Microbial Signature in Adipose Tissue of Crohn’s Disease Patients

 ,
,  , , , ,
, , , ,
Abstract
1. Introduction
2. Material and Methods
2.1. Study Design
2.2. RNA Extraction from Tissues
2.3. PCR Amplification and Analysis of 16S-rRNA Sequences
2.4. Bioinformatic Analysis
2.5. Statistical Analysis
2.6. Ethics Approval and Consent to Participate
3. Results
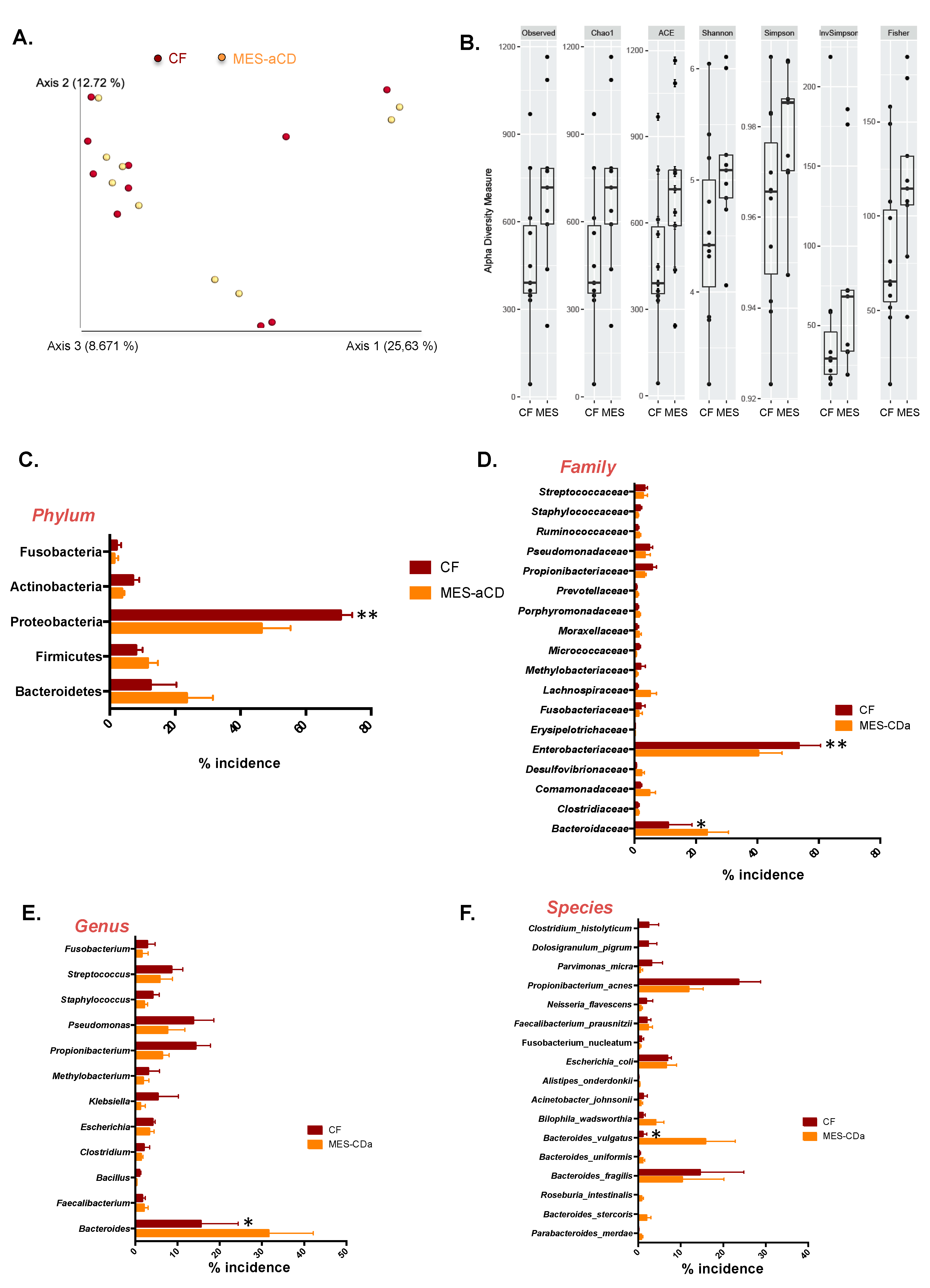
3.1. Microbiome Signature of Mesenteric Adipose Tissue in Patients with Crohn’s Disease
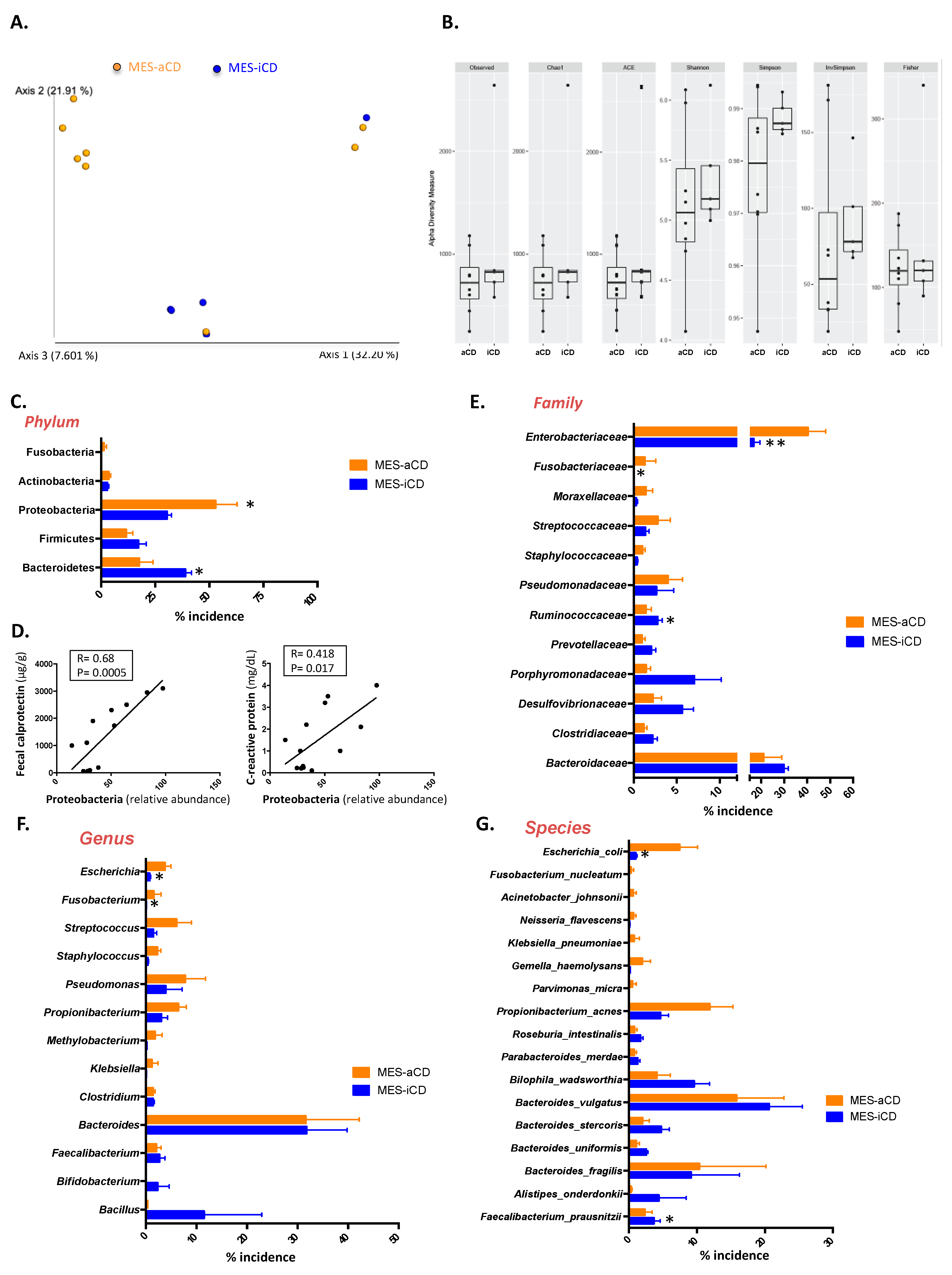
3.2. The Microbiome Signature in Mesenteric Adipose Tissue in Crohn’s Disease Is Related to the Clinical Status
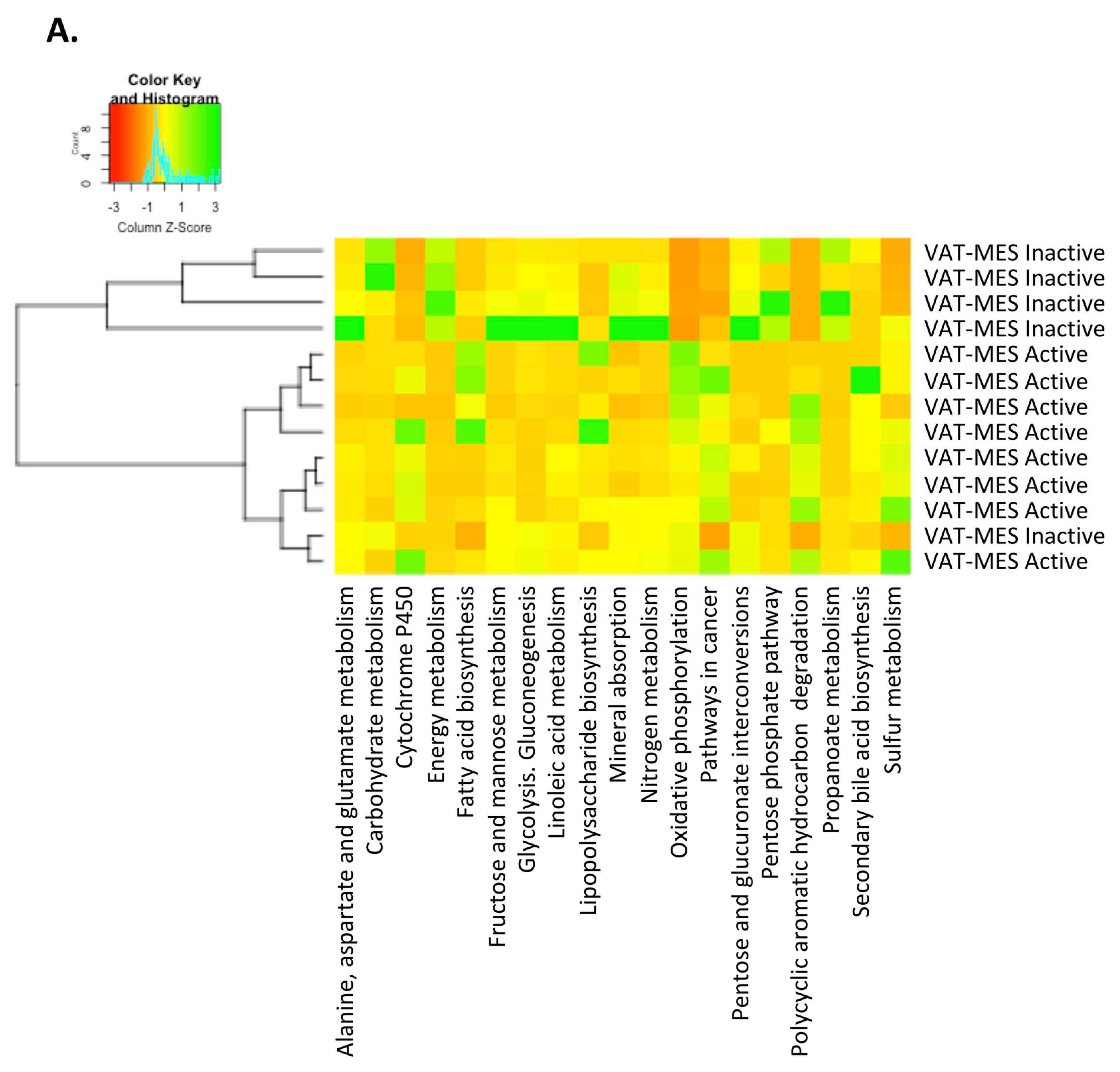
3.3. Functional Differences in the Gut Microbiota in Mesenteric Adipose Tissue between Active and Inactive Crohn’s Disease
4. Discussion
5. Conclusions
List of Abbreviations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B.; Mazmanian, S.K. Intestinal microbes in inflammatory bowel diseases. Am. J. Gastroenterol. Suppl. 2012, 1, 15–21. [Google Scholar] [CrossRef]
- Mosca, A.; Leclerc, M.; Hugot, J.P. Gut microbiota diversity and human diseases: Should we reintroduce key predators in our ecosystem? Front. Microbiol. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef]
- McIlrath, D.C. Diverting ileostomy or colostomy in the management of Crohn’s disease of the colon. Arch. Surg. 1971, 103, 308–310. [Google Scholar] [CrossRef]
- Zelas, P.; Jagelman, D.G. Loop ileostomy in the management of Crohn’s colitis in the debilitated patient. Ann. Surg. 1980, 191, 164–168. [Google Scholar] [CrossRef]
- Harper, P.H.; Truelove, S.C.; Lee, E.C.G.; Kettlewell, M.G.; Jewell, D.P. Split ileostomy and ileocolostomy for Crohn’s disease of the colon and ulcerative colitis: A 20 year survey. Gut 1983, 24, 106–113. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, W.; Gong, J.; Shen, B. The role of the mesentery in Crohn’s disease. Lancet Gastroenterol. Hepatol. 2017, 2, 244–245. [Google Scholar] [CrossRef]
- Buskens, C.J.; de Groof, E.J.; Bemelman, W.A.; Wildenberg, M.E. The role of the mesentery in Crohn’s disease. Lancet Gastroenterol. Hepatol. 2017, 2, 245–246. [Google Scholar] [CrossRef]
- Coffey, J.C.; O’Leary, D.P. The mesentery: Structure, function, and role in disease. Lancet Gastroenterol. Hepatol. 2016, 1, 238–247. [Google Scholar] [CrossRef]
- Siegmund, B. Mesenteric fat in Crohn’s disease: The hot spot of inflammation? Gut 2012, 61, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Büning, C.; Von Kraft, C.; Hermsdorf, M.; Gentz, E.; Wirth, E.K.; Valentini, L.; Haas, V. Visceral adipose tissue in patients with Crohn’s disease correlates with disease activity, inflammatory markers, and outcome. Inflamm. Bowel Dis. 2015, 21, 2590–2597. [Google Scholar] [CrossRef]
- Connelly, T.M.; Juza, R.M.; Sangster, W.; Sehgal, R.; Tappouni, R.F.; Messaris, E. Volumetric fat ratio and not body mass index is predictive of ileocolectomy outcomes in Crohn’s disease patients. Dig. Surg. 2014, 31, 219–224. [Google Scholar] [CrossRef]
- Gonçalves, P.; Magro, F.; Martel, F. Metabolic inflammation in inflammatory bowel disease: Crosstalk between adipose tissue and bowel. Inflamm. Bowel Dis. 2015, 21, 453–467. [Google Scholar] [CrossRef]
- Zulian, A.; Cancello, R.; Ruocco, C.; Gentilini, D.; Di Blasio, A.M.; Danelli, P.; Micheletto, G.; Cesana, E.; Invitti, C. Differences in visceral fat and fat bacterial colonization between ulcerative colitis and Crohn’s disease. An in vivo and in vitro study. PLoS ONE 2013, 8, e78495. [Google Scholar] [CrossRef]
- Pachón-Peña, G.; Serena, C.; Ejarque, M.; Petriz, J.; Duran, X.; Oliva-Olivera, W.; Simó, R.; Tinahones, F.J.; Fernández-Veledo, S.; Vendrell, J. Obesity determines the immunophenotypic profile and functional characteristics of human mesenchymal stem cells from adipose tissue. Stem Cells Transl. Med. 2016, 5, 464–475. [Google Scholar] [CrossRef]
- Serena, C.; Keiran, N.; Ceperuelo-Mallafre, V.; Ejarque, M.; Fradera, R.; Roche, K.; Nuñez-Roa, C.; Vendrell, J.; Fernández-Veledo, S. Obesity and type 2 diabetes alters the immune properties of human adipose derived stem cells. Stem Cells 2016, 34, 2559–2573. [Google Scholar] [CrossRef]
- Serena, C.; Keiran, N.; Madeira, A.; Maymó-Masip, E.; Ejarque, M.; Terrón-Puig, M.; Espin, E.; Martí, M.; Borruel, N.; Guarner, F.; et al. Crohn’s disease disturbs the immune properties of human adipose-derived stem cells related to inflammasome activation. Stem Cell Rep. 2017, 9, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Burcelin, R.; Serino, M.; Chabo, C.; Garidou, L.; Pomié, C.; Courtney, M.; Amar, J.; Bouloumié, A. Metagenome and metabolism: The tissue microbiota hypothesis. Diabetes Obes. Metab. 2013, 15, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Zulian, A.; Cancello, R.; Cesana, E.; Rizzi, E.; Consolandi, C.; Severgnini, M.; Panizzo, V.; Di Blasio, A.M.; Micheletto, G.; Invitti, C. Adipose tissue microbiota in humans: An open issue. Int. J. Obes. 2016, 40, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- de Goffau, M.C.; Lager, S.; Sovio, U.; Gaccioli, F.; Cook, E.; Peacock, S.J.; Parkhill, J.; Charnock-Jones, D.S.; Smith, G.C.S. Human placenta has no microbiome but can contain potential pathogens. Nature 2019, 572, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Anhê, F.F.; Jensen, B.A.H.; Varin, T.V.; Servant, F.; Van Blerk, S.; Richard, D.; Marceau, S.; Surette, M.; Biertho, L.; Lelouvier, B.; et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat. Metab. 2020, 2, 233–242. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Scharl, M.; Sempere, L.; Holler, E.; Zapater, P.; Almenta, I.; González-Navajas, J.M.; Such, J.; Wiest, R.; Rogler, G.; et al. Genetic susceptibility to increased bacterial translocation influences the response to biological therapy in patients with Crohn’s disease. Gut 2014, 63, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Laffineur, G.; Lescut, D.; Vincent, P.; Quandalle, P.; Wurtz, A.; Colombel, J.F. Bacterial translocation in Crohn disease. Gastroenterol. Clin. Biol. 1992, 16, 777–781. [Google Scholar]
- Best, W.R.; Becktel, J.M.; Singleton, J.W.; Kern, F. Development of a Crohn’s disease activity index. Gastroenterology 1976, 70, 439–444. [Google Scholar] [CrossRef]
- Van Assche, G.; Dignass, A.; Reinisch, W.; van der Woude, C.J.; Sturm, A.; De Vos, M.; Guslandi, M.; Oldenburg, B.; Dotan, I.; Marteau, P.; et al. The second European evidence-based Consensus on the diagnosis and management of Crohn’s disease: Special situations. J. Crohns Colitis 2010, 4, 63–101. [Google Scholar] [CrossRef]
- Leiva-Gea, I.; Sánchez-Alcoholado, L.; Martín-Tejedor, B.; Castellano-Castillo, D.; Moreno-Indias, I.; Urda-Cardona, A.; Tinahones, F.J.; Fernández-García, J.C.; Queipo-Ortuño, M.I. Gut microbiota differs in composition and functionality between children with type 1 diabetes and MODY2 and healthy control subjects: A case-control study. Diabetes Care 2018, 41, 2385–2395. [Google Scholar] [CrossRef]
- Bhute, S.; Pande, P.; Shetty, S.A.; Shelar, R.; Mane, S.; Kumbhare, S.V.; Gawali, A.; Makhani, H.; Navandar, M.; Dhotre, D.; et al. Molecular characterization and meta-analysis of gut microbial communities illustrate enrichment of prevotella and megasphaera in Indian subjects. Front. Microbiol. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alcoholado, L.; Castellano-Castillo, D.; Jordán-Martínez, L.; Moreno-Indias, I.; Cardila-Cruz, P.; Elena, D.; Muñoz-Garcia, A.J.; Queipo-Ortuño, M.I.; Jimenez-Navarro, M. Role of gut microbiota on cardio-metabolic parameters and immunity in coronary artery disease patients with and without type-2 diabetes mellitus. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Hieken, T.J.; Chen, J.; Hoskin, T.L.; Walther-Antonio, M.; Johnson, S.; Ramaker, S.; Xiao, J.; Radisky, D.C.; Knutson, K.L.; Kalari, K.R.; et al. The microbiome of aseptically collected human breast tissue in benign and malignant disease. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chiang, H.I.; Jiang, S.B.; Nagarajan, H.; Zengler, K.; Gallo, R.L. The microbiome extends to subepidermal compartments of normal skin. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Pedicino, D.; Severino, A.; Ucci, S.; Bugli, F.; Flego, D.; Giglio, A.F.; Trotta, F.; Ruggio, A.; Lucci, C.; Iaconelli, A.; et al. Epicardial adipose tissue microbial colonization and inflammasome activation in acute coronary syndrome. Int. J. Cardiol. 2017, 236, 95–99. [Google Scholar] [CrossRef]
- Lluch, J.; Servant, F.; Païssé, S.; Valle, C.; Valière, S.; Kuchly, C.; Vilchez, G.; Donnadieu, C.; Courtney, M.; Burcelin, R.; et al. The characterization of novel tissue microbiota using an optimized 16S metagenomic sequencing pipeline. PLoS ONE 2015, 10, 1–22. [Google Scholar] [CrossRef]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef]
- Antony, K.M.; Ma, J.; Mitchell, K.B.; Racusin, D.A.; Versalovic, J.; Aagaard, K. The preterm placental microbiome varies in association with excess maternal gestational weight gain. Am. J. Obstet. Gynecol. 2015, 212, 653.e1–653.e16. [Google Scholar] [CrossRef]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Cani, P.D.; Van Hul, M. Microbial signatures in metabolic tissues: A novel paradigm for obesity and diabetes? Nat. Metab. 2020, 2, 211–212. [Google Scholar] [CrossRef]
- Gophna, U.; Sommerfeld, K.; Gophna, S.; Doolittle, W.F.; Van Zanten, S.J.O.V. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J. Clin. Microbiol. 2006, 44, 4136–4141. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, K.G.; Walsh, D.; Walsh, L.; Mirapeix, R.; Lamers, W.; Dockery, P.; McDermott, K.; Coffey, J.C. AB060. 207. Digital reconstruction of human mesentery development. Mesentery Peritoneum 2019, 3, AB060. [Google Scholar] [CrossRef]
- Coffey, C.J.; Kiernan, M.G.; Sahebally, S.M.; Jarrar, A.; Burke, J.P.; Kiely, P.A.; Shen, B.; Waldron, D.; Peirce, C.; Moloney, M.; et al. Inclusion of the mesentery in ileocolic resection for Crohn’s disease is associated with reduced surgical recurrence. J. Crohns Colitis 2018, 12, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of enterobacteriaceae. Cell Host Microbe 2007, 2, 119–129. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.-L.; Barnich, N.; Bringer, M.-A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.-F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef]
- Sokol, H.; Seksik, P.; Rigottier-Gois, L.; Lay, C.; Lepage, P.; Podglajen, I.; Marteau, P.; Doré, J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 106–111. [Google Scholar] [CrossRef]
- Chassaing, B.; Darfeuille–Michaud, A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011, 140, 1720–1728.e3. [Google Scholar] [CrossRef]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Benjamin, J.L.; Hedin, C.R.H.; Koutsoumpas, A.; Ng, S.C.; McCarthy, N.E.; Prescott, N.J.; Pessoa-Lopes, P.; Mathew, C.G.; Sanderson, J.; Hart, A.L.; et al. Smokers with active Crohn’s disease have a clinically relevant dysbiosis of the gastrointestinal microbiota. Inflamm. Bowel Dis. 2012, 18, 1092–1100. [Google Scholar] [CrossRef]
- Ohkusa, T.; Yoshida, T.; Sato, N.; Watanabe, S.; Tajiri, H.; Okayasu, I. Commensal bacteria can enter colonic epithelial cells and induce proinflammatory cytokine secretion: A possible pathogenic mechanism of ulcerative colitis. J. Med. Microbiol. 2009, 58, 535–545. [Google Scholar] [CrossRef]
- Ohkusa, T.; Sato, N.; Ogihara, T.; Morita, K.; Ogawa, M.; Okayasu, I. Fusobacterium varium localized in the colonic mucosa of patients with ulcerative colitis stimulates species-specific antibody. J. Gastroenterol. Hepatol. 2002, 17, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Ohkusa, T.; Okayasu, I.; Ogihara, T.; Morita, K.; Ogawa, M.; Sato, N. Induction of experimental ulcerative colitis by Fusobacterium varium isolated from colonic mucosa of patients with ulcerative colitis. Gut 2003, 52, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Kaplan, G.G.; Beck, P.L.; Rioux, K.; Panaccione, R.; Devinney, R.; Lynch, T.; Allen-Vercoe, E. Invasive potential of gut mucosa-derived fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm. Bowel Dis. 2011, 17, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Torres, J.; Hu, N.; Medrano-Guzman, R.; Herrera-Goepfert, R.; Humphrys, M.S.; Wang, L.; Wang, C.; Ding, T.; Ravel, J.; et al. Molecular characterization of the human stomach microbiota in Gastric Cancer Patients. Front. Cell. Infect. Microbiol. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Rocha, C.; Kabakchiev, B.; Borowski, K.; Turpin, W.; Boland, K.; Milgrom, R.; Stempak, J.M.; Smith, M.I.; Silverberg, M.S. 257—Higher abundance of bile acid-metabolizing microbiota is associated with type of disease, biopsy location and mucosal inflammation in inflammatory bowel disease patients. Gastroenterology 2019, 156, S-49. [Google Scholar] [CrossRef]
- Carbonero, F.; Benefiel, A.C.; Alizadeh-Ghamsari, A.H.; Gaskins, H.R. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 2012, 3, 448. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Llopis, M.; Antolin, M.; Carol, M.; Borruel, N.; Casellas, F.; Martinez, C.; Espín-Basany, E.; Guarner, F.; Malagelada, J.R. Lactobacillus casei downregulates commensals’ inflammatory signals in Crohn’s disease mucosa. Inflamm. Bowel Dis. 2009, 15, 275–283. [Google Scholar] [CrossRef]
- Varela, E.; Manichanh, C.; Gallart, M.; Torrejón, A.; Borruel, N.; Casellas, F.; Guarner, F.; Antolin, M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2013, 38, 151–161. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wang, H.; Liao, W.D.; Peng, C.; Shu, X.; Zhu, X.; Zhu, Z.H. Characteristics of mucosa-associated gut microbiota during treatment in Crohn’s disease. World J. Gastroenterol. 2019, 25, 2204–2216. [Google Scholar] [CrossRef] [PubMed]
- Iyadorai, T.; Mariappan, V.; Vellasamy, K.M.; Wanyiri, J.W.; Roslani, A.C.; Lee, G.K.; Sears, C.; Vadivelu, J. Prevalence and association of pks+ Escherichia coli with colorectal cancer in patients at the University Malaya Medical Centre, Malaysia. PLoS ONE 2020, 15, e0228217. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, J.; Yao, H.; Hu, H. Fusobacterium and colorectal cancer. Front. Oncol. 2018, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- Allen-Vercoe, E.; Jobin, C. Fusobacterium and enterobacteriaceae: Important players for CRC? Immunol. Lett. 2014, 162, 54–61. [Google Scholar] [CrossRef]
- Sebastian, S.; Hernández, V.; Myrelid, P.; Kariv, R.; Tsianos, E.; Toruner, M.; Marti-Gallostra, M.; Spinelli, A.; van der Meulen-de Jong, A.E.; Yuksel, E.S.; et al. Colorectal cancer in inflammatory bowel disease: Results of the 3rd ECCO pathogenesis scientific workshop (I). J. Crohns Colitis 2014, 8, 5–18. [Google Scholar] [CrossRef]
- dos Santos, S.C.D.; Barbosa, L.A.R. Crohn’s disease: Risk factor for colorectal cancer. J. Coloproctol. 2017, 37, 55–62. [Google Scholar] [CrossRef][Green Version]
- Freeman, H.J. Colorectal cancer risk in Crohn’s disease. World J. Gastroenterol. 2008, 14, 1810. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Control | Inactive CD | Active CD | |
|---|---|---|---|
| n | 8 | 5 | 8 |
| Sex (male/female) | 4/4 | 2/3 | 4/4 |
| Age (years) | 46.1 (37.2–55.1) | 47.6 (35.2–60.1) | 45.8 (35.4–58.5) |
| BMI (kg/m2) | 25.8 (20.2–29.1) | 21.59 (20.1–24.1) | 23.8 (20.5–27.3) |
| Glucose (mg/dL) | 96.6 ± 6.3 | 94.6 ± 5.4 | 84.83 ± 4.7a |
| Smoking, n (%) | 4 (50) | 3 (60) | 4 (50) |
| Cholesterol (mg/dL) | 131 ± 11.2 | 118.2 ± 10.9 | 119.2 ± 15 |
| HDL (mg/dL) | 30.7 ± 4.5 | 31.13 ± 3.2 | 31.4 ± 6.4 |
| Triglycerides (mg/dL) | 186 (167.2–205.5) | 186 (170.3–199.2) | 119.6 (100.9–140) a,b |
| Insulin (µIU/mL) | 2.05 ± 1.9 | 4.98 ± 1.1 a | 6.12 ± 2.3 a |
| HOMA-IR | 0.7 ± 0.4 | 1.18 ± 0.5 | 1.44 ± 0.7 a,b |
| Age at diagnosis (years) | 26.5 ± 6.6 | 28.1 ± 8.5 | |
| Time in remission (months) | 29 ± 9.3 | ||
| Indication of surgery | CoH | CoH | 5SCD/5FCD |
| Immunomodulator use | 5/5 | 10/10 | |
| Biological agent treatment | 2/5 | 4/10 | |
| Steroid treatment | 4/5 | 7/10 | |
| C-reactive protein (mg/dL) | 0.05 ± 0.04 | 0.3 ± 0.13 a | 3.4 ± 1.6 a,b |
| Fecal calprotectin (µg/g) | 81.25 ± 20.3 | 2132.4 ± 159.1 b |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serena, C.; Queipo-Ortuño, M.; Millan, M.; Sanchez-Alcoholado, L.; Caro, A.; Espina, B.; Menacho, M.; Bautista, M.; Monfort-Ferré, D.; Terrón-Puig, M.; et al. Microbial Signature in Adipose Tissue of Crohn’s Disease Patients. J. Clin. Med. 2020, 9, 2448. https://doi.org/10.3390/jcm9082448
Serena C, Queipo-Ortuño M, Millan M, Sanchez-Alcoholado L, Caro A, Espina B, Menacho M, Bautista M, Monfort-Ferré D, Terrón-Puig M, et al. Microbial Signature in Adipose Tissue of Crohn’s Disease Patients. Journal of Clinical Medicine. 2020; 9(8):2448. https://doi.org/10.3390/jcm9082448
Chicago/Turabian StyleSerena, Carolina, Maribel Queipo-Ortuño, Monica Millan, Lidia Sanchez-Alcoholado, Aleidis Caro, Beatriz Espina, Margarita Menacho, Michelle Bautista, Diandra Monfort-Ferré, Margarida Terrón-Puig, and et al. 2020. "Microbial Signature in Adipose Tissue of Crohn’s Disease Patients" Journal of Clinical Medicine 9, no. 8: 2448. https://doi.org/10.3390/jcm9082448
APA StyleSerena, C., Queipo-Ortuño, M., Millan, M., Sanchez-Alcoholado, L., Caro, A., Espina, B., Menacho, M., Bautista, M., Monfort-Ferré, D., Terrón-Puig, M., Núñez-Roa, C., Maymó-Masip, E., Rodriguez, M. M., Tinahones, F. J., Espin, E., Martí, M., Fernández-Veledo, S., & Vendrell, J. (2020). Microbial Signature in Adipose Tissue of Crohn’s Disease Patients. Journal of Clinical Medicine, 9(8), 2448. https://doi.org/10.3390/jcm9082448