudolphus, A.; Seersholm, N.; et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am. J. Respir. Crit. Care Med. 1999, 160, 1468–1472. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.R.; Burdon, J.G.; Piitulainen, E.; Sandhaus, R.A.; Seersholm, N.; Stocks, J.M.; Stoel, B.C.; Huang, L.; Yao, Z.; Edelman, J.M.; et al. Intravenous augmentation treatment and lung density in severe alpha1 antitrypsin deficiency (RAPID): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 386, 360–368. [Google Scholar] [CrossRef]
- McElvaney, N.G. Alpha-1 Antitrypsin Therapy in Cystic Fibrosis and the Lung Disease Associated with Alpha-1 Antitrypsin Deficiency. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. S2), S191–S196. [Google Scholar] [CrossRef]
- Gadek, J.E.; Klein, H.G.; Holland, P.V.; Crystal, R.G. Replacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjects. J. Clin. Investig. 1981, 68, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Gadek, J.E.; Crystal, R.G. Experience with replacement therapy in the destructive lung disease associated with severe alpha-1-antitrypsin deficiency. Am. Rev. Respir. Dis. 1983, 127, S45–S46. [Google Scholar] [CrossRef]
- Wewers, M.D.; Casolaro, M.A.; Sellers, S.E.; Swayze, S.C.; McPhaul, K.M.; Wittes, J.T.; Crystal, R.G. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N. Engl. J. Med. 1987, 316, 1055–1062. [Google Scholar] [CrossRef]
- Hubbard, R.C.; Sellers, S.; Czerski, D.; Stephens, L.; Crystal, R.G. Biochemical efficacy and safety of monthly augmentation therapy for alpha 1-antitrypsin deficiency. JAMA J. Am. Med. Assoc. 1988, 260, 1259–1264. [Google Scholar] [CrossRef]
- American Thoracic, S.; European Respiratory, S. American Thoracic Society/European Respiratory Society statement: Standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 2003, 168, 818–900. [Google Scholar] [CrossRef]
- Sorrells, S.; Camprubi, S.; Griffin, R.; Chen, J.; Ayguasanosa, J. SPARTA clinical trial design: Exploring the efficacy and safety of two dose regimens of alpha1-proteinase inhibitor augmentation therapy in alpha1-antitrypsin deficiency. Respir. Med. 2015, 109, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.A.; Vergalla, J.; Steer, C.J.; Bradley-Moore, P.R.; Vierling, J.M. Metabolism of intact and desialylated alpha 1-antitrypsin. Clin. Sci. Mol. Med. 1978, 55, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Stolk, J.; Tov, N.; Chapman, K.R.; Fernandez, P.; MacNee, W.; Hopkinson, N.S.; Piitulainen, E.; Seersholm, N.; Vogelmeier, C.F.; Bals, R.; et al. Efficacy and safety of inhaled α1-antitrypsin in patients with severe α1-antitrypsin deficiency and frequent exacerbations of COPD. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 2019, 54. [Google Scholar] [CrossRef] [PubMed]
- Straus, S.D.; Fells, G.A.; Wewers, M.D.; Courtney, M.; Tessier, L.H.; Tolstoshev, P.; Lecocq, J.P.; Crystal, R.G. Evaluation of recombinant DNA-directed E. coli produced alpha 1-antitrypsin as an anti-neutrophil elastase for potential use as replacement therapy of alpha 1-antitrypsin deficiency. Biochem. Biophys. Res. Commun. 1985, 130, 1177–1184. [Google Scholar] [CrossRef]
- Flotte, T.R.; Trapnell, B.C.; Humphries, M.; Carey, B.; Calcedo, R.; Rouhani, F.; Campbell-Thompson, M.; Yachnis, A.T.; Sandhaus, R.A.; McElvaney, N.G.; et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing alpha1-antitrypsin: Interim results. Hum. Gene Ther. 2011, 22, 1239–1247. [Google Scholar] [CrossRef]
- Amann, T.; Hansen, A.H.; Kol, S.; Hansen, H.G.; Arnsdorf, J.; Nallapareddy, S.; Voldborg, B.; Lee, G.M.; Andersen, M.R.; Kildegaard, H.F. Glyco-engineered CHO cell lines producing alpha-1-antitrypsin and C1 esterase inhibitor with fully humanized N-glycosylation profiles. Metab. Eng. 2019, 52, 143–152. [Google Scholar] [CrossRef]
- Morifuji, Y.; Xu, J.; Karasaki, N.; Iiyama, K.; Morokuma, D.; Hino, M.; Masuda, A.; Yano, T.; Mon, H.; Kusakabe, T.; et al. Expression, Purification, and Characterization of Recombinant Human alpha1-Antitrypsin Produced Using Silkworm-Baculovirus Expression System. Mol. Biotechnol. 2018, 60, 924–934. [Google Scholar] [CrossRef]
- Seersholm, N.; Wencker, M.; Banik, N.; Viskum, K.; Dirksen, A.; Kok-Jensen, A.; Konietzko, N. Does alpha1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) alpha1-AT study group. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 1997, 10, 2260–2263. [Google Scholar] [CrossRef]
- Wencker, M.; Banik, N.; Buhl, R.; Seidel, R.; Konietzko, N. Long-term treatment of alpha1-antitrypsin deficiency-related pulmonary emphysema with human alpha1-antitrypsin. Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL)-alpha1-AT-study group. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 1998, 11, 428–433. [Google Scholar] [CrossRef]
- Wencker, M.; Fuhrmann, B.; Banik, N.; Konietzko, N. Longitudinal follow-up of patients with alpha(1)-protease inhibitor deficiency before and during therapy with IV alpha(1)-protease inhibitor. Chest 2001, 119, 737–744. [Google Scholar] [CrossRef]
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am. J. Respir. Crit. Care Med. 1998, 158, 49–59. [Google Scholar] [CrossRef]
- Stoller, J.K.; Fallat, R.; Schluchter, M.D.; O’Brien, R.G.; Connor, J.T.; Gross, N.; O’Neil, K.; Sandhaus, R.; Crystal, R.G. Augmentation therapy with alpha1-antitrypsin: Patterns of use and adverse events. Chest 2003, 123, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, A.R.; Rouhani, F.; Li, N.; Schreck, P.; Brantly, M.L. Alpha-1-antitrypsin augmentation therapy in deficient individuals enrolled in the Alpha-1 Foundation DNA and Tissue Bank. Int. J. Chronic Obstr. Pulm. Dis. 2009, 4, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Barros-Tizon, J.C.; Torres, M.L.; Blanco, I.; Martinez, M.T.; Investigators of the rEXA Study Group. Reduction of severe exacerbations and hospitalization-derived costs in alpha-1-antitrypsin-deficient patients treated with alpha-1-antitrypsin augmentation therapy. Ther. Adv. Respir. Dis. 2012, 6, 67–78. [Google Scholar] [CrossRef]
- Stockley, R.A.; Parr, D.G.; Piitulainen, E.; Stolk, J.; Stoel, B.C.; Dirksen, A. Therapeutic efficacy of alpha-1 antitrypsin augmentation therapy on the loss of lung tissue: An integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respir. Res. 2010, 11, 136. [Google Scholar] [CrossRef]
- McElvaney, N.G.; Burdon, J.; Holmes, M.; Glanville, A.; Wark, P.A.; Thompson, P.J.; Hernandez, P.; Chlumsky, J.; Teschler, H.; Ficker, J.H.; et al. Long-term efficacy and safety of alpha1 proteinase inhibitor treatment for emphysema caused by severe alpha1 antitrypsin deficiency: An open-label extension trial (RAPID-OLE). Lancet Respir. Med. 2017, 5, 51–60. [Google Scholar] [CrossRef]
- Chapman, K.R.; Chorostowska-Wynimko, J.; Koczulla, A.R.; Ferrarotti, I.; McElvaney, N.G. Alpha 1 antitrypsin to treat lung disease in alpha 1 antitrypsin deficiency: Recent developments and clinical implications. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 419–432. [Google Scholar] [CrossRef]
- Esteves Brandão, M.; Conde, B.; Seixas, S.; Clotilde Silva, J.; Fernandes, A. Pulmonary Emphysema in a Child With Alpha-1 Antitrypsin Deficiency: Evaluation of 2 Years of Intravenous Augmentation Therapy. Arch. Bronconeumol. 2019, 55, 502–504. [Google Scholar] [CrossRef]
- Menga, G.; Fernandez Acquier, M.; Echazarreta, A.L.; Sorroche, P.B.; Lorenzon, M.V.; Fernandez, M.E.; Saez, M.S.; grupo de estudio, D.A. Prevalence of Alpha-1 Antitrypsin Deficiency in COPD Patients in Argentina. The DAAT.AR Study. Arch. Bronconeumol. 2019. [Google Scholar] [CrossRef]
- Janciauskiene, S.; DeLuca, D.S.; Barrecheguren, M.; Welte, T.; Miravitlles, M.; Ancochea, J.; Badiola, C.; Sánchez, G.; Duran, E.; Río, F.G.; et al. Serum Levels of Alpha1-antitrypsin and Their Relationship with COPD in the General Spanish Population. Arch. Bronconeumol. 2020, 56, 76–83. [Google Scholar] [CrossRef]
- Lopez-Campos, J.L.; Casas-Maldonado, F.; Torres-Duran, M.; Medina-Gonzálvez, A.; Rodriguez-Fidalgo, M.L.; Carrascosa, I.; Calle, M.; Osaba, L.; Rapun, N.; Drobnic, E.; et al. Results of a diagnostic procedure based on multiplex technology on dried blood spots and buccal swabs for subjects with suspected alpha1 antitrypsin deficiency. Arch. Bronconeumol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Campos, J.L.; Carrasco Hernandez, L.; Marquez-Martin, E.; Ortega Ruiz, F.; Martinez Delgado, B. Diagnostic Performance of a Lateral Flow Assay for the Detection of Alpha-1-Antitrypsin Deficiency. Arch. Bronconeumol. 2020, 56, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.; Turner, A. What Do Alpha-1 Antitrypsin Levels Tell Us About Chronic Inflammation in COPD? Arch. Bronconeumol. 2020, 56, 72–73. [Google Scholar] [CrossRef] [PubMed]
- Sandhaus, R.A.; Turino, G.; Brantly, M.L.; Campos, M.; Cross, C.E.; Goodman, K.; Hogarth, D.K.; Knight, S.L.; Stocks, J.M.; Stoller, J.K.; et al. The Diagnosis and Management of Alpha-1 Antitrypsin Deficiency in the Adult. Chronic Obstr. Pulm. Dis. 2016, 3, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.P.; Mineiro, M.A.; Costa, F.; Gomes, J.; Santos, C.; Antunes, C.; Maia, D.; Melo, R.; Canotilho, M.; Magalhaes, E.; et al. Portuguese consensus document for the management of alpha-1-antitrypsin deficiency. Pulmonology 2018, 24 (Suppl. S1), 1–21. [Google Scholar] [CrossRef]
- Piitulainen, E.; Montero, L.C.; Nystedt-Duzakin, M.; Stoel, B.C.; Sveger, T.; Wollmer, P.; Tanash, H.A.; Diaz, S. Lung Function and CT Densitometry in Subjects with alpha-1-Antitrypsin Deficiency and Healthy Controls at 35 Years of Age. COPD 2015, 12, 162–167. [Google Scholar] [CrossRef]
- Stockley, R.A.; Edgar, R.G.; Pillai, A.; Turner, A.M. Individualized lung function trends in alpha-1-antitrypsin deficiency: A need for patience in order to provide patient centered management? Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 1745–1756. [Google Scholar] [CrossRef]
- Dawkins, P.A.; Dawkins, C.L.; Wood, A.M.; Nightingale, P.G.; Stockley, J.A.; Stockley, R.A. Rate of progression of lung function impairment in alpha1-antitrypsin deficiency. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 2009, 33, 1338–1344. [Google Scholar] [CrossRef]
- Clark, V.C. Liver Transplantation in Alpha-1 Antitrypsin Deficiency. Clin. Liver Dis. 2017, 21, 355–365. [Google Scholar] [CrossRef]
- Strange, C.; Stoller, J.K.; Sandhaus, R.A.; Dickson, R.; Turino, G. Results of a survey of patients with alpha-1 antitrypsin deficiency. Respir. Int. Rev. Thorac. Dis. 2006, 73, 185–190. [Google Scholar] [CrossRef]
- Wang, Y.; Cobanoglu, M.C.; Li, J.; Hidvegi, T.; Hale, P.; Ewing, M.; Chu, A.S.; Gong, Z.; Muzumdar, R.; Pak, S.C.; et al. An analog of glibenclamide selectively enhances autophagic degradation of misfolded alpha1-antitrypsin Z. PLoS ONE 2019, 14, e0209748. [Google Scholar] [CrossRef]
- Tang, Y.; Blomenkamp, K.S.; Fickert, P.; Trauner, M.; Teckman, J.H. NorUDCA promotes degradation of alpha1-antitrypsin mutant Z protein by inducing autophagy through AMPK/ULK1 pathway. PLoS ONE 2018, 13, e0200897. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.I.; Blomenkamp, K.; Peterson, R.M.; Subbotin, V.M.; Schwabe, C.; Hamilton, J.; Chu, Q.; Christianson, D.R.; Hegge, J.O.; Kolbe, J.; et al. Development of an RNAi therapeutic for alpha-1-antitrypsin liver disease. JCI Insight 2020. [Google Scholar] [CrossRef]
- Karadagi, A.; Johansson, H.; Zemack, H.; Salipalli, S.; Mork, L.M.; Kannisto, K.; Jorns, C.; Gramignoli, R.; Strom, S.; Stokkeland, K.; et al. Exogenous alpha 1-antitrypsin down-regulates SERPINA1 expression. PLoS ONE 2017, 12, e0177279. [Google Scholar] [CrossRef] [PubMed]
- Soy, D.; de la Roza, C.; Lara, B.; Esquinas, C.; Torres, A.; Miravitlles, M. Alpha-1-antitrypsin deficiency: Optimal therapeutic regimen based on population pharmacokinetics. Thorax 2006, 61, 1059–1064. [Google Scholar] [CrossRef]
- Vidal Pla, R.; Padullés Zamora, N.; Sala Piñol, F.; Jardí Margaleff, R.; Rodríguez Frías, F.; Montoro Ronsano, J.B. Pharmacokinetics of alpha1-antitrypsin replacement therapy in severe congenital emphysema. Arch. Bronconeumol. 2006, 42, 553–556. [Google Scholar] [CrossRef]
- Greulich, T.; Chlumsky, J.; Wencker, M.; Vit, O.; Fries, M.; Chung, T.; Shebl, A.; Vogelmeier, C.; Chapman, K.R.; McElvaney, N.G.; et al. Safety of biweekly alpha1-antitrypsin treatment in the RAPID programme. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 2018, 52. [Google Scholar] [CrossRef]
- Campos, M.A.; Kueppers, F.; Stocks, J.M.; Strange, C.; Chen, J.; Griffin, R.; Wang-Smith, L.; Brantly, M.L. Safety and pharmacokinetics of 120 mg/kg versus 60 mg/kg weekly intravenous infusions of alpha-1 proteinase inhibitor in alpha-1 antitrypsin deficiency: A multicenter, randomized, double-blind, crossover study (SPARK). COPD 2013, 10, 687–695. [Google Scholar] [CrossRef]
- Esquinas, C.; Miravitlles, M. Are There Differences Between the Available Treatments for Emphysema Associated with Alpha-1 Antitrypsin Deficiency? Arch. Bronconeumol. 2018, 54, 451–452. [Google Scholar] [CrossRef]
- Boerema, D.J.; An, B.; Gandhi, R.P.; Papineau, R.; Regnier, E.; Wilder, A.; Molitor, A.; Tang, A.P.; Kee, S.M. Biochemical comparison of four commercially available human alpha1-proteinase inhibitors for treatment of alpha1-antitrypsin deficiency. Biologicals 2017, 50, 63–72. [Google Scholar] [CrossRef]
- Cowden, D.I.; Fisher, G.E.; Weeks, R.L. A pilot study comparing the purity, functionality and isoform composition of alpha-1-proteinase inhibitor (human) products. Curr. Med. Res. Opin. 2005, 21, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Tirado-Conde, G.; Lara, B.; Miravitlles, M. Augmentation therapy for emphysema due to alpha-1-antitrypsin deficiency. Ther. Adv. Respir. Dis. 2008, 2, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.; Dirksen, A.; Turner, A.M. Treatment of lung disease. In Alpha1-Antitrypsin Deficiency; Strnad, P., Brantly, M.L., Bals, R., Eds.; ERS: Lausanne, Switzerland, 2019; pp. 78–92. [Google Scholar]
- McElvaney, O.J.; Carroll, T.P.; Franciosi, A.N.; Sweeney, J.; Hobbs, B.D.; Kowlessar, V.; Gunaratnam, C.; Reeves, E.P.; McElvaney, N.G. Consequences of Abrupt Cessation of Alpha1-Antitrypsin Replacement Therapy. N. Engl. J. Med. 2020, 382, 1478–1480. [Google Scholar] [CrossRef] [PubMed]
- Kleinerova, J.; Ging, P.; Rutherford, C.; Lawrie, I.; Winward, S.; Eaton, D.; Redmond, K.C.; Egan, J.J. The withdrawal of replacement therapy and outcomes in alpha-1 antitrypsin deficiency lung transplant recipients. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 2019, 53. [Google Scholar] [CrossRef]
- Murphy, M.P.; McEnery, T.; McQuillan, K.; McElvaney, O.F.; McElvaney, O.J.; Landers, S.; Coleman, O.; Bussayajirapong, A.; Hawkins, P.; Henry, M.; et al. alpha1 Antitrypsin therapy modulates the neutrophil membrane proteome and secretome. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 2020, 55. [Google Scholar] [CrossRef]
- Tanash, H.A.; Riise, G.C.; Hansson, L.; Nilsson, P.M.; Piitulainen, E. Survival benefit of lung transplantation in individuals with severe alpha1-anti-trypsin deficiency (PiZZ) and emphysema. J. Heart Lung Transpl. 2011, 30, 1342–1347. [Google Scholar] [CrossRef]
- Stone, H.M.; Edgar, R.G.; Thompson, R.D.; Stockley, R.A. Lung Transplantation in Alpha-1-Antitrypsin Deficiency. COPD 2016, 13, 146–152. [Google Scholar] [CrossRef]
- Thabut, G. Estimating the Survival Benefit of Lung Transplantation: Considering the Disease Course during the Wait. Ann. Am. Thorac. Soc. 2017, 14, 163–164. [Google Scholar] [CrossRef]
- King, M.B.; Campbell, E.J.; Gray, B.H.; Hertz, M.I. The proteinase-antiproteinase balance in alpha-1-proteinase inhibitor-deficient lung transplant recipients. Am. J. Respir. Crit. Care Med. 1994, 149, 966–971. [Google Scholar] [CrossRef]
- Iskender, I.; Sakamoto, J.; Nakajima, D.; Lin, H.; Chen, M.; Kim, H.; Guan, Z.; Del Sorbo, L.; Hwang, D.; Waddell, T.K.; et al. Human alpha1-antitrypsin improves early post-transplant lung function: Pre-clinical studies in a pig lung transplant model. J. Heart Lung Transpl. 2016, 35, 913–921. [Google Scholar] [CrossRef]
- Emtiazjoo, A.M.; Hu, H.; Lu, L.; Brantly, M.L. Alpha-1 Antitrypsin Attenuates Acute Lung Allograft Injury in a Rat Lung Transplant Model. Transpl. Direct 2019, 5, e458. [Google Scholar] [CrossRef] [PubMed]
- Miravitlles, M.; Dirksen, A.; Ferrarotti, I.; Koblizek, V.; Lange, P.; Mahadeva, R.; McElvaney, N.G.; Parr, D.; Piitulainen, E.; Roche, N.; et al. European Respiratory Society statement: Diagnosis and treatment of pulmonary disease in alpha1-antitrypsin deficiency. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 2017, 50, 1700610. [Google Scholar] [CrossRef] [PubMed]
- Piras, B.; Ferrarotti, I.; Lara, B.; Martinez, M.T.; Bustamante, A.; Ottaviani, S.; Pirina, P.; Luisetti, M.; Miravitlles, M. Clinical phenotypes of Italian and Spanish patients with alpha1-antitrypsin deficiency. Eur. Respir. J. Off. J. Eur. Soc. Clin. Respir. Physiol. 2013, 42, 54–64. [Google Scholar] [CrossRef]
- Matamala, N.; Lara, B.; Gómez-Mariano, G.; Martínez, S.; Vázquez-Domízquez, I.; Otero-Sobrino, Á.; Muñoz-Callejas, A.; Sánchez, E.; Esquinas, C.; Bustamante, A.; et al. miR-320c Regulates SERPINA1 Expression and Is Induced in Patients With Pulmonary Disease. Arch. Bronconeumol. 2020. [Google Scholar] [CrossRef]
- Matamala, N.; Lara, B.; Gomez-Mariano, G.; Martinez, S.; Retana, D.; Fernandez, T.; Silvestre, R.A.; Belmonte, I.; Rodriguez-Frias, F.; Vilar, M.; et al. Characterization of Novel Missense Variants of SERPINA1 Gene Causing Alpha-1 Antitrypsin Deficiency. Am. J. Respir. Cell Mol. Biol. 2018, 58, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Brecher, A.S.; Thevananther, S.; Franco-Saenz, R. Acetaldehyde inhibits the anti-elastase activity of alpha 1-antitrypsin. Alcohol 1994, 11, 181–185. [Google Scholar] [CrossRef]
- Gadek, J.E.; Fells, G.A.; Crystal, R.G. Cigarette smoking induces functional antiprotease deficiency in the lower respiratory tract of humans. Science 1979, 206, 1315–1316. [Google Scholar] [CrossRef]
- Cohen, M.D.; Sisco, M.; Baker, K.; Chen, L.C.; Schlesinger, R.B. Rapid communication: Effect of inhaled chromium on pulmonary A1AT. Inhal. Toxicol. 2002, 14, 765–771. [Google Scholar] [CrossRef]
- Carter, R.I.; Mumford, R.A.; Treonze, K.M.; Finke, P.E.; Davies, P.; Si, Q.; Humes, J.L.; Dirksen, A.; Piitulainen, E.; Ahmad, A.; et al. The fibrinogen cleavage product Aα-Val360, a specific marker of neutrophil elastase activity in vivo. Thorax 2011, 66, 686–691. [Google Scholar] [CrossRef]
- Stephenson, S.E.; Wilson, C.L.; Crothers, K.; Attia, E.F.; Wongtrakool, C.; Petrache, I.; Schnapp, L.M. Impact of HIV infection on α(1)-antitrypsin in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L583–L592. [Google Scholar] [CrossRef]
- Cook, L.; Burdon, J.; Brenton, S.; Janus, E.D.; Knight, K. Alpha-1-antitrypsin PLowell: A normally functioning variant present in low concentration. Aust. New Zealand J. Med. 1995, 25, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Laffranchi, M.; Elliston, E.L.K.; Gangemi, F.; Berardelli, R.; Lomas, D.A.; Irving, J.A.; Fra, A. Characterisation of a type II functionally-deficient variant of alpha-1-antitrypsin discovered in the general population. PLoS ONE 2019, 14, e0206955. [Google Scholar] [CrossRef]
- Fujita, J.; Nelson, N.L.; Daughton, D.M.; Dobry, C.A.; Spurzem, J.R.; Irino, S.; Rennard, S.I. Evaluation of elastase and antielastase balance in patients with chronic bronchitis and pulmonary emphysema. Am. Rev. Respir. Dis. 1990, 142, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Abboud, R.T.; Vimalanathan, S. Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int. J. Tuberc. Lung Dis. Off. J. Int. Union Tuberc. Lung Dis. 2008, 12, 361–367. [Google Scholar]
- Barrecheguren, M.; Torres-Duran, M.; Casas-Maldonado, F.; Miravitlles, M. Spanish Implementation of the New International Alpha-1 Anitrypsin Deficiency International Registry: The European Alpha-1 Research Collaboration (EARCO). Arch. Bronconeumol. 2020. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | N | Age | FEV1 | Mutations | |||
|---|---|---|---|---|---|---|---|
| Eligibility Criteria | Cohort Results * | Eligibility Criteria | Cohort Result * | Eligibility Criteria | Cohort Result | ||
| Observational studies with control | |||||||
| Seersholm N, et al.; WALT. Eur Respir J, 1997 [18] | 295 | NR | 46 (8) | FEV1 < 65% or decline > 120 mL/year | 37 (14)% | Severe AATD (NR) | NR |
| NHLBI Registry. AJRCCM, 1998 [21] | 927 | ≥18 years | 46 (11) | NR | <35%: 43.6% 35–49%: 21.1% 50–79%: 16.2% ≥80%: 19.1% | AAT ≤ 11 µM or ZZ | NR |
| Wencker M, et al.; WALT. Eur Respir J, 1998 [19] | 443 | >18 years | 45 (7) | FEV1 < 65% or decline > 120 mL/year | Ex-smokers: 35.5 (14.8)% Non-smokers: 42.2 (18.2)% | AAT < 50 mg/dL (nephelometry) AAT < 80 mg/dL (immunodiffusion) | ZZ: 88.9% SZ: 7.1% Other/unknown: 4.0% |
| Wencker M, et al.; WALT. Chest, 2001 [20] | 96 | NR | 44 (8) | FEV1 < 65% or decline > 120 mL/year | 41 (17.3)% | AAT < 35% | ZZ: 85% SZ: 8% Other: 3% |
| Stoller JK, et al.; NHLBI. Chest, 2003 [22] | 747 | NR | 48 (9) | NR | 37 (18)% | AAT ≤ 11 µM or ZZ or null | NR |
| Tonelli AR, et al. Int J COPD, 2009 [23] | 164 | NR | 61.3 (0.7) | NR | 43 (2)% | ZZ | ZZ |
| Barros-Tizón JC, et al. Ther Adv Respir Dis, 2012 [24] | 127 | >18 years | 51.7 (9.1) | NR | 1.25 (0.50) L | AAT ≤ 11 µM (50 mg/dL) and ZZ. rare or null | ZZ: 93.6% SZ: 0.8% Other: 5.6% |
| Clinical trials | |||||||
| Dirksen A, et al.; Danish–Dutch study. AJRCCM, 1999 [3] | 56 | NR | Danish: 50.4 (1.62) Dutch 45.1 (1.17) | FEV1 30–80% | Danish: 1.5 (0.9) L Dutch: 1.6 (0.1) L | ZZ | ZZ |
| Dirksen A, et al.; EXACTLE. Eur Respir J, 2009 [2] | 77 | ≥18 years | 54.7 (8.4) | NR | 46.3 (19.6)% | AAT ≤ 11 µM | ZZ or Zn |
| Chapman KR, et al.; RAPID. Lancet, 2015 [4] | 180 | 18–65 | 53.8 (6.9) | FEV1 35–70% | 47.4 (12.1)% | AAT ≤ 11 µM | ZZ: 93% Other: 7% |
| Prolastin | Zemaira/Respreeza | |
|---|---|---|
| Origin * | Human donor plasma | Human donor plasma |
| Presentations * | 1 gr + 40 mL solvent (25 mg/mL) | 1 gr + 20 mL solvent (50 mg/mL) 4 gr + 76 mL solvent (50 mg/mL) 5 gr + 95 mL solvent (50 mg/mL) |
| Excipients * | Powder: Sodium chloride Sodium dihydrogen phosphate Solvent: Water for injections | Powder: Sodium chloride Sodium dihydrogen phosphate monohydrate Mannitol Solvent: Water for injections |
| Purity (AAT/proteins) [49] | 76.9% | 97.4% |
| Specific activity (active AAT/proteins) [49] | 64% | 86.2% |
| Infusion velocity * | 0.08 mL/kg/min | 0.08 mL/kg/min |
| Time of infusion 60 mg/kg dose † | 30 min | 15 min |
| Time of infusion 120 mg/kg dose ‡ | 60 min | 30 min |
| Lung density decline reduction [4,25] | Versus basal: −1.73 g/L/year Versus placebo: 1.01 g/L/year | Versus basal: −1.45 g/L/year Versus placebo: 0.74 g/L/year |
| In Favor | Against |
|---|---|
| The biochemical efficacy is expected to be the same as in non-transplanted AATD patients. | There are no formal trials on its clinical efficacy in lung density deterioration after transplant. |
| Augmentation therapy is safe and well-tolerated, and patients get used to it as part of their lives. It is not expected to create an additional burden. | Lung transplant patients already have to cope with a considerable amount of medication with potential adverse effects that determine their lives, without adding another treatment of unproven efficacy in this context. |
| AATD lung-transplant patients are generally younger, with a longer life expectancy, so it is vital to take all the necessary measures to protect the transplanted lung. | Emphysema due to AATD is a slow, progressive disease. It may take decades until clinically relevant emphysema is developed in the new lung. |
| The number of lung donors is limited, so every transplant has an opportunity cost, since it could have been received by another patient. Therefore, it is unethical not to take all possible steps to preserve the transplanted lung. | It has not been proven that the risk of rejection is increased if recipients do not receive augmentation therapy. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Campos, J.L.; Carrasco Hernandez, L.; Caballero Eraso, C. Implications of a Change of Paradigm in Alpha1 Antitrypsin Deficiency Augmentation Therapy: From Biochemical to Clinical Efficacy. J. Clin. Med. 2020, 9, 2526. https://doi.org/10.3390/jcm9082526
López-Campos JL, Carrasco Hernandez L, Caballero Eraso C. Implications of a Change of Paradigm in Alpha1 Antitrypsin Deficiency Augmentation Therapy: From Biochemical to Clinical Efficacy. Journal of Clinical Medicine. 2020; 9(8):2526. https://doi.org/10.3390/jcm9082526
Chicago/Turabian StyleLópez-Campos, José Luis, Laura Carrasco Hernandez, and Candelaria Caballero Eraso. 2020. "Implications of a Change of Paradigm in Alpha1 Antitrypsin Deficiency Augmentation Therapy: From Biochemical to Clinical Efficacy" Journal of Clinical Medicine 9, no. 8: 2526. https://doi.org/10.3390/jcm9082526
APA StyleLópez-Campos, J. L., Carrasco Hernandez, L., & Caballero Eraso, C. (2020). Implications of a Change of Paradigm in Alpha1 Antitrypsin Deficiency Augmentation Therapy: From Biochemical to Clinical Efficacy. Journal of Clinical Medicine, 9(8), 2526. https://doi.org/10.3390/jcm9082526