Large-Scale Drug Screen Identifies FDA-Approved Drugs for Repurposing in Sickle-Cell Disease

 ,
, 
Abstract
1. Introduction
2. Experimental Section
2.1. Cell Culture
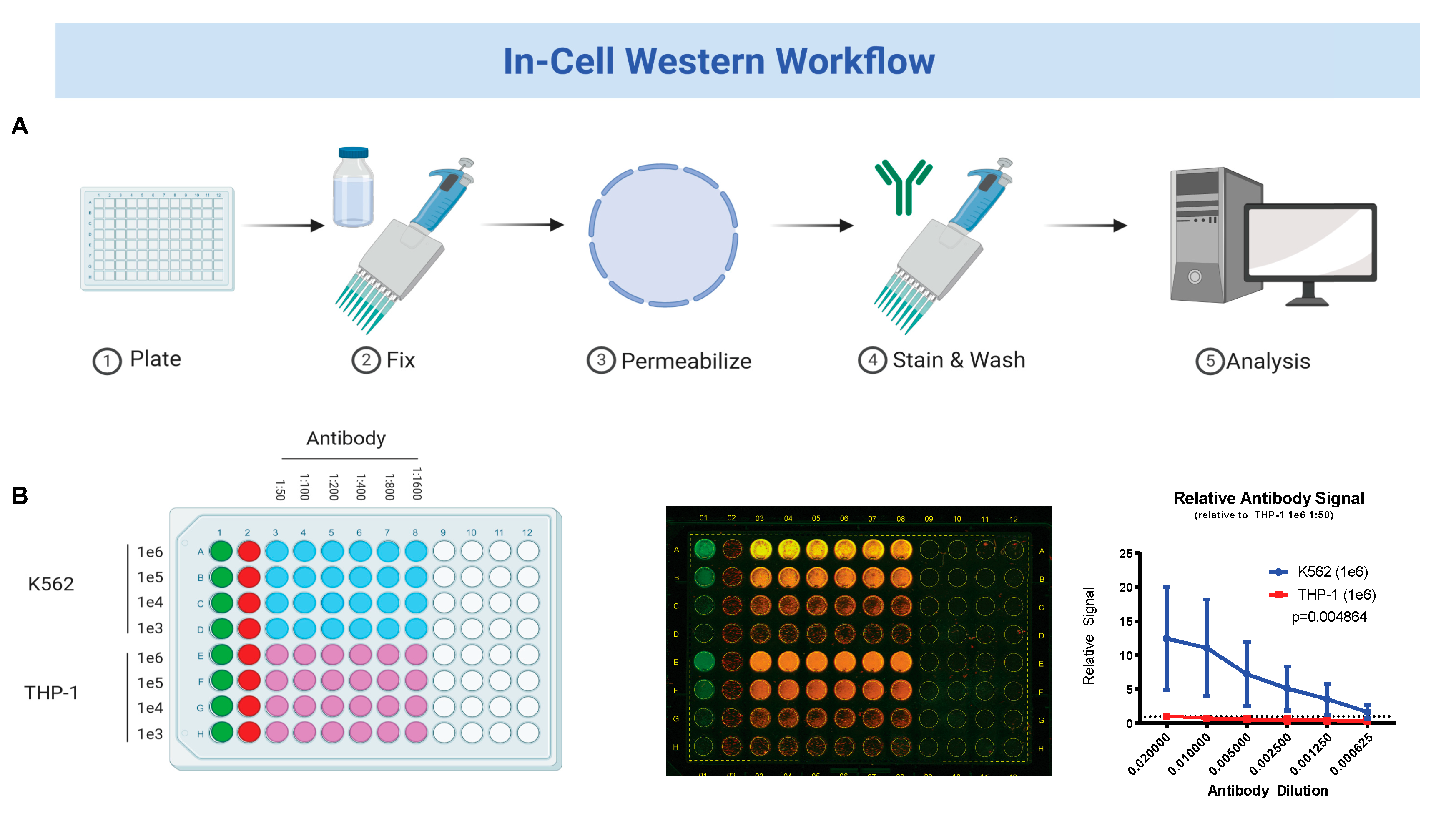
2.2. In-Cell Western Blot
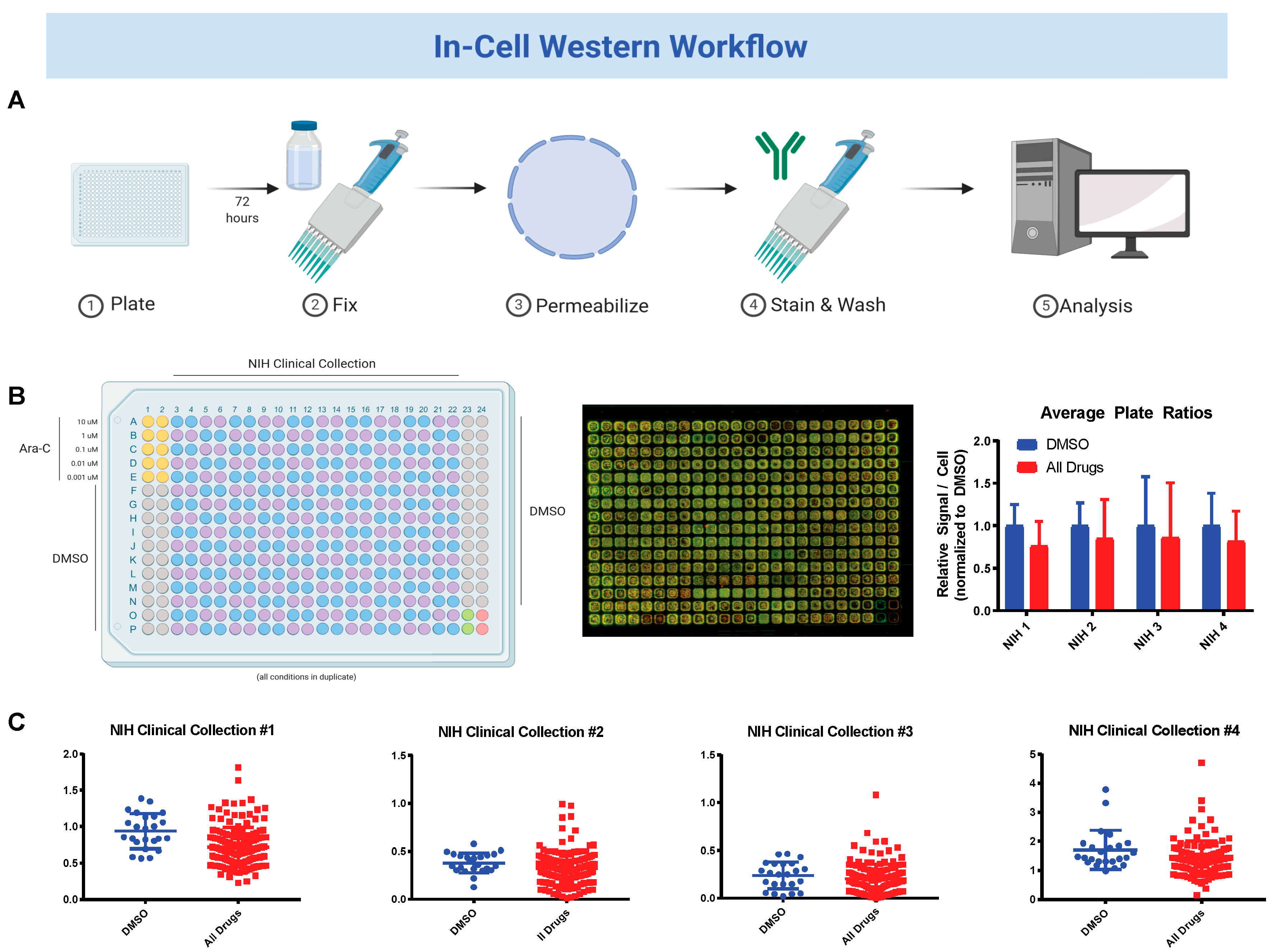
2.3. National Institute of Health (NIH) Clinical Collection (NCC) Screen
2.4. MTS Viability Assays
2.5. Immunoblot
2.6. Circular Chromosome Conformation Capture (4C)
2.7. Statistical Analysis
3. Results
3.1. Fetal Hemoglobin Expression on In-Cell Western Correlates with Traditional Western Blot Analysis
3.2. Sequential Drug Screens Show New Candidates for Sickle-Cell Disease (SCD)
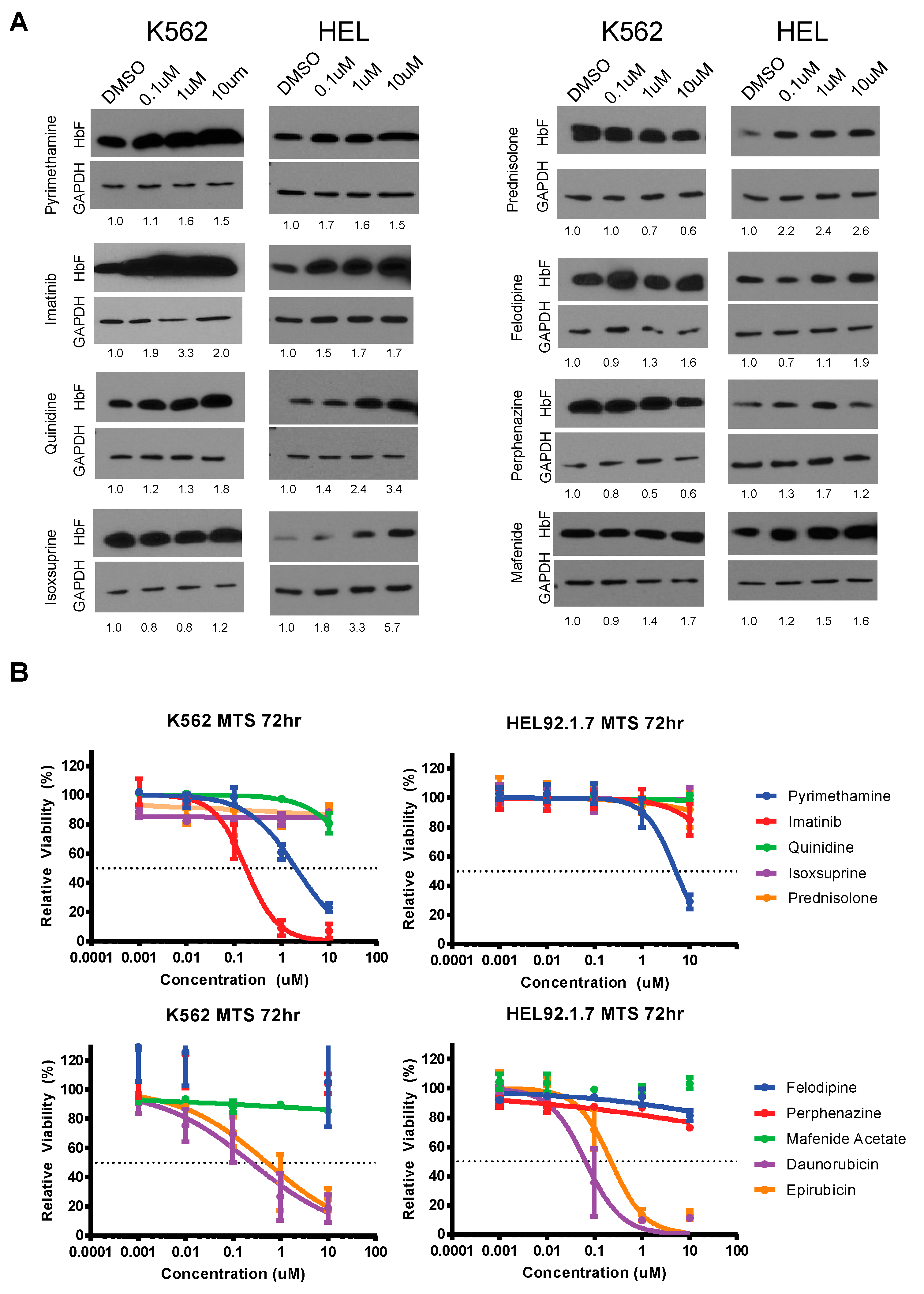
3.3. Validation and Cytotoxicity Evaluation of NCC Screen Results
3.4. Changes in Cis Interactions at the HBG2 Promoter Induced by Imatinib in K562 and HEL92.1.7 Cell Lines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of sickle cell disease. Annu. Rev. Pathol. 2019, 14, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.; Glass, S.; Smith, S.; Heinlein, M.; Lapalombella, R.; Desai, P.C. High-throughput mirna analysis suggests pro-inflammatory profile in sickle cell disease. Blood 2018, 132, 1077. [Google Scholar] [CrossRef]
- Conran, N.; Belcher, J.D. Inflammation in sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 263–299. [Google Scholar] [CrossRef]
- Agrawal, R.K.; Patel, R.K.; Shah, V.; Nainiwal, L.; Trivedi, B. Hydroxyurea in sickle cell disease: Drug review. Indian J. Hematol. Blood Transfus. 2014, 30, 91–96. [Google Scholar] [CrossRef]
- Food and Drug Administration. Drug Approval Package: Endari (L-glutamine): Summary Review; Food and Drug Administration: Silver Spring, MD, USA, 2017.
- Kutlar, A.; Kanter, J.; Liles, D.K.; Alvarez, O.A.; Cancado, R.D.; Friedrisch, J.R.; Knight-Madden, J.M.; Bruederle, A.; Shi, M.; Zhu, Z.; et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: A SUSTAIN study analysis. Am. J. Hematol. 2019, 94, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.R.; Riley, T.T. Profile of crizanlizumab and its potential in the prevention of pain crises in sickle cell disease: Evidence to date. J. Blood Med. 2019, 10, 307–311. [Google Scholar] [CrossRef]
- Han, J.; Saraf, S.L.; Gordeuk, V.R. Systematic Review of Voxelotor: A First-in-Class Sickle Hemoglobin Polymerization Inhibitor for Management of Sickle Cell Disease. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 525–534. [Google Scholar] [CrossRef]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, C.M.; Dever, D.P.; Davis, T.H.; Camarena, J.; Srifa, W.; Zhang, Y.; Paikari, A.; Chang, A.K.; Porteus, M.H.; et al. Highly efficient editing of the beta-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019, 47, 7955–7972. [Google Scholar] [CrossRef]
- Metais, J.Y.; Doerfler, P.A.; Mayuranathan, T.; Bauer, D.E.; Fowler, S.C.; Hsieh, M.M.; Katta, V.; Keriwala, S.; Lazzarotto, C.R.; Luk, K.; et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019, 3, 3379–3392. [Google Scholar] [CrossRef]
- Singh, R.; Jordan, R.; Hanlon, C. Economic Impact of Sickle Cell Hospitalization. Blood 2014, 124, 5971. [Google Scholar] [CrossRef]
- Antwi-Boasiako, C.; Frimpong, E.; Ababio, G.K.; Dzudzor, B.; Ekem, I.; Gyan, B.; Sodzi-Tettey, N.A.; Antwi, D.A. Sickle cell disease: Reappraisal of the role of foetal haemoglobin levels in the frequency of vaso-occlusive crisis. Ghana Med. J. 2015, 49, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Lettre, G.; Bauer, D.E. Fetal haemoglobin in sickle-cell disease: From genetic epidemiology to new therapeutic strategies. Lancet 2016, 387, 2554–2564. [Google Scholar] [CrossRef]
- Bethlenfalvay, N.C.; Motulsky, A.G.; Ringelhann, B.; Lehmann, H.; Humbert, J.R.; Konotey-Ahulu, F.I. Hereditary persistence of fetal hemoglobin, beta thalassemia, and the hemoglobin delta-beta locus: Further family data and genetic interpretations. Am. J. Hum. Genet. 1975, 27, 140–154. [Google Scholar]
- Huisman, T.H.; Schroeder, W.A.; Stamatoyannopoulos, G.; Bouver, N.; Shelton, J.R.; Shelton, J.B.; Apell, G. Nature of fetal hemoglobin in the Greek type of hereditary persistence of fetal hemoglobin with and without concurrent beta-thalassemia. J. Clin. Investig. 1970, 49, 1035–1040. [Google Scholar] [CrossRef]
- Manning, L.R.; Russell, J.E.; Padovan, J.C.; Chait, B.T.; Popowicz, A.; Manning, R.S.; Manning, J.M. Human embryonic, fetal, and adult hemoglobins have different subunit interface strengths. Correlation with lifespan in the red cell. Protein Sci. 2007, 16, 1641–1658. [Google Scholar] [CrossRef]
- Molokie, R.; Lavelle, D.; Gowhari, M.; Pacini, M.; Krauz, L.; Hassan, J.; Ibanez, V.; Ruiz, M.A.; Ng, K.P.; Woost, P.; et al. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: A randomized phase 1 study. PLoS Med. 2017, 14, e1002382. [Google Scholar] [CrossRef]
- Jalali Far, M.A.; Dehghani Fard, A.; Hajizamani, S.; Mossahebi-Mohammadi, M.; Yaghooti, H.; Saki, N. Thalidomide is more efficient than sodium butyrate in enhancing GATA-1 and EKLF gene expression in erythroid progenitors derived from HSCs with beta-globin gene mutation. Int. J. Hematol. Oncol. Stem Cell Res. 2016, 10, 37–41. [Google Scholar]
- Guo, L.; Chen, J.; Wang, Q.; Zhang, J.; Huang, W. Oridonin enhances gammaglobin expression in erythroid precursors from patients with betathalassemia via activation of p38 MAPK signaling. Mol. Med. Rep. 2020, 21, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zuo, Y.; Zhang, X.; Ye, Y.; Bao, X.; Huang, H.; Tepakhan, W.; Wang, L.; Ju, J.; Chen, G.; et al. A genetic variant ameliorates beta-thalassemia severity by epigenetic-mediated elevation of human fetal hemoglobin expression. Am. J. Hum. Genet. 2017, 101, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, A.; Phylactides, M.; Katsantoni, E.; Vougas, K.; Garbis, S.D.; Fanis, P.; Sitarou, M.; Thein, S.L.; Kleanthous, M. Proteomic studies for the investigation of gamma-globin induction by decitabine in human primary erythroid progenitor cultures. J. Clin. Med. 2020, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Le, C.Q.; Myers, G.; Habara, A.; Jearawiriyapaisarn, N.; Murphy, G.J.; Chui, D.H.K.; Steinberg, M.H.; Engel, J.D.; Cui, S. Inhibition of LSD1 by small molecule inhibitors stimulates fetal hemoglobin synthesis. Blood 2019, 133, 2455–2459. [Google Scholar] [CrossRef]
- LE, C.; Myers, G.; Habara, A.; Chui, D.H.K.; Steinberg, M.H.; Engel, J.D.; Cui, S. LSD1 inhibitors induce fetal hemoglobin in primary human erythroid cells. Blood 2018, 132, 1066. [Google Scholar] [CrossRef]
- Cui, S.; Lim, K.C.; Shi, L.; Lee, M.; Jearawiriyapaisarn, N.; Myers, G.; Campbell, A.; Harro, D.; Iwase, S.; Trievel, R.C.; et al. The LSD1 inhibitor RN-1 induces fetal hemoglobin synthesis and reduces disease pathology in sickle cell mice. Blood 2015, 126, 386–396. [Google Scholar] [CrossRef]
- Rivers, A.; Vaitkus, K.; Jagadeeswaran, R.; Ruiz, M.A.; Ibanez, V.; Ciceri, F.; Cavalcanti, F.; Molokie, R.E.; Saunthararajah, Y.; Engel, J.D.; et al. Oral administration of the LSD1 inhibitor ORY-3001 increases fetal hemoglobin in sickle cell mice and baboons. Exp. Hematol. 2018, 67, 60–64.e2. [Google Scholar] [CrossRef]
- Shi, L.; Cui, S.; Engel, J.D.; Tanabe, O. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat. Med. 2013, 19, 291–294. [Google Scholar] [CrossRef]
- Papayannopoulou, T.; Torrealba de Ron, A.; Veith, R.; Knitter, G.; Stamatoyannopoulos, G. Arabinosylcytosine induces fetal hemoglobin in baboons by perturbing erythroid cell differentiation kinetics. Science 1984, 224, 617–619. [Google Scholar] [CrossRef]
- Veith, R.; Galanello, R.; Papayannopoulou, T.; Stamatoyannopoulos, G. Stimulation of F-cell production in patients with sickle-cell anemia treated with cytarabine or hydroxyurea. N. Engl. J. Med. 1985, 313, 1571–1575. [Google Scholar] [CrossRef]
- Barbarani, G.; Fugazza, C.; Strouboulis, J.; Ronchi, A.E. The pleiotropic effects of GATA1 and KLF1 in physiological erythropoiesis and in dyserythropoietic disorders. Front. Physiol. 2019, 10, 91. [Google Scholar] [CrossRef]
- Haas, N.; Riedt, T.; Labbaf, Z.; Bassler, K.; Gergis, D.; Frohlich, H.; Gutgemann, I.; Janzen, V.; Schorle, H. Kit transduced signals counteract erythroid maturation by MAPK-dependent modulation of erythropoietin signaling and apoptosis induction in mouse fetal liver. Cell Death Differ. 2015, 22, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Kodeboyina, S.; Liu, L.; Dzandu, J.; Sangerman, J.; Ofori-Acquah, S.F.; Pace, B.S. Role of STAT3 and GATA-1 interactions in gamma-globin gene expression. Exp. Hematol. 2009, 37, 889–900. [Google Scholar] [CrossRef]
- Mattè, A.; Federti, E.; Rechiutti, A.; Panataleo, A.; Russo, R.; Turrini, F.; Siciliano, A.; Tharaux, P.L.; El Nemer, W.; Cedrick, S.; et al. Imatinib protects against hypoxia/reoxygenation induced lung and kidney injury in a humanized mouse model for SCD. Blood 2018, 132, 725. [Google Scholar] [CrossRef]
- Close, J.L.; Lottenberg, R. Effectiveness of imatinib therapy for a patient with sickle cell anemia and chronic myelocytic leukemia. Blood 2009, 114, 2559. [Google Scholar] [CrossRef]
- Zitvogel, L.; Rusakiewicz, S.; Routy, B.; Ayyoub, M.; Kroemer, G. Immunological off-target effects of imatinib. Nat. Rev. Clin. Oncol. 2016, 13, 431–446. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Fold Change | Indication |
|---|---|---|
| Daunorubicin Hydrochloride | 4.55 | Acute Myeloid Leukemia |
| Albendazole | 2.86 | Anthelmintic |
| Chloroxine | 2.76 | Seborrheic Dermatitis |
| Raltitrexed | 2.62 | Colorectal Cancer |
| Epirubicin Hydrochloride | 2.58 | Breast Cancer |
| Floxuridine | 2.50 | Hepatic Cancer |
| Pindolol | 2.50 | Hypertension |
| Methylprednisolone | 2.32 | Rheumatoid Arthritis |
| Fexofenadine Hydrochloride | 2.28 | Seasonal Allergic Rhinitis |
| 6-Azauridine | 2.25 | Psoriasis |
| Pyrimethamine | 2.23 | Malarial Infection |
| Mafenide Acetate | 2.11 | Bacterial Infection |
| Prednisolone | 2.09 | Congenital Adrenal Hyperplasia |
| Ribavirin | 2.02 | Chronic Hepatitis C |
| Quinidine Hydrochloride | 1.99 | Cardiac Dysrhythmia |
| Ebselen | 1.95 | Type 2 Diabetes Mellitus * |
| Tegaserod Maleate | 1.93 | Irritable Bowel Syndrome |
| Delta1-Hydrocortisone 21-hemisuccinate Sodium | 1.90 | Corticosteroid-responsive Dematoses |
| Enalapril Maleate | 1.81 | Congestive Heart Failure |
| Granisetron Hydrochloride | 1.75 | Nausea |
| Drug Name | Fold Change | Indication |
|---|---|---|
| Daunorubicin Hydrochloride | 5.76 | Acute Myeloid Leukemia |
| Perphenazine | 5.16 | Schizophrenia |
| Acyclovir | 5.14 | Viral Infection |
| Cefazolin Sodium | 4.71 | Bacterial Infection |
| Epirubicin Hydrochloride | 4.59 | Breast Cancer |
| Atenolol | 4.49 | Hypertension |
| Methoxsalen | 4.44 | Cutaneous T-Cell Lymphoma |
| Ampiroxicam | 4.34 | Arthritis |
| Prednisolone | 4.32 | Rheumatoid Arthritis |
| Oxytetracycline hydrochloride | 4.30 | Bacterial Infection |
| Pfizerpen | 4.14 | Bacterial Infection |
| Tetracycline | 3.95 | Bacterial Infection |
| Pitavastatin | 3.92 | Elevated Cholesterol |
| Benazepril Hydrochloride | 3.80 | Elevated Blood Pressure |
| Triamcinolone Acetonide | 3.67 | Eczema |
| Phenelzine | 3.52 | Depression |
| Digoxin | 3.50 | Heart Failure |
| Dibenzyline | 3.28 | Pheochromocytoma |
| Buspar | 3.28 | Anxiety |
| Fluoxetine Hydrochloride | 3.25 | Depression |
| Drug Name | K562 | iRBC | Indication |
|---|---|---|---|
| Daunorubicin Hydrochloride | 4.55 | 5.76 | Acute Myeloid Leukemia |
| Epirubicin Hydrochloride | 2.58 | 4.59 | Breast Cancer |
| Pyrimethamine | 2.23 | 2.01 | Malarial Infection |
| Mafenide Acetate | 2.11 | 2.71 | Bacterial Infection |
| Prednisolone | 2.09 | 4.32 | Immunosuppression |
| Quinidine Hydrochloride | 1.99 | 2.08 | Malarial Infection |
| Perphenazine | 1.71 | 5.16 | Schizophrenia |
| Felodipine | 1.62 | 1.96 | Hypertension |
| Duvadilan | 1.60 | 2.58 | Cerebral Vascular Insufficiency |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cannon, M.; Phillips, H.; Smith, S.; Williams, K.; Brinton, L.; Gregory, C.; Landes, K.; Desai, P.; Byrd, J.; Lapalombella, R. Large-Scale Drug Screen Identifies FDA-Approved Drugs for Repurposing in Sickle-Cell Disease. J. Clin. Med. 2020, 9, 2276. https://doi.org/10.3390/jcm9072276
Cannon M, Phillips H, Smith S, Williams K, Brinton L, Gregory C, Landes K, Desai P, Byrd J, Lapalombella R. Large-Scale Drug Screen Identifies FDA-Approved Drugs for Repurposing in Sickle-Cell Disease. Journal of Clinical Medicine. 2020; 9(7):2276. https://doi.org/10.3390/jcm9072276
Chicago/Turabian StyleCannon, Matthew, Hannah Phillips, Sidney Smith, Katie Williams, Lindsey Brinton, Charles Gregory, Kristina Landes, Payal Desai, John Byrd, and Rosa Lapalombella. 2020. "Large-Scale Drug Screen Identifies FDA-Approved Drugs for Repurposing in Sickle-Cell Disease" Journal of Clinical Medicine 9, no. 7: 2276. https://doi.org/10.3390/jcm9072276
APA StyleCannon, M., Phillips, H., Smith, S., Williams, K., Brinton, L., Gregory, C., Landes, K., Desai, P., Byrd, J., & Lapalombella, R. (2020). Large-Scale Drug Screen Identifies FDA-Approved Drugs for Repurposing in Sickle-Cell Disease. Journal of Clinical Medicine, 9(7), 2276. https://doi.org/10.3390/jcm9072276