New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges


Abstract
1. Introduction
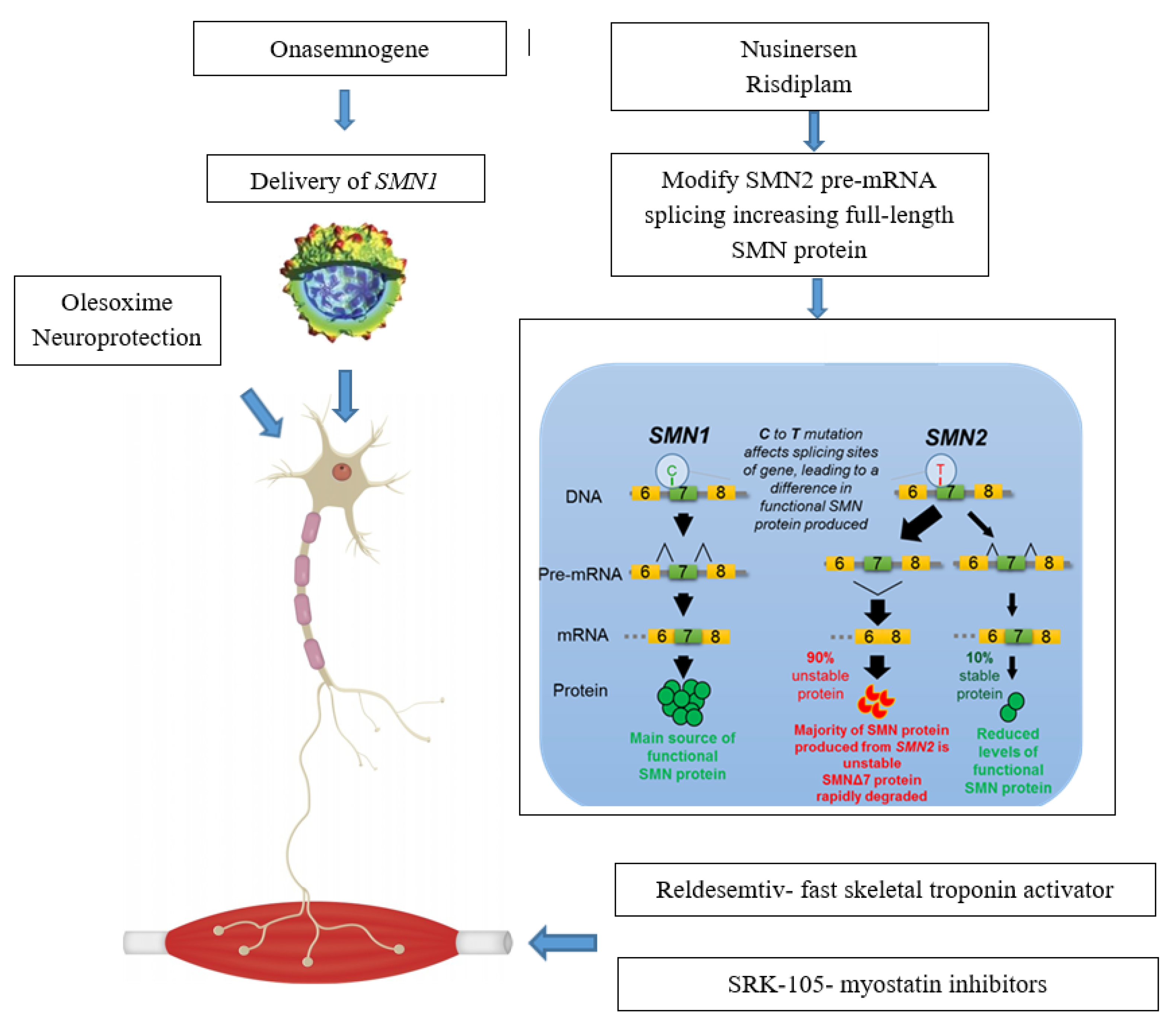
2. SMN—Dependent Gene Therapies
2.1. Splicing Modification of SMN2
2.1.1. Nusinersen
2.1.2. Small Molecules
2.2. SMN1 Gene Replacement
3. Treatments Targeting Survival Motor Neuron (SMN)—Independent Factors
3.1. Muscle Enhancing Therapies
3.2. Future Prospectives in SMN Independent Therapeutic Targets
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Bertini, E.; Iannaccone, S.T. Childhood spinal muscular atrophy: Controversies and challenges. Lancet Neurol. 2012, 11, 443–452. [Google Scholar] [CrossRef]
- Farrar, M.A.; Vucic, S.; Johnston, H.M.; du Sart, D.; Kiernan, M.C. Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J. Pediatr. 2013, 162, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Meyer, O.H.; Simonds, A.K.; Schroth, M.K.; Graham, R.J.; Kirschner, J.; Iannaccone, S.T.; Crawford, T.O.; Woods, S.; et al. Diagnosis and management ofspinal muscular atrophy: Part 2: Pulmonary and acutecare; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul. Disord. 2018, 28, 197–207. [Google Scholar] [CrossRef]
- Sugarman, E.A.; Nagan, N.; Zhu, H. Pan-ethnic carrier screening and Prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72,400 specimens. Eur. J. Hum. Genet. 2012, 20, 27–32. [Google Scholar] [CrossRef]
- Ogino, S.; Wilson, R.B.; Gold, B. New insights on the evolution of the SMN1 and SMN2 region: Simulation and meta-analysis for allele and haplotype frequency calculations. Eur. J. Hum. Genet. 2004, 12, 1015–1023. [Google Scholar] [CrossRef]
- Ingrid, E.C.; Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy–A literature review. Orphanet J. Rare Dis. 2017, 2, 124. [Google Scholar] [CrossRef]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Chen, T.H. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? Int. J. Mol. Sci. 2020, 21, 3297. [Google Scholar] [CrossRef] [PubMed]
- Messina, S. New Directions for SMA Therapy. J. Clin. Med. 2018, 7, 251. [Google Scholar] [CrossRef]
- Wadman, R.I.; van der Pol, W.L.; Bosboom, W.M.; Asselman, F.L.; van den Berg, L.H.; Iannaccone, S.T.; Vrancken, A.F. Drug treatment for spinal muscular atrophy type I. Cochrane Database Syst. Rev. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Wadman, R.I.; van der Pol, W.L.; Bosboom, W.M.; Asselman, F.L.; van den Berg, L.H.; Iannaccone, S.T.; Vrancken, A.F. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst. Rev. 2020, 1. [Google Scholar] [CrossRef]
- Ramdas, S.; Servais, L. New treatments in spinal muscular atrophy: An overview of currently available data. Expert Opin. Pharmacother. 2020, 21, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Schorling, D.C.; Pechmann, A.; Kirschner, J. Advances in Treatment of Spinal Muscular Atrophy—New Phenotypes, New Challenges, New Implications for Care. J. Neuromuscul. Dis. 2020, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol. Cell Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef]
- Rigo, F.; Hua, Y.; Krainer, A.R.; Bennett, C.F. Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol. 2012, 199, 21–25. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet Lond. Engl. 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, J.; Darras, B.; Farrar, M.; Mercuri, E.; Chiriboga, C.; Kuntz, N.; Shieh, P.; Tulinius, M.; Montes, J.; Reyna, S.; et al. Interim report on the safety and efficacy of longer-term treatment with nusinersen in later-onset spinal muscular atrophy (SMA): Results from the SHINE study. Presented at the World-Muscle-Society Conference 2019. Neuromuscul. Disord. 2019, 29, S184. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen Initiated in Infants During the Presymptomatic Stage of Spinal Muscular Atrophy: Interim Efficacy and Safety Results From the Phase 2 NURTURE Study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef]
- Messina, S.; Pane, M.; Sansone, V.; Bruno, C.; Catteruccia, M.; Vita, G.; Palermo, C.; Albamonte, E.; Pedemonte, M.; Bertini, E.; et al. Expanded access program with Nusinersen in SMA type I in Italy: Strengths and pitfalls of a successful experience. Neuromuscul. Disord. 2017, 27, 1084–1086. [Google Scholar] [CrossRef] [PubMed]
- Farrar, M.A.; Teoh, H.L.; Carey, K.A.; Cairns, A.; Forbes, R.; Herbert, K.; Holland, S.; Kristi, K.J.; Menezes, M.P.; Morrison, M.; et al. Nusinersen for SMA: Expanded access programme. J. Neurol. Neurosurg. Psychiatry 2018, 89, 937–942. [Google Scholar] [CrossRef]
- Pechmann, A.; Langer, T.; Schorling, D.; Stein, S.; Vogt, S.; Schara, U.; Kölbel, H.; Schwartz, O.; Hahn, A.; Giese, K.; et al. Evaluation of children with SMA type 1 under treatment with nusinersen within the expanded access program in Germany. J. Neuromuscul. Dis. 2018, 5, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Aragon-Gawinska, K.; Seferian, A.M.; Daron, A.; Gargaun, E.; Vuillerot, C.; Cances, C.; Ropars, J.; Chouchane, M.; Cuppen, Y.; Hughes, I.; et al. Nusinersen in patients older than 7 months with spinal muscular atrophy type 1: A cohort study. Neurology 2018, 91, 1312–1318. [Google Scholar] [CrossRef]
- Pane, M.; Palermo, C.; Messina, S.; Sansone, V.A.; Bruno, C.; Catteruccia, M.; Sframeli, M.; Albamonte, E.; Pedemonte, M.; D’Amico, A.; et al. Nusinersen in type 1 SMA infants, children and young adults: Preliminary results on motor function. Neuromuscul. Disord. 2018, 28, 582–585. [Google Scholar] [CrossRef]
- Pane, M.; Coratti, G.; Sansone, V.A.; Messina, S.; Bruno, C.; Catteruccia, M.; Sframeli, M.; Albamonte, E.; Pedemonte, M.; D’Amico, A.; et al. Nusinersen in type 1 spinal muscular atrophy: Twelve-month real-world data. Ann. Neurol. 2019, 86, 443–451. [Google Scholar] [CrossRef]
- Sivaramakrishnan, M.; McCarthy, K.D.; Campagne, S.; Huber, S.; Meier, S.; Augustin, A.; Heckel, T.; Meistermann, H.; Hug, M.N.; Birrer, P.; et al. Binding to SMN2 pre-mRNA-protein Complex Elicits Specificity for Small Molecule Splicing Modifiers. Nat. Commun. 2017, 8, 1476. [Google Scholar] [CrossRef]
- Poirier, A.; Weetall, M.; Heinig, K.; Bucheli, F.; Schoenlein, K.; Alsenz, J.; Bassett, S.; Ullah, M.; Senn, C.; Ratni, H.; et al. Risdiplam Distributes and Increases SMN Protein in Both the Central Nervous System and Peripheral Organs. Pharmacol. Res. Perspect. 2018, 29, e00447. [Google Scholar] [CrossRef]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 ( SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 9, 6501–6517. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Gillingwater, T.H. Spinal Muscular Atrophy: Going beyond the Motor Neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Yeo, C.J.J.; Darras, B.T. Overturning the Paradigm of Spinal Muscular Atrophy as just a Motor Neuron Disease. Pediatr. Neurol. 2020. [Google Scholar] [CrossRef]
- Sturm, S.; Günther, A.; Jaber, B.; Jordan, P.; Al Kotbi, N.; Parkar, N.; Cleary, Y.; Frances, N.; Bergauer, T.; Heinig, K.; et al. A Phase 1 Healthy Male Volunteer Single Escalating Dose Study of the Pharmacokinetics and Pharmacodynamics of Risdiplam (RG7916, RO7034067), a SMN2 Splicing Modifier. Br. J. Clin. Pharmacol. 2019, 85, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Baranello, G.; Servais, L.; Day, J.W.; Deconinck, N.; Mercuri, E.; Klein, A.; Darras, B.; Masson, R.; Kletzl, H.; Cleary, Y.; et al. FIREFISH Part 1: 1-Year Results on Motor Function in Babies with Type 1 SMA. Presented at the American Academy of Neurology Conference 2019. Neurology 2019, 92, 3. [Google Scholar]
- Baranello, G.; Servais, L.; Day, J.W.; Deconinck, N.; Mercuri, E.; Klein, A.; Darras, B.; Masson, R.; Kletzl, H.; Cleary, Y.; et al. FIREFISH Part 1: 16-Month safety and exploratory outcomes of risdiplam (RG7916) treatment in infants with Type 1 spinal muscular atrophy (SMA). Presented at the World-Muscle-Society Conference 2019. Neuromuscul. Disord. 2019, 29, S184. [Google Scholar] [CrossRef]
- Servais, L.; Baranello, G.; Masson, R.; Mazurkiewicz-Bełdzińska, M.; Rose, K.; Vlodavets, D.; Xiong, H.; Zanoteli, E.; El-Khairi, M.; Fuerst-Recktenwald, S.; et al. FIREFISH Part 2: Efficacy and Safety of Risdiplam (RG7916) in Infants with Type 1 Spinal Muscular Atrophy (SMA). Presented at the American Academy of Neurology Conference 2020. Neurology 2020, 94, 1302. [Google Scholar]
- Mercuri, E.; Barisic, N.; Boespflug-Tanguy, O.; Deconinck, N.; Kostera-Pruszczyk, A.; Masson, R.; Mazzone, E.; Nascimento, R.; Osorio, A.; Saito, K.; et al. SUNFISH Part 2: Efficacy and safety of risdiplam (RG7916) in patients with Type 2 or non-ambulant Type 3 spinal muscular atrophy (SMA) Presented at the American Academy of Neurology Conference 2020. Neurology 2020, 94, 1260. [Google Scholar]
- Roche Press Release 07/04/2020: Roche Provides Regulatory Update on Risdiplam for the Treatment of Spinal Muscular Atrophy (SMA). Available online: https://www.roche.com/media/releases/med-cor-2020-04-07.htm (accessed on 30 May 2020).
- Le, T.T.; McGovern, V.L.; Alwine, I.E.; Wang, X.; Massoni-Laporte, A.; Rich, M.M.; Burghes, A.H.M. Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 2011, 20, 3578–3591. [Google Scholar] [CrossRef]
- Valori, C.F.; Ning, K.; Wyles, M.; Mead, R.J.; Grierson, A.J.; Shaw, P.J.; Azzouz, M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2010, 2, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.M.; et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an AAV vector expressing human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Sproule, D.M.; Al-Zaidy, S.A.; Shell, R. AVXS-101 phase 1 gene therapy clinical trial in SMA type 1: Experience with pre-existing anti-AAV9 antibody in the SMA1 population (S13.001). Neurology 2017, 88, 1. [Google Scholar]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Kolb, S.J.; Coffey, C.S.; Yankey, J.W.; Krosschell, K.; Arnold, W.D.; Rutkove, S.B.; Swoboda, K.J.; Reyna, S.P.; Sakonju, A.; Darras, B.T. Natural history of infantile-onset spinal muscular atrophy. Ann. Neurol. 2017, 82, 883–891. [Google Scholar] [CrossRef]
- Al-Zaidy, S.A.; Kolb, S.J.; Lowes, L.; Alfano, L.N.; Shell, R.; Church, K.R.; Nagendran, S.; Sproule, D.M.; Feltner, D.E.; Wells, C.; et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: Comparative Study with a Prospective Natural History Cohort. J. Neuromuscul. Dis. 2019, 6, 307–317. [Google Scholar] [CrossRef]
- Lowes, L.P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; et al. Impact of age andmotor function in a phase 1/2A study of infants with SMA Type 1 receiving single-dose gene replacement therapy. Pediatr. Neurol. 2019, 19, 30280–30282. [Google Scholar] [CrossRef]
- Day, J.D.; Chiriboga, C.A.; Crawford, T.O.; Darras, B.T.; Finkel, R.S.; Connolly, A.M.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.N.; Shieh, P.B.; et al. Onasemnogene Abeparvovec-xioi Gene-Replacement Therapy for Spinal Muscular Atrophy Type 1 (SMA1): Phase 3 US Study (STR1VE) Update (1828) Presented at the American Academy of Neurology Conference 2020. Neurology 2020, 94, 1828. [Google Scholar]
- Press Release Novartis 2019. Available online: https://www.novartis.com/news/media-releases/avexis-receives-fda-approval-zolgensma-first-and-only-gene-therapy-pediatric-patients-spinal-muscular-atrophy-sma (accessed on 30 May 2020).
- Press Release Novartis 2020. Available online: https://www.globenewswire.com/news-release/2020/05/19/2035354/0/en/AveXis-receives-EC-approval-and-activates-Day-One-access-program-for-Zolgensma-the-only-gene-therapy-for-spinal-muscular-atrophy-SMA.html (accessed on 30 May 2020).
- Hwee, D.T.; Kennedy, A.; Ryans, J.; Russell, A.J.; Jia, Z.; Hinken, A.C.; Morgans, D.J.; Malik, F.I.; Jasper, J.R. Fast skeletal muscle troponin activator tirasemtiv increases muscle function and performance in the B6SJL-SOD1G93A ALS mouse model. PLoS ONE 2014, 9, e96921. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.J.; Hwee, D.T.; Kim, L.H.; Durham, N.; Yang, H.T.; Hinken, A.C.; Kennedy, A.R.; Terjung, R.L.; Jasper, J.R.; Malik, F.I.; et al. Fast skeletal muscle troponin activator CK-2066260 increases fatigue resistance by reducing the energetic cost of muscle contraction. J. Physiol. 2019, 597, 4615–4625. [Google Scholar] [CrossRef] [PubMed]
- Andrews, A.J.; Miller, T.M.; Vijayakumar, V.; Stoltz, R.; James, J.K.; Meng, L.; Wolff, A.A.; Malik, F. CK-2127107 Amplifies Skeletal Muscle Response to Nerve Activation in Humans. Muscle Nerve 2018, 57, 729–734. [Google Scholar] [CrossRef]
- Rudnicki, S.A.; Andrews, J.A.; Malik, F.I. CY 5021 A phase 2, double-blind, randomized, placebo-controlled, multiple-dose study of reldesemtiv 2 ascending-dose cohorts of patients with Spinal Muscular Atrophy (SMA). In Proceedings of the Cure SMA 2018, Dallas, TX, USA, 16 June 2018. [Google Scholar]
- Feng, Z.; Ling, K.K.; Zhao, X.; Zhou, C.; Karp, G.; Welch, E.M.; Naryshkin, N.; Ratni, H.; Chen, K.S.; Metzger, F.; et al. Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum. Mol. Genet. 2016, 25, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef]
- Garcera, A.; Bahi, N.; Periyakaruppiah, A.; Arumugam, S.; Soler, R.M. Survival motor neuron protein reduction deregulates autophagy in spinal cord motoneurons in vitro. Cell. Death Dis. 2013, 4, e686. [Google Scholar] [CrossRef]
- Periyakaruppiah, A.; de la Fuente, S.; Arumugam, S.; Bahí, N.; Garcera, A.; Soler, R.M. Autophagy modulators regulate survival motor neuron protein stability in motoneurons. Exp. Neurol. 2016, 283, 287–297. [Google Scholar] [CrossRef]
- Piras, A.; Boido, M. Autophagy inhibition: A new therapeutic target in spinal muscular atrophy. Neural Regen. Res. 2018, 13, 813–814. [Google Scholar] [CrossRef]
- Oliván, S.; Calvo, A.C.; Rando, A.; Herrando-Grabulosa, M.; Manzano, R.; Zaragoza, P.; Tizzano, E.F.; Aquilera, J.; Osta, R. Neuroprotective effect of non-viral gene therapy treatment based on tetanus toxin C-fragment in a severe mouse model of spinal muscular atrophy. Front. Mol. Neurosci. 2016, 9, 76. [Google Scholar] [CrossRef]
- Parker, G.C.; Li, X.; Anguelov, R.A.; Toth, G.; Cristescu, A.; Acsadi, G. Survival motor neuron protein regulates apoptosis in an in vitro model of spinal muscular atrophy. Neurotox. Res. 2008, 13, 39–48. [Google Scholar] [CrossRef]
- Genabai, N.K.; Ahmad, S.; Zhang, Z.; Jiang, X.; Gabaldon, C.A.; Gangwani, L. Genetic inhibition of JNK3 ameliorates spinal muscular atrophy. Hum. Mol. Genet. 2015, 15, 6986–7004. [Google Scholar] [CrossRef] [PubMed]
- Schellino, R.; Boido, M.; Borsello, T.; Vercelli, A. Pharmacological c-Jun NH2-Terminal Kinase (JNK) Pathway Inhibition Reduces Severity of Spinal Muscular Atrophy Disease in Mice. Front. Mol. Neurosci. 2018, 11, 308. [Google Scholar] [CrossRef]
- Boido, M.; De Amicis, E.; Valsecchi, V.; Trevisan, M.; Ala, U.; Ruegg, M.A.; Hettwer, S.; Vercelli, A. Increasing Agrin Function Antagonizes Muscle Atrophy and Motor Impairment in Spinal Muscular Atrophy. Front. Cell Neurosci. 2018, 12, 17. [Google Scholar] [CrossRef]
- Kim, J.K.; Caine, C.; Awano, T.; Herbst, R.; Monani, U.R. Motor neuronal repletion of the NMJ organizer, Agrin, modulates the severity of the spinal muscular atrophy disease phenotype in model mice. Hum. Mol. Genet. 2017, 26, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Vita, G.L.; Aguennouz, M.; Sframeli, M.; Romeo, S.; Rodolico, C.; Vita, G. Activation of NF-κB pathway in Duchenne muscular dystrophy: Relation to age. Acta Myol. 2011, 30, 16–23. [Google Scholar] [PubMed]
- Mincheva, S.; Garcera, A.; Gou-Fabregas, M.; Encinas, M.; Dolcet, X.; Soler, R.M. The canonical nuclear factor-kappa B pathway regulates cell survival in a developmental model of spinal cord Motoneurons. J. Neurosci. 2011, 31, 6493–6503. [Google Scholar] [CrossRef]
- Arumugam, S.; Mincheva-Tasheva, S.; Periyakaruppiah, A.; de la Fuente, S.; Soler, R.M.; Garcera, A. Regulation of Survival Motor Neuron Protein by the Nuclear Factor-Kappa B Pathway in Mouse Spinal Cord Motoneurons. Mol. Neurobiol. 2018, 55, 5019–5030. [Google Scholar] [CrossRef]
- Dangouloff, T.; Servais, L. Clinical evidence supporting early treatment in spinal muscular atrophy: Current perspectives. Therap. Clin. Risk Manag. 2019, 15, 1153–1161. [Google Scholar] [CrossRef]
- Pera, M.C.; Coratti, G.; Berti, B.; D’Amico, A.; Sframeli, M.; Albamonte, E.; de Sanctis, R.; Messina, S.; Catteruccia, M.; Brigati, G.; et al. Diagnostic journey in Spinal Muscular Atrophy: Is it still an odyssey? PLoS ONE 2020, 15, e0230677. [Google Scholar] [CrossRef] [PubMed]
- Kraszewski, J.N.; Kay, D.M.; Stevens, C.F.; Koval, C.; Haser, B.; Ortiz, V.; Albertorio, A.; Cohen, L.L.; Jain, R.; Andrew, S.P.; et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet. Med. 2018, 20, 608–613. [Google Scholar] [CrossRef]
- Vill, K.; Kolbel, H.; Schwartz, O.; Blaschek, A.; Olgemoller, B.; Harms, E.; Burggraf, S.; Roschinger, W.; Durner, J.; Glaser, D.; et al. One year of newborn screening for SMA – results of a German Pilot Project. J. Neuromuscul. Dis. 2019, 6, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Boemer, F.; Caberg, J.H.; Dideberg, V.; Dardenne, D.; Bours, V.; Hiligsmann, M.; Dangouloff, T.; Servais, L. Newborn screening for SMA in Southern Belgium. Neuromuscul. Dis. 2019, 29, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Chiang, S.C.; Weng, W.C.; Lee, N.C.; Lin, C.J.; Hsieh, W.S.; Lee, W.T.; Jong, Y.J.; Ko, T.M.; Hwu, W.L. Presymptomatic diagnosis of Spinal Muscular Atrophy through newborn screening. J. Pediatr. 2017, 190, 124–129.e1. [Google Scholar] [CrossRef] [PubMed]
- Czibere, L.; Burggraf, S.; Fleige, T.; Gluck, B.; Keitel, L.M.; Landt, O.; Durner, J.; Roschinger, W.; Hohenfellner, K.; Wirth, B.; et al. High-throughput genetic newborn screening for spinal muscular atrophy by rapid nucleic acid extraction from dried blood spots and 384-well qPCR. Eur. J. Hum. Gen. 2020, 28, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Glascock, J.; Sampson, J.; Haidet-Phillips, A.; Connolly, A.; Darras, B.; Day, J.; Finkel, R.; Howell, R.R.; Klinger, K.; Kuntz, N.; et al. Treatment algorithm for infants diagnosed with Spinal Muscular Atrophy through Newborn Screening. J. Neuromuscul. Dis. 2018, 5, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Müller-Felber, W.; Vill, K.; Schwartz, O.; Gläser, D.; Nennstiel, U.; Wirth, B.; Burggraf, S.; Röschinger, W.; Becker, M.; Durner, J.; et al. Infants Diagnosed with Spinal Muscular Atrophy and 4 SMN2 Copies through Newborn Screening—Opportunity or Burden? J. Neuromuscul. Dis. 2020, 7, 109–117. [Google Scholar] [CrossRef]
- Institute for Quality and Efficiency in Health Care (IQWiG). Newborn Screening for 5q-Linked Spinal Muscular Atrophy: IQWiG Reports. Commission No. S18-02; Institute for Quality and Efficiency in Health Care (IQWiG): Cologne, Germany, 2020. [Google Scholar]
- Wirth, B.; Garbes, L.; Riessland, M. How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr. Opin. Genet. Dev. 2013, 23, 330–338. [Google Scholar] [CrossRef]
- Kariyawasam, D.S.T.; D’Silva, A.; Lin, C.; Ryan, M.M.; Farrar, M.A. Biomarkers and the development of a personalized medicine approach in Spinal Muscular Atrophy. Front. Neurol. 2019, 10, 898. [Google Scholar] [CrossRef]
- Olsson, B.; Alberg, L.; Cullen, N.C.; Michael, E.; Wahlgren, L.; Kroksmark, A.K.; Rostasy, K.; Blennow, K.; Zetterberg, H.; Tulinius, M. NFL is a marker of treatment response in children with SMA treated with nusinersen. J. Neurol. 2019, 266, 2129–2136. [Google Scholar] [CrossRef]
- Darras, B.T.; Crawford, T.O.; Finkel, R.S.; Mercuri, E.; De Vivo, D.C.; Oskoui, M.; Tizzano, E.F.; Ryan, M.M.; Muntoni, F.; Zhao, G.; et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 932–944. [Google Scholar] [CrossRef]
- Totzeck, A.; Stolte, B.; Kizina, K.; Bolz, S.; Schlag, M.; Thimm, A.; Kleinschnitz, C.; Hagenacker, T. Neurofilament Heavy Chain and Tau Protein are not elevated in cerebrospinal fluid of adult patients with Spinal Muscular Atrophy during loading with Nusinersen. Int. J. Mol. Sci. 2019, 20, 5397. [Google Scholar] [CrossRef]
- Faravelli, I.; Meneri, M.; Saccomanno, D.; Velardo, D.; Abati, E.; Gagliardi, D.; Parente, V.; Petrozzi, L.; Ronchi, D.; Stocchetti, D.; et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: An explorative study on Spinal Muscular Atrophy type 3 patients. J. Cell. Mol. Med. 2020, 24, 3034–3039. [Google Scholar] [CrossRef]
- Bonanno, S.; Marcuzzo, S.; Malacarne, C.; Giagnorio, E.; Masson, R.; Zanin, R.; Arnoldi, M.T.; Andreetta, F.; Simoncini, O.; Venerando, A.; et al. Circulating MyomiRs as Potential Biomarkers to Monitor Response to Nusinersen in Pediatric SMA Patients. Biomedicines 2020, 26, 21. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Latzer, P.; Schmid, D.; Warnken, U.; Saffari, A.; Ziegler, A.; Kollmer, J.; Möhlenbruch, M.; Ulfert, C.; Herweh, C.; et al. Cerebrospinal fluid proteomic profiling in nusinersen-treated patients with spinal muscular atrophy. J. Neurochem. 2020, 153, 650–661. [Google Scholar] [CrossRef]
- Dabbous, O.; Maru, B.; Jansen, J.P.; Lorenzi, M.; Cloutier, M.; Guérin, A.; Pivneva, I.; Wu, E.Q.; Arjunji, R.; Feltner, D.; et al. Survival, motor function, and motor milestones: Comparison of AVXS-101 relative to Nusinersen for the treatment of infants with Spinal Muscular Atrophy Type 1. Adv. Ther. 2019, 36, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Connock, M.; Andronis, L.; Auguste, P.; Dussart, C.; Armoiry, X. Will the US$5 million onasemnogene abeparvosec treatment for spinal muscular atrophy represent ‘value for money’ for the NHS? A rapid inquiry into suggestions that it may be cost-effective. Expert. Opin. Biol. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Malone, D.C.; Dean, R.; Arjunji, R.; Arjunji, R.; Jensen, I.; Cyr, P.; Miller, B.; Maru, B.; Sproule, D.M.; Feltner, D.E.; et al. Cost-effectiveness analysis of using onasemnogene abeparvocec (AVXS-101) in spinal muscular atrophy type 1 patients. J. Mark. Access Health Policy 2019, 7, 1601484. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. International SMA Patient Registry—Full Text View. Available online: https://clinicaltrials.gov/ct2/show/NCT00466349 (accessed on 30 May 2020).
- TREAT-NMD: National SMA Registries. Available online: http://www.treat-nmd.eu/sma/patient-registries/sma/ (accessed on 30 May 2020).
- Pechmann, A.; Konig, K.; Bernert, G.; Schachtrup, K.; Schara, U.; Schorling, D.; Schwersenz, I.; Stein, S.; Tassoni, A.; Vogt, S.; et al. SMArtCARE—A platform to collect real-life outcome data of patients with spinal muscular atrophy. Orphanet J. Rare Dis. 2019, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Finkel, R.; Scoto, M.; Hall, S.; Eaton, S.; Rashid, A.; Balashkina, J.; Coratti, G.; Pera, M.C.; Samsuddin, S.; et al. Development of an academic disease registry for spinal muscular atrophy. Neuromuscul. Disord. 2019, 29, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Frongia, A.L.; Antonaci, L.; Pera, M.C.; Coratti, G.; Pane, M.; Pasternak, A.; Civitello, M.; Montes, J.; Mayhew, A.; et al. A critical review of patient and parent caregiver oriented tools to assess health-related quality of life, activity of daily living and caregiver burden in spinal muscular atrophy. Neuromuscul. Disord. 2019, 29, 940–950. [Google Scholar] [CrossRef]
- Mercuri, E.; Messina, S.; Montes, J.; Muntoni, F.; Sansone, V.A.; All Participants and the SMA PROM Working Group. Patient and parent oriented tools to assess health-related quality of life, activity of daily living and caregiver burden in SMA. Rome, 13 July 2019. Neuromuscul. Disord. 2020. [Google Scholar] [CrossRef] [PubMed]
- Polido, G.J.; de Miranda, M.M.V.; Carvas, N.; Mendonça, R.H.; Caromano, F.A.; Reed, U.C.; Zanoteli, E.; Voos, M.C. Cognitive performance of children with spinal muscular atrophy: A systematic review. Dement. Neuropsychol. 2019, 13, 436–443. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Approach /Compound | Sponsor | Mechanism | Trials’ Phase (SMA Type) | Administration | FDA Approval |
|---|---|---|---|---|---|
| Splicing modifiers of SMN2 gene | |||||
| Nusinersen | Ionis-Biogen | ASO | I, II and III (I, II, III) | Intrathecal | X |
| Risdiplam | Roche | Small molecule | I, II and III (I, II, III) | Oral | pending |
| Albuterol | Beta-adrenergic agonist | Off-label | Oral | ||
| Replacing SMN1 gene | |||||
| Onasemnogene abeparvosec | Novartis-Avexis | AAV-9-vector construct | I, II and III (I, II) | Intravenous | X |
| Onasemnogene abeparvosec | Novartis-Avexis | AAV-9-vector construct | I | Intrathecal | |
| Muscle enhancing | |||||
| Reldesemtiv | Cytokinetics | Troponin activator | I and II (II, III, IV) | Oral | |
| SRK-105 | Scholar Rock | Myostatin inhibitor | I and II (II, III) | Intravenous | |
| Neuroprotection | |||||
| Olesoxime | Hoffmann-La Roche | Anti-apoptotic agent | I and II (II, III) (development ended in 2018) | Oral | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messina, S.; Sframeli, M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 2020, 9, 2222. https://doi.org/10.3390/jcm9072222
Messina S, Sframeli M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. Journal of Clinical Medicine. 2020; 9(7):2222. https://doi.org/10.3390/jcm9072222
Chicago/Turabian StyleMessina, Sonia, and Maria Sframeli. 2020. "New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges" Journal of Clinical Medicine 9, no. 7: 2222. https://doi.org/10.3390/jcm9072222
APA StyleMessina, S., & Sframeli, M. (2020). New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. Journal of Clinical Medicine, 9(7), 2222. https://doi.org/10.3390/jcm9072222