Genetic Study in Korean Pediatric Patients with Steroid-Resistant Nephrotic Syndrome or Focal Segmental Glomerulosclerosis

 , , , ,
, , , ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Strategy of Mutational Studies
2.3. Targeted Exome Sequencing (TES) and Variant Calling
2.4. Whole-Exome Sequencing (WES) and Variant Calling
2.5. Statistical Analysis
3. Results
3.1. Phenotypes
3.2. Genotypes
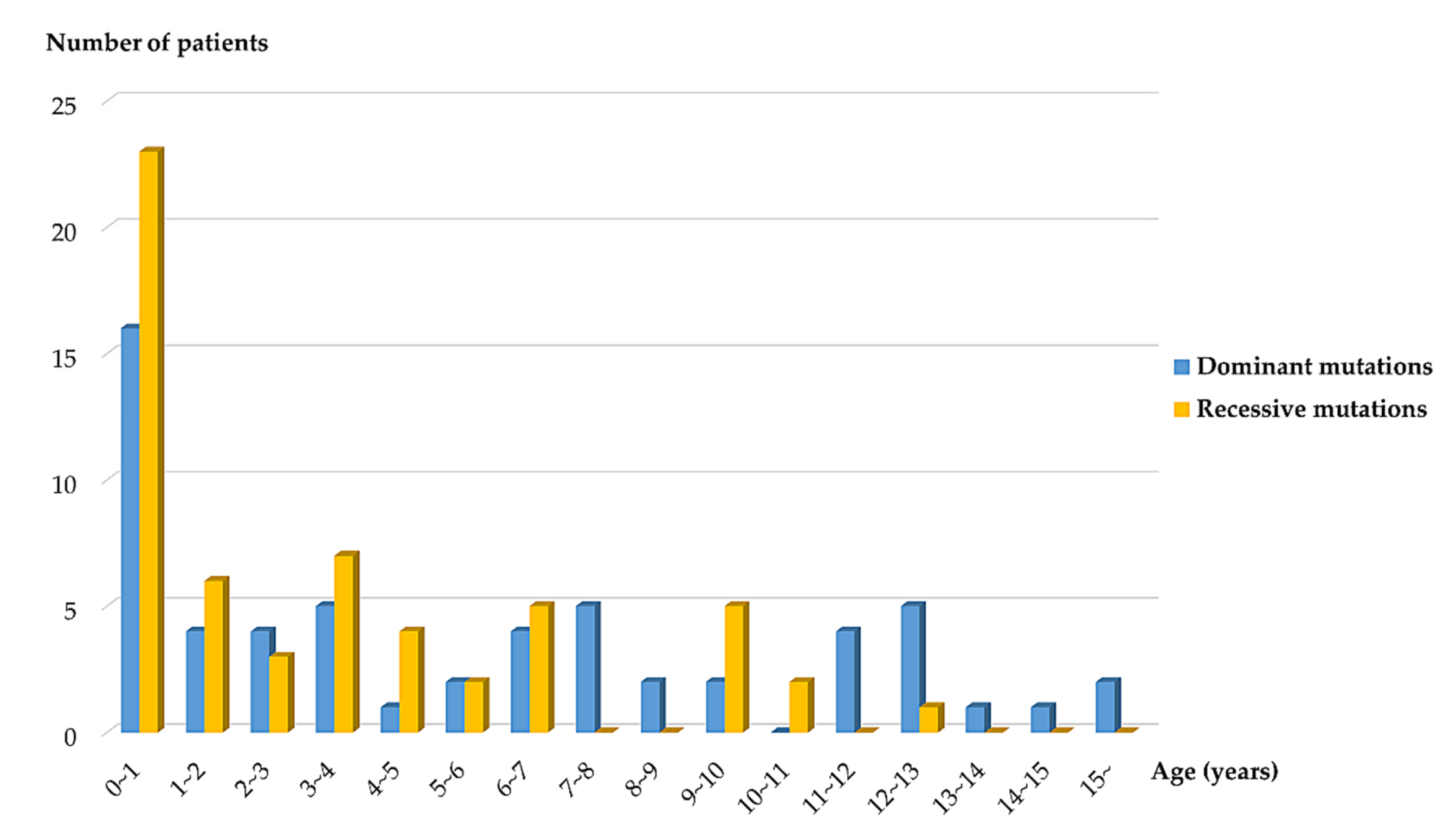
3.3. Genotype-Phenotype Correlations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Noone, D.G.; Iijima, K.; Parekh, R. Idiopathic nephrotic syndrome in children. Lancet 2018, 392, 61–74. [Google Scholar] [CrossRef]
- Bierzynska, A.; McCarthy, H.J.; Soderquest, K.; Sen, E.S.; Colby, E.; Ding, W.Y.; Nabhan, M.M.; Kerecuk, L.; Hegde, S.; Hughes, D.; et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. 2017, 91, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, A.; Bodria, M.; Ozaltin, F.; Gheisari, A.; Melk, A.; Azocar, M.; Anarat, A.; Caliskan, S.; Emma, F.; Gellermann, J.; et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: The PodoNet registry cohort. Clin. J. Am. Soc. Nephrol. 2015, 10, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, C.E.; Lovric, S.; Ashraf, S.; Pabst, W.L.; Gee, H.Y.; Kohl, S.; Engelmann, S.; Vega-Warner, V.; Fang, H.; Halbritter, J.; et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 2015, 26, 1279–1289. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, Y.; Mao, J.; Yu, Z.; Yi, Z.; Yu, L.; Sun, J.; Wei, X.; Ding, F.; Zhang, H.; et al. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 2017, 32, 1181–1192. [Google Scholar] [CrossRef]
- Nagano, C.; Yamamura, T.; Horinouchi, T.; Aoto, Y.; Ishiko, S.; Sakakibara, N.; Shima, Y.; Nakanishi, K.; Nagase, H.; Iijima, K.; et al. Comprehensive genetic diagnosis of Japanese patients with severe proteinuria. Sci. Rep. 2020, 10, 270. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Heeringa, S.F. Specific podocin mutations determine age of onset of nephrotic syndrome all the way into adult life. Kidney Int. 2009, 75, 669–671. [Google Scholar] [CrossRef]
- Harita, Y. Application of next-generation sequencing technology to diagnosis and treatment of focal segmental glomerulosclerosis. Clin. Exp. Nephrol. 2018, 22, 491–500. [Google Scholar] [CrossRef]
- Groopman, E.E.; Rasouly, H.M.; Gharavi, A.G. Genomic medicine for kidney disease. Nat. Rev. Nephrol. 2018, 14, 83–104. [Google Scholar] [CrossRef]
- Lovric, S.; Ashraf, S.; Tan, W.; Hildebrandt, F. Genetic testing in steroid-resistant nephrotic syndrome: When and how? Nephrol. Dial. Transplant. 2016, 31, 1802–1813. [Google Scholar] [CrossRef]
- Preston, R.; Stuart, H.M.; Lennon, R. Genetic testing in steroid-resistant nephrotic syndrome: Why, who, when and how? Pediatr. Nephrol. 2019, 34, 195–210. [Google Scholar] [CrossRef]
- Hinkes, B.G.; Mucha, B.; Vlangos, C.N.; Gbadegesin, R.; Liu, J.; Hasselbacher, K.; Hangan, D.; Ozaltin, F.; Zenker, M.; Hildebrandt, F.; et al. Nephrotic syndrome in the first year of life: Two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 2007, 119, e907–e919. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, K.H.; Lee, H.; Kang, H.G.; Moon, K.C.; Shin, J.I.; Hahn, H.; Park, Y.S.; Pai, K.S.; Cho, B.S.; et al. Genetic basis of congenital and infantile nephrotic syndromes. Am. J. Kidney Dis. 2011, 58, 1042–1043. [Google Scholar] [CrossRef]
- Cheong, H.I. Genetic tests in children with steroid-resistant nephrotic syndrome. Kidney Res. Clin. Pract. 2020, 39, 7–16. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2015 Clinical Practice Guideline for glomerulonephritis. Kidney Int. Suppl. 2012, 2, 139–274. [Google Scholar]
- Cheong, H.I.; Chae, J.H.; Kim, J.S.; Park, H.W.; Ha, I.S.; Hwang, Y.S.; Lee, H.S.; Choi, Y. Hereditary glomerulopathy associated with a mitochondrial tRNA(Leu) gene mutation. Pediatr. Nephrol. 1999, 13, 477–480. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kang, H.G.; Lee, M.; Lee, K.B.; Hughes, M.; Kwon, B.S.; Lee, S.; McNagny, K.M.; Ahn, Y.H.; Ko, J.M.; Ha, I.S.; et al. Loss of podocalyxin causes a novel syndromic type of congenital nephrotic syndrome. Exp. Mol. Med. 2017, 49, e414. [Google Scholar] [CrossRef]
- Warejko, J.K.; Tan, W.; Daga, A.; Schapiro, D.; Lawson, J.A.; Shril, S.; Lovric, S.; Ashraf, S.; Rao, J.; Hermle, T.; et al. Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 53–62. [Google Scholar] [CrossRef]
- Boute, N.; Gribouval, O.; Roselli, S.; Benessy, F.; Lee, H.; Fuchshuber, A.; Dahan, K.; Gubler, M.C.; Niaudet, P.; Antignac, C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet. 2000, 24, 349–354. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Sudhakar, A.; Le, T.C.; Nguyen, T.; Yao, J.; Schwimmer, J.A.; Schachter, A.D.; Poch, E.; Abreu, P.F.; Appel, G.B.; et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J. Clin. Investig. 2002, 110, 1659–1666. [Google Scholar] [CrossRef]
- Park, E.; Ahn, Y.H.; Kang, H.G.; Yoo, K.H.; Won, N.H.; Lee, K.B.; Moon, K.C.; Seong, M.W.; Gwon, T.R.; Park, S.S.; et al. COQ6 mutations in children with steroid-resistant focal segmental glomerulosclerosis and sensorineural hearing loss. Am. J. Kidney Dis. 2017, 70, 139–144. [Google Scholar] [CrossRef]
- Park, E.; Kang, H.G.; Choi, Y.H.; Lee, K.B.; Moon, K.C.; Jeong, H.J.; Nagata, M.; Cheong, H.I. Focal segmental glomerulosclerosis and medullary nephrocalcinosis in children with ADCK4 mutations. Pediatr. Nephrol. 2017, 32, 1547–1554. [Google Scholar] [CrossRef]
- Park, E.; Ahn, Y.H.; Kang, H.G.; Miyake, N.; Tsukaguchi, H.; Cheong, H.I. NUP107 mutations in children with steroid-resistant nephrotic syndrome. Nephrol. Dial. Transplant. 2017, 32, 1013–1017. [Google Scholar]
- Lee, S.E.; Min, S.I.; Kim, Y.S.; Ha, J.; Ha, I.S.; Cheong, H.I.; Kim, S.J.; Choi, Y.; Kang, H.G. Recurrence of idiopathic focal segmental glomerulosclerosis after kidney transplantation: Experience of a Korean tertiary center. Pediatr. Transplant. 2014, 18, 369–376. [Google Scholar] [CrossRef]
- Bergmann, C. Advances in renal genetic diagnosis. Cell Tissue Res. 2017, 369, 93–104. [Google Scholar] [CrossRef]
- Lipska, B.S.; Ranchin, B.; Iatropoulos, P.; Gellermann, J.; Melk, A.; Ozaltin, F.; Caridi, G.; Seeman, T.; Tory, K.; Jankauskiene, A.; et al. Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int. 2014, 85, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.H.; Park, E.J.; Kang, H.G.; Kim, S.H.; Cho, H.Y.; Shin, J.I.; Lee, J.H.; Park, Y.S.; Kim, K.S.; Ha, I.S.; et al. Genotype-phenotype analysis of pediatric patients with WT1 glomerulopathy. Pediatr. Nephrol. 2017, 32, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef] [PubMed]
- Montini, G.; Malaventura, C.; Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008, 358, 2849–2850. [Google Scholar] [CrossRef] [PubMed]
- Jungraithmayr, T.C.; Hofer, K.; Cochat, P.; Chernin, G.; Cortina, G.; Fargue, S.; Grimm, P.; Knueppel, T.; Kowarsch, A.; Neuhaus, T.; et al. Screening for NPHS2 mutations may help predict FSGS recurrence after transplantation. J. Am. Soc. Nephrol. 2011, 22, 579–585. [Google Scholar] [CrossRef]
- National Cancer Institute. NCI Dictionary of Genetics Terms. Available online: https://www.cancer.gov/publications/dictionaries/genetics-dictionary/def/phenocopy (accessed on 27 February 2020).
- Landini, S.; Mazzinghi, B.; Becherucci, F.; Allinovi, M.; Provenzano, A.; Palazzo, V.; Ravaglia, F.; Artuso, R.; Bosi, E.; Stagi, S.; et al. Reverse phenotyping after whole-exome sequencing in steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2020, 15, 89–100. [Google Scholar] [CrossRef]
- Schaefer, F. It’s in your genes: Exome sequencing enables precision diagnostics in proteinuric kidney diseases. Clin. J. Am. Soc. Nephrol. 2020, 15, 10–12. [Google Scholar] [CrossRef]
- Kaplan, J.M.; Kim, S.H.; North, K.N.; Rennke, H.; Correia, L.A.; Tong, H.Q.; Mathis, B.J.; Rodríguez-Pérez, J.C.; Allen, P.G.; Beggs, A.H.; et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000, 24, 251–256. [Google Scholar] [CrossRef]
- Kranz, C.; Denecke, J.; Lehle, L.; Sohlbach, K.; Jeske, S.; Meinhardt, F.; Rossi, R.; Gudowius, S.; Marquardt, T. Congenital disorder of glycosylation type Ik (CDG-Ik): A defect of mannosyltransferase I. Am. J. Hum. Genet. 2004, 74, 545–551. [Google Scholar] [CrossRef][Green Version]
- Gbadegesin, R.A.; Hall, G.; Adeyemo, A.; Hanke, N.; Tossidou, I.; Burchette, J.; Wu, G.; Homstad, A.; Sparks, M.A.; Gomez, J.; et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J. Am. Soc. Nephrol. 2014, 25, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Friedman, D.J.; Ross, M.D.; Lecordier, L.; Uzureau, P.; Freedman, B.I.; Bowden, D.W.; Langefeld, C.D.; Oleksyk, T.K.; Uscinski Knob, A.L.; et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010, 329, 841–845. [Google Scholar] [CrossRef]
- Akilesh, S.; Suleiman, H.; Yu, H.; Stander, M.C.; Lavin, P.; Gbadegesin, R.; Antignac, C.; Pollak, M.; Kopp, J.B.; Winn, M.P.; et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J. Clin. Investig. 2011, 121, 4127–4137. [Google Scholar] [CrossRef]
- Gee, H.Y.; Saisawat, P.; Ashraf, S.; Hurd, T.W.; Vega-Warner, V.; Fang, H.; Beck, B.B.; Gribouval, O.; Zhou, W.; Diaz, K.A.; et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J. Clin. Investig. 2013, 123, 3243–3253. [Google Scholar] [CrossRef]
- Dear, T.N.; Meier, N.T.; Hunn, M.; Boehm, T. Gene structure, chromosomal localization, and expression pattern of Capn12, a new member of the calpain large subunit gene family. Genomics 2000, 68, 152–160. [Google Scholar] [CrossRef]
- Karamatic Crew, V.; Burton, N.; Kagan, A.; Green, C.A.; Levene, C.; Flinter, F.; Brady, R.L.; Daniels, G.; Anstee, D.J. CD151, the first member of the tetraspanin (TM4) superfamily detected on erythrocytes, is essential for the correct assembly of human basement membranes in kidney and skin. Blood 2004, 104, 2217–2223. [Google Scholar] [CrossRef]
- Löwik, M.M.; Groenen, P.J.; Pronk, I.; Lilien, M.R.; Goldschmeding, R.; Dijkman, H.B.; Levtchenko, E.N.; Monnens, L.A.; van den Heuvel, L.P. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int. 2007, 72, 1198–1203. [Google Scholar] [CrossRef]
- Sethi, S.; Fervenza, F.C.; Zhang, Y.; Smith, R.J. Secondary focal and segmental glomerulosclerosis associated with single-nucleotide polymorphisms in the genes encoding complement factor H and C3. Am. J. Kidney Dis. 2012, 60, 316–321. [Google Scholar] [CrossRef]
- Voskarides, K.; Damianou, L.; Neocleous, V.; Zouvani, I.; Christodoulidou, S.; Hadjiconstantinou, V.; Ioannou, K.; Athanasiou, Y.; Patsias, C.; Alexopoulos, E.; et al. COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J. Am. Soc. Nephrol. 2007, 18, 3004–3016. [Google Scholar] [CrossRef]
- Slajpah, M.; Gorinsek, B.; Berginc, G.; Vizjak, A.; Ferluga, D.; Hvala, A.; Meglic, A.; Jaksa, I.; Furlan, P.; Gregoric, A.; et al. Sixteen novel mutations identified in COL4A3, COL4A4, and COL4A5 genes in Slovenian families with Alport syndrome and benign familial hematuria. Kidney Int. 2007, 71, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Sacconi, S.; Murer, L.; Zacchello, G.; Franceschini, L.; Laverda, A.M.; Basso, G.; Quinzii, C.; Angelini, C.; Hirano, M.; et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: A CoQ10-responsive condition. Neurology 2005, 65, 606–608. [Google Scholar] [CrossRef] [PubMed]
- Ebarasi, L.; Ashraf, S.; Bierzynska, A.; Gee, H.Y.; McCarthy, H.J.; Lovric, S.; Sadowski, C.E.; Pabst, W.; Vega-Warner, V.; Fang, H.; et al. Defects of CRB2 cause steroid-resistant nephrotic syndrome. Am. J. Hum. Genet. 2015, 96, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Ovunc, B.; Otto, E.A.; Vega-Warner, V.; Saisawat, P.; Ashraf, S.; Ramaswami, G.; Fathy, H.M.; Schoeb, D.; Chernin, G.; Lyons, R.H.; et al. Exome sequencing reveals cubilin mutation as a single-gene cause of proteinuria. J. Am. Soc. Nephrol. 2011, 22, 1815–1820. [Google Scholar] [CrossRef] [PubMed]
- Ozaltin, F.; Li, B.; Rauhauser, A.; An, S.W.; Soylemezoglu, O.; Gonul, I.I.; Taskiran, E.Z.; Ibsirlioglu, T.; Korkmaz, E.; Bilginer, Y.; et al. DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. J. Am. Soc. Nephrol. 2013, 24, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Izu, A.; Yanagida, H.; Sugimoto, K.; Fujita, S.; Sakata, N.; Wada, N.; Okada, M.; Takemura, T. Pathogenesis of focal segmental glomerular sclerosis in a girl with the partial deletion of chromosome 6p. Tohoku J. Exp. Med. 2011, 223, 187–192. [Google Scholar] [CrossRef]
- Gee, H.Y.; Ashraf, S.; Wan, X.; Vega-Warner, V.; Esteve-Rudd, J.; Lovric, S.; Fang, H.; Hurd, T.W.; Sadowski, C.E.; Allen, S.J.; et al. Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am. J. Hum. Genet. 2014, 94, 884–890. [Google Scholar] [CrossRef]
- Castelletti, F.; Donadelli, R.; Banterla, F.; Hildebrandt, F.; Zipfel, P.F.; Bresin, E.; Otto, E.; Skerka, C.; Renieri, A.; Todeschini, M.; et al. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc. Natl. Acad. Sci. USA 2008, 105, 2538–2543. [Google Scholar] [CrossRef]
- Moudgil, A.; Perriello, P.; Loechelt, B.; Przygodzki, R.; Fitzerald, W.; Kamani, N. Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: An unusual cause of proteinuria in infancy. Pediatr. Nephrol. 2007, 22, 1799–1802. [Google Scholar] [CrossRef]
- Okamoto, K.; Tokunaga, K.; Doi, K.; Fujita, T.; Suzuki, H.; Katoh, T.; Watanabe, T.; Nishida, N.; Mabuchi, A.; Takahashi, A.; et al. Common variation in GPC5 is associated with acquired nephrotic syndrome. Nat. Genet. 2011, 43, 459–463. [Google Scholar] [CrossRef]
- Barua, M.; Brown, E.J.; Charoonratana, V.T.; Genovese, G.; Sun, H.; Pollak, M.R. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 2013, 83, 316–322. [Google Scholar] [CrossRef]
- Has, C.; Spartà, G.; Kiritsi, D.; Weibel, L.; Moeller, A.; Vega-Warner, V.; Waters, A.; He, Y.; Anikster, Y.; Esser, P.; et al. Integrin α3 mutations with kidney, lung, and skin disease. N. Engl. J. Med. 2012, 366, 1508–1514. [Google Scholar] [CrossRef]
- Kambham, N.; Tanji, N.; Seigle, R.L.; Markowitz, G.S.; Pulkkinen, L.; Uitto, J.; D’Agati, V.D. Congenital focal segmental glomerulosclerosis associated with beta4 integrin mutation and epidermolysis bullosa. Am. J. Kidney Dis. 2000, 36, 190–196. [Google Scholar] [CrossRef]
- Gee, H.Y.; Zhang, F.; Ashraf, S.; Kohl, S.; Sadowski, C.E.; Vega-Warner, V.; Zhou, W.; Lovric, S.; Fang, H.; Nettleton, M.; et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J. Clin. Investig. 2015, 125, 2375–2384. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, R.; Hoffman, M.; Cliften, P.; Seshan, S.; Liapis, H.; Jain, S. Targeted exome sequencing integrated with clinicopathological information reveals novel and rare mutations in atypical, suspected and unknown cases of Alport syndrome or proteinuria. PLoS ONE 2013, 8, e76360. [Google Scholar] [CrossRef]
- Zenker, M.; Aigner, T.; Wendler, O.; Tralau, T.; Müntefering, H.; Fenski, R.; Pitz, S.; Schumacher, V.; Royer-Pokora, B.; Wühl, E.; et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum. Mol. Genet. 2004, 13, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Thong, K.M.; Xu, Y.; Cook, J.; Takou, A.; Wagner, B.; Kawar, B.; Ong, A.C. Cosegregation of focal segmental glomerulosclerosis in a family with familial partial lipodystrophy due to a mutation in LMNA. Nephron Clin. Pract. 2013, 124, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, S.D.; Zhou, G.; Baldini, A.; Winterpacht, A.; Zabel, B.; Cole, W.; Johnson, R.L.; Lee, B. Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat. Genet. 1998, 19, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Shohat, M.; Magal, N.; Shohat, T.; Chen, X.; Dagan, T.; Mimouni, A.; Danon, Y.; Lotan, R.; Ogur, G.; Sirin, A.; et al. Phenotype-genotype correlation in familial Mediterranean fever: Evidence for an association between Met694Val and amyloidosis. Eur. J. Hum. Genet. 1999, 7, 287–292. [Google Scholar] [CrossRef]
- Sanna-Cherchi, S.; Burgess, K.E.; Nees, S.N.; Caridi, G.; Weng, P.L.; Dagnino, M.; Bodria, M.; Carrea, A.; Allegretta, M.A.; Kim, H.R.; et al. Exome sequencing identified MYO1E and NEIL1 as candidate genes for human autosomal recessive steroid-resistant nephrotic syndrome. Kidney Int. 2011, 80, 389–396. [Google Scholar] [CrossRef]
- Brunelli, S.M.; Meyers, K.E.; Guttenberg, M.; Kaplan, P.; Kaplan, B.S. Cobalamin C deficiency complicated by an atypical glomerulopathy. Pediatr. Nephrol. 2002, 17, 800–803. [Google Scholar] [CrossRef]
- Yasukawa, T.; Suzuki, T.; Ueda, T.; Ohta, S.; Watanabe, K. Modification defect at anticodon wobble nucleotide of mitochondrial tRNAs(Leu)(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J. Biol. Chem. 2000, 275, 4251–4257. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, F.; Vogel, H.; Hawkins, E.P.; Vladutiu, G.D.; Liu, L.L.; Wong, L.J. Novel homoplasmic mutation in the mitochondrial tRNATyr gene associated with atypical mitochondrial cytopathy presenting with focal segmental glomerulosclerosis. Am. J. Med. Genet. A 2003, 123, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Smith, M.W.; Nelson, G.W.; Johnson, R.C.; Freedman, B.I.; Bowden, D.W.; Oleksyk, T.; McKenzie, L.M.; Kajiyama, H.; Ahuja, T.S.; et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat. Genet. 2008, 40, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Mele, C.; Iatropoulos, P.; Donadelli, R.; Calabria, A.; Maranta, R.; Cassis, P.; Buelli, S.; Tomasoni, S.; Piras, R.; Krendel, M.; et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N. Engl. J. Med. 2011, 365, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Kestilä, M.; Lenkkeri, U.; Männikkö, M.; Lamerdin, J.; McCready, P.; Putaala, H.; Ruotsalainen, V.; Morita, T.; Nissinen, M.; Herva, R.; et al. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol. Cell 1998, 1, 575–582. [Google Scholar] [CrossRef]
- Miyake, N.; Tsukaguchi, H.; Koshimizu, E.; Shono, A.; Matsunaga, S.; Shiina, M.; Mimura, Y.; Imamura, S.; Hirose, T.; Okudela, K.; et al. Biallelic Mutations in Nuclear Pore Complex Subunit NUP107 Cause Early-Childhood-Onset Steroid-Resistant Nephrotic Syndrome. Am. J. Hum. Genet. 2015, 97, 555–566. [Google Scholar] [CrossRef]
- Esposito, T.; Lea, R.A.; Maher, B.H.; Moses, D.; Cox, H.C.; Magliocca, S.; Angius, A.; Nyholt, D.R.; Titus, T.; Kay, T.; et al. Unique X-linked familial FSGS with co-segregating heart block disorder is associated with a mutation in the NXF5 gene. Hum. Mol. Genet. 2013, 22, 3654–3666. [Google Scholar] [CrossRef] [PubMed]
- Kerti, A.; Csohány, R.; Wagner, L.; Jávorszky, E.; Maka, E.; Tory, K. NPHS2 homozygous p.R229Q variant: Potential modifier instead of causal effect in focal segmental glomerulosclerosis. Pediatr. Nephrol. 2013, 28, 2061–2064. [Google Scholar] [CrossRef]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; Dimauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef]
- Gbadegesin, R.; Hinkes, B.G.; Hoskins, B.E.; Vlangos, C.N.; Heeringa, S.F.; Liu, J.; Loirat, C.; Ozaltin, F.; Hashmi, S.; Ulmer, F.; et al. Mutations in PLCE1 are a major cause of isolated diffuse mesangial sclerosis (IDMS). Nephrol. Dial. Transplant. 2008, 23, 1291–1297. [Google Scholar] [CrossRef]
- Ozaltin, F.; Ibsirlioglu, T.; Taskiran, E.Z.; Baydar, D.E.; Kaymaz, F.; Buyukcelik, M.; Kilic, B.D.; Balat, A.; Iatropoulos, P.; Asan, E.; et al. Disruption of PTPRO causes childhood-onset nephrotic syndrome. Am. J. Hum. Genet. 2011, 89, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Berkovic, S.F.; Dibbens, L.M.; Oshlack, A.; Silver, J.D.; Katerelos, M.; Vears, D.F.; Lüllmann-Rauch, R.; Blanz, J.; Zhang, K.W.; Stankovich, J.; et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am. J. Hum. Genet. 2008, 82, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Boerkoel, C.F.; Takashima, H.; John, J.; Yan, J.; Stankiewicz, P.; Rosenbarker, L.; André, J.L.; Bogdanovic, R.; Burguet, A.; Cockfield, S.; et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat. Genet. 2002, 30, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wang, Z.; Pan, X.; Wang, W.; Chen, X.; Ren, H.; Hao, C.; Han, B.; Chen, N. Functional analysis of promoter mutations in the ACTN4 and SYNPO genes in focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2010, 25, 824–835. [Google Scholar] [CrossRef]
- Santín, S.; Ars, E.; Rossetti, S.; Salido, E.; Silva, I.; García-Maset, R.; Giménez, I.; Ruíz, P.; Mendizábal, S.; Luciano Nieto, J.; et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2009, 24, 3089–3096. [Google Scholar] [CrossRef]
- Huynh Cong, E.; Bizet, A.A.; Boyer, O.; Woerner, S.; Gribouval, O.; Filhol, E.; Arrondel, C.; Thomas, S.; Silbermann, F.; Canaud, G.; et al. A homozygous missense mutation in the ciliary gene TTC21B causes familial FSGS. J. Am. Soc. Nephrol. 2014, 25, 2435–2443. [Google Scholar] [CrossRef]
- Colin, E.; Huynh Cong, E.; Mollet, G.; Guichet, A.; Gribouval, O.; Arrondel, C.; Boyer, O.; Daniel, L.; Gubler, M.C.; Ekinci, Z.; et al. Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome. Am. J. Hum. Genet. 2014, 95, 637–648. [Google Scholar] [CrossRef]
- Barbaux, S.; Niaudet, P.; Gubler, M.C.; Grünfeld, J.P.; Jaubert, F.; Kuttenn, F.; Fékété, C.N.; Souleyreau-Therville, N.; Thibaud, E.; Fellous, M.; et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat. Genet. 1997, 17, 467–470. [Google Scholar] [CrossRef]
- Pelletier, J.; Bruening, W.; Kashtan, C.E.; Mauer, S.M.; Manivel, J.C.; Striegel, J.E.; Houghton, D.C.; Junien, C.; Habib, R.; Fouser, L.; et al. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 1991, 67, 437–447. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
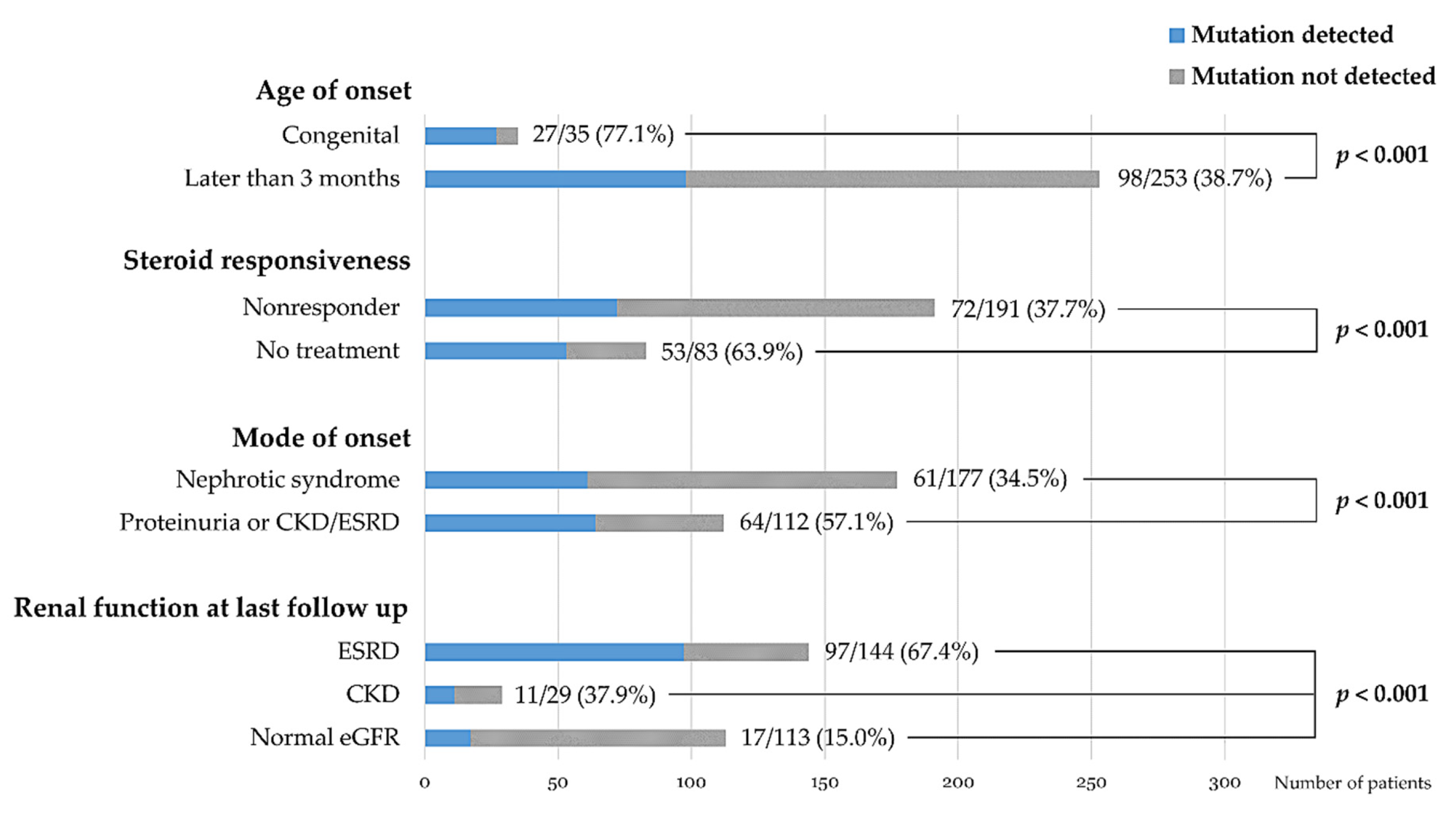
| Phenotype | Mutation (+) Patients (n = 127) | Mutation (−) Patients (n = 164) | Total Patients (n = 291) | Mutation Detection Rate | p Value |
|---|---|---|---|---|---|
| Age at onset | |||||
| Congenital | 27 (21.3%) | 8 (4.9%) | 35 (12.0%) | 77.1% | <0.001 b |
| Infantile | 11 (8.7%) | 14 (8.5%) | 25 (8.6%) | 44.0% | |
| 1–6 years | 40 (31.5%) | 74 (45.1%) | 114 (39.2%) | 35.1% | |
| 7–12 years | 32 (25.2%) | 46 (28.0%) | 78 (26.8%) | 41.0% | |
| ≥13 years | 15 (11.8%) | 21 (12.8%) | 36 (12.4%) | 41.7% | |
| Data unavailable | 2 (1.6%) | 1 (0.6%) | 3 (1.0%) | ||
| Sex ratio a (Male:female) | 71:56 | 91:73 | 162:129 | ||
| Family history (+) | 24 (18.9%) | 24 (14.6%) | 48 (16.5%) | 50.0% | |
| Mode of onset | |||||
| Nephrotic syndrome | 61 (48.0%) | 116 (70.7%) | 177 (60.8%) | 34.5% | <0.001 c |
| Proteinuria | 53 (41.7%) | 40 (24.4%) | 93 (32.0%) | 57.0% | |
| CKD/ESRD | 11 (8.7%) | 8 (4.9%) | 19 (6.5%) | 57.9% | |
| Data unavailable | 2 (1.6%) | 0 | 2 (0.7%) | ||
| Steroid responsiveness | |||||
| Responder | 0 | 15 (9.1%) | 15 (5.2%) | 0% | |
| Non-responder | 72 (56.7%) | 119 (72.6%) | 191 (65.6%) | 37.7% | |
| Initial non-responder | 71 | 108 | 179 | 39.7% | |
| Late non-responder | 1 | 11 | 12 | 8.3% | |
| No treatment | 53 (41.7%) | 30 (18.3%) | 83 (28.5%) | 63.9% | <0.001 d |
| Data unavailable | 2 (1.6%) | 0 | 2 (0.7%) | ||
| Renal biopsy | |||||
| FSGS | 78 (61.4%) | 87 (53.0%) | 165 (56.7%) | 47.3% | |
| Minimal change disease | 0 | 31 (18.9%) | 31 (10.7%) | 0% | |
| Others | 20 (15.7%) | 20 (12.2%) | 40 (13.7%) | 50.0% | |
| Not done | 27 (21.3%) | 23 (14.0%) | 50 (17.2%) | 54.0% | |
| Data unavailable | 2 (1.6%) | 3 (1.8%) | 5 (1.7%) | ||
| Renal function at the last FU | <0.001 e | ||||
| Normal eGFR | 17 (13.4%) | 96 (58.5%) | 113 (38.8%) | 15.0% | |
| CKD stages 2–4 | 11 (8.7%) | 18 (11.0%) | 29 (10.0%) | 37.9% | |
| ESRD | 97 (76.4%) | 47 (28.7%) | 144 (49.5%) | 67.4% | |
| Data unavailable | 2 (1.6%) | 3 (1.8%) | 5 (1.7%) | ||
| Duration (years) from onset to ESRD (n = 144) | 3.6 ± 4.3 | 5.0 ± 4.8 | 4.1 ± 4.5 | 0.091 | |
| Recurrence after renal transplantation (n = 100) | 0/64 (0%) | 9/36 (25.0%) | 9/100 (9.0%) | <0.0001 | |
| Gene | Mode of Inheritance | No. of Patients (%) | % of Total Patients (n = 291) |
|---|---|---|---|
| SRNS/FSGS gene | |||
| WT1 | AD | 30 (23.6%) | 10.3% |
| COQ6 | AR | 11 (8.7%) | 3.8% |
| NPHS1 | AR | 11 (8.7%) | 3.8% |
| NUP107 | AR | 9 (7.1%) | 3.1% |
| COQ8B | AR | 8 (6.3%) | 2.7% |
| MYH9 | AD | 6 (4.7%) | 2.1% |
| INF2 | AD | 6 (4.7%) | 2.1% |
| PAX2 | AD | 5 (3.9%) | 1.7% |
| NPHS2 | AR | 4 (3.1%) | 1.4% |
| MAFB | AD | 4 (3.1%) | 1.4% |
| LAMB2 | AR | 3 (2.4%) | 1.0% |
| SMARCAL1 | AR | 3 (2.4%) | 1.0% |
| MT-TL1 | Mitochondrial | 3 (2.4%) | 1.0% |
| ACTN4 | AD | 1 (0.8%) | 0.3% |
| LMX1B | AD | 1 (0.8%) | 0.3% |
| ANLN | AD | 1 (0.8%) | 0.3% |
| TRPC6 | AD | 1 (0.8%) | 0.3% |
| TP53RK | AR | 1 (0.8%) | 0.3% |
| PODXL | AR | 1 (0.8%) | 0.3% |
| DGKE | AR | 1 (0.8%) | 0.3% |
| FOXP3 | X-linked | 1 (0.8%) | 0.3% |
| LCAT COQ2 | AR AR | 1 (0.8%) 1 (0.8%) | 0.3% 0.3% |
| Subtotal | 113 (89.0%) | 38.8% | |
| Phenocopying gene | |||
| COL4A5 | X-linked | 6 (4.7%) | 2.1% |
| COL4A4 | AD/AR | 4 (3.1%) | 1.4% |
| WDR19 | AR | 3 (2.4%) | 1.0% |
| COL4A3 | AD | 1 (0.8%) | 0.3% |
| Subtotal | 14 (11.0%) | 4.8% | |
| Total | 127 (100%) | 43.6% | |
| Trautmann et al., 2015 [3] | Sadowski et al., 2015 [4] | Bierzynska et al., 2017 [2] | Wang et al., 2017 [5] | Warejko et al., 2018 [23] | Nagano et al., 2020 [6] | This Study | |
|---|---|---|---|---|---|---|---|
| Country | International | International | United Kingdom | China | International | Japan | Korea |
| Modality | GP (31 genes) | GP (27 genes) | WES (53 genes) | GP (28 genes) | WES | GP (60 genes) | Sanger/GP (57 genes) c |
| Detection rate a | 277/1174 (23.6%) | 526/1783 (29.5%) | 49/187 (26.2%) | 34/120 (28.3%) | 85/300 (28.3%) | 69/230 (30.0%) | 127/291 (43.6%) |
| Commonly mutated genes b | NPHS2 138 (49.8%) | NPHS2 177 (33.7%) | NPHS1 14 (28.6%) | COQ8B 8 (23.5%) | NPHS1 13 (15.3%) | WT1 17 (24.6%) | WT1 30 (23.6%) |
| WT1 48 (17.3%) | NPHS1 131 (24.9%) | NPHS2 12 (24.5%) | NPHS1 7 (20.6%) | PLCE1 11 (12.9%) | NPHS1 8 (11.6%) | COQ6 11 (8.7%) | |
| NPHS1 41 (14.8%) | WT1 85 (16.2%) | WT1 4 (8.2%) | WT1 7 (20.6%) | NPHS2 8 (9.4%) | INF2 8 (11.6%) | NPHS1 11 (8.7%) | |
| SMARCAL1 12 (4.3%) | PLCE1 37 (7.0%) | NUP107 4 (8.2%) | NPHS2 4 (11.8%) | SMARCAL1 8 (9.4%) | TRPC6 7 (10.1%) | NUP107 9 (7.1%) | |
| PLCE1 10 (3.6%) | LAMB2 20 (3.8%) | TRPC6 3 (6.1%) | LMX1B 2 (5.9%) | LAMB2 6 (7.1%) | LAMB2 6 (8.7%) | COQ8B 8 (6.3%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, E.; Lee, C.; Kim, N.K.D.; Ahn, Y.H.; Park, Y.S.; Lee, J.H.; Kim, S.H.; Cho, M.H.; Cho, H.; Yoo, K.H.; et al. Genetic Study in Korean Pediatric Patients with Steroid-Resistant Nephrotic Syndrome or Focal Segmental Glomerulosclerosis. J. Clin. Med. 2020, 9, 2013. https://doi.org/10.3390/jcm9062013
Park E, Lee C, Kim NKD, Ahn YH, Park YS, Lee JH, Kim SH, Cho MH, Cho H, Yoo KH, et al. Genetic Study in Korean Pediatric Patients with Steroid-Resistant Nephrotic Syndrome or Focal Segmental Glomerulosclerosis. Journal of Clinical Medicine. 2020; 9(6):2013. https://doi.org/10.3390/jcm9062013
Chicago/Turabian StylePark, Eujin, Chung Lee, Nayoung K. D. Kim, Yo Han Ahn, Young Seo Park, Joo Hoon Lee, Seong Heon Kim, Min Hyun Cho, Heeyeon Cho, Kee Hwan Yoo, and et al. 2020. "Genetic Study in Korean Pediatric Patients with Steroid-Resistant Nephrotic Syndrome or Focal Segmental Glomerulosclerosis" Journal of Clinical Medicine 9, no. 6: 2013. https://doi.org/10.3390/jcm9062013
APA StylePark, E., Lee, C., Kim, N. K. D., Ahn, Y. H., Park, Y. S., Lee, J. H., Kim, S. H., Cho, M. H., Cho, H., Yoo, K. H., Shin, J. I., Kang, H. G., Ha, I.-S., Park, W.-Y., & Cheong, H. I. (2020). Genetic Study in Korean Pediatric Patients with Steroid-Resistant Nephrotic Syndrome or Focal Segmental Glomerulosclerosis. Journal of Clinical Medicine, 9(6), 2013. https://doi.org/10.3390/jcm9062013