Gene Expression Profiling in Fibromyalgia Indicates an Autoimmune Origin of the Disease and Opens New Avenues for Targeted Therapy


Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. PBMCs Isolation
2.3. Microarray Analysis
2.4. Protein-Protein Interaction (PPI) Network Construction and Network Clustering
2.5. Gene Functional Classification and Enrichment Analysis
2.6. Real-Time RT-PCR
2.7. FACS Analysis
2.8. Detection of Soluble Mediators in Sera of FM Patients and Healthy Controls
2.9. Sjögren’s Autoantibodies Detection in FM patients’ Sera
2.10. Statistical Analysis
3. Results
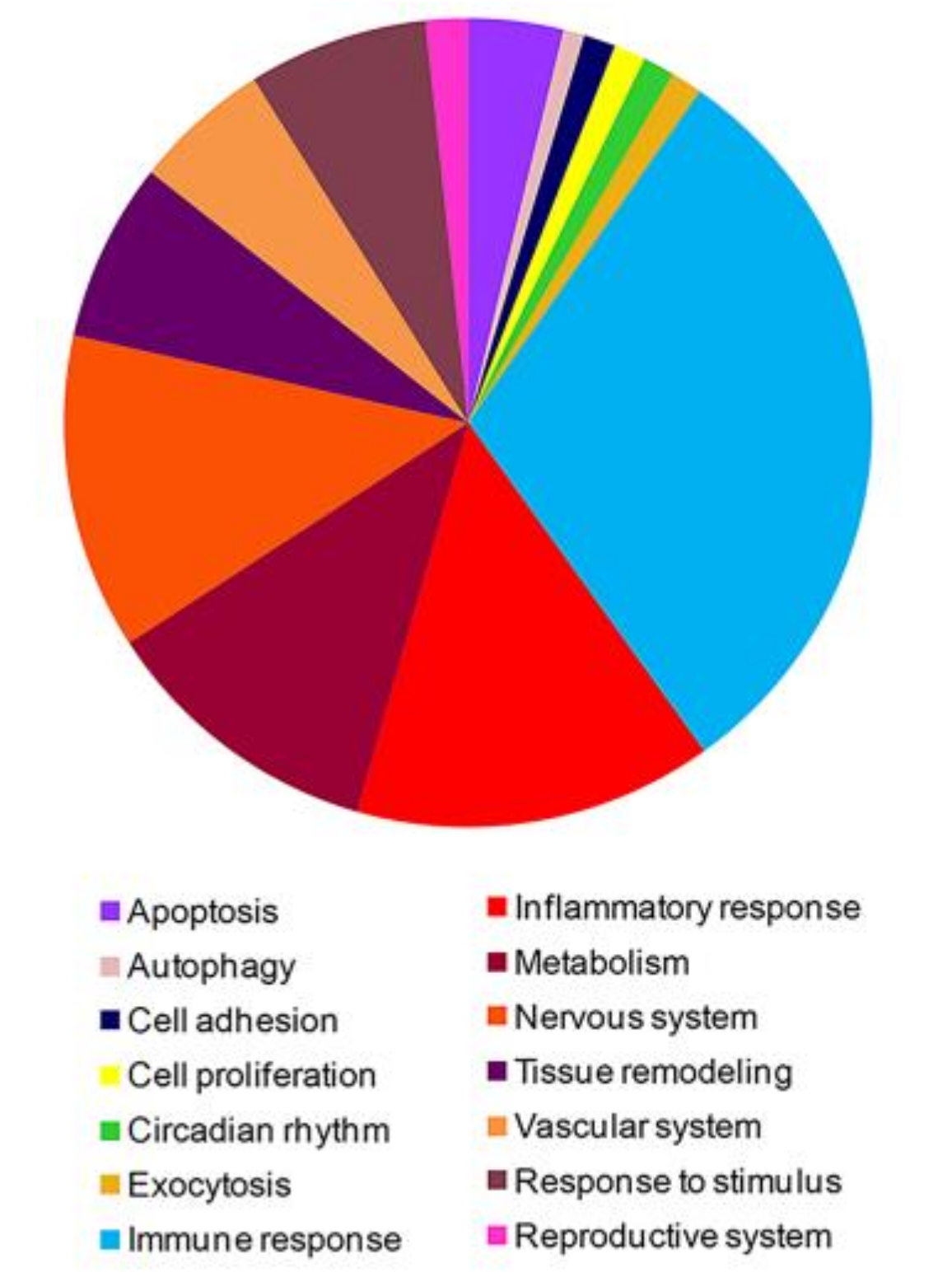
3.1. High-Throughput Gene Expression Profiling in Peripheral Blood Mononuclear Cells of FM Patients
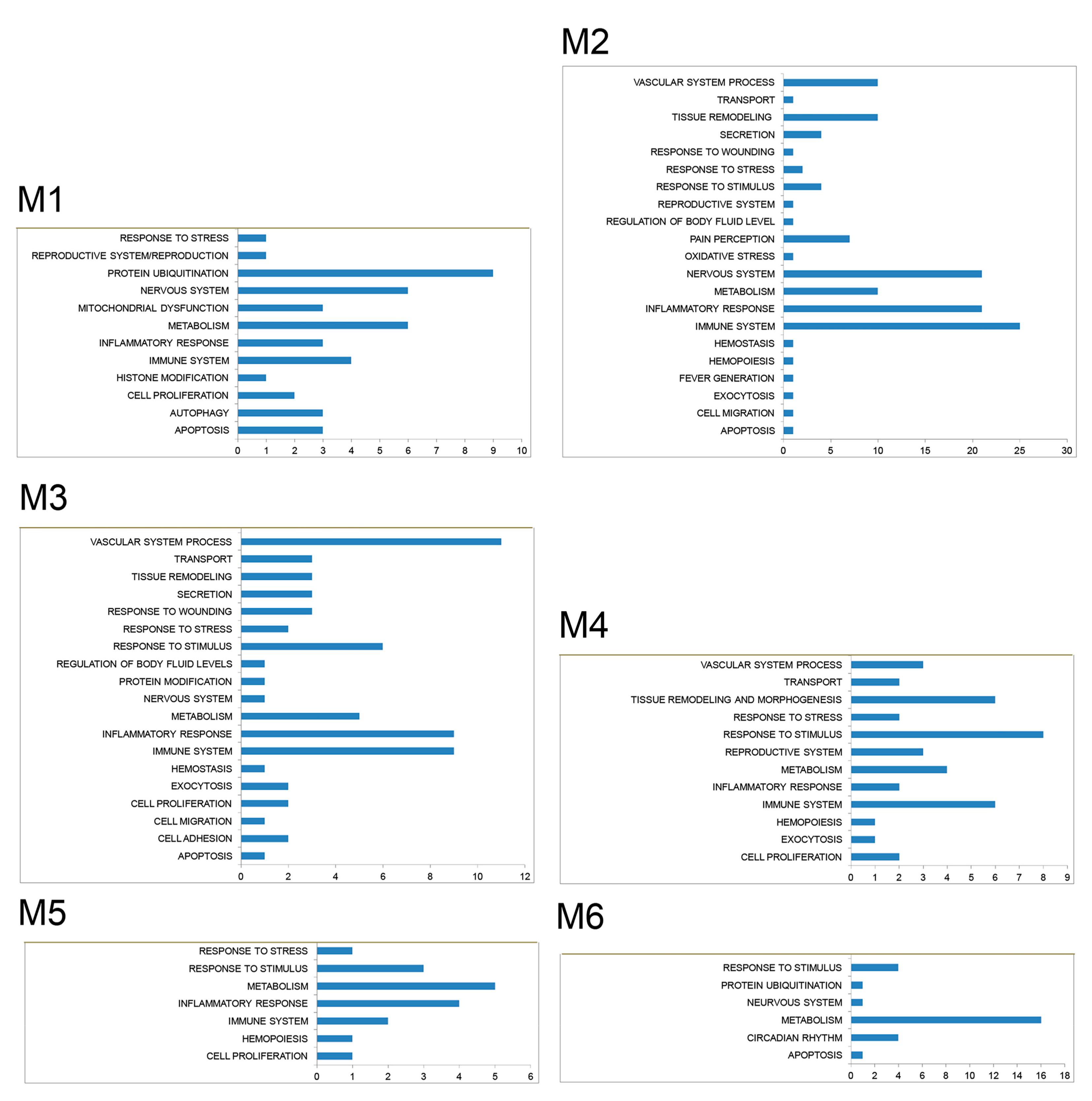
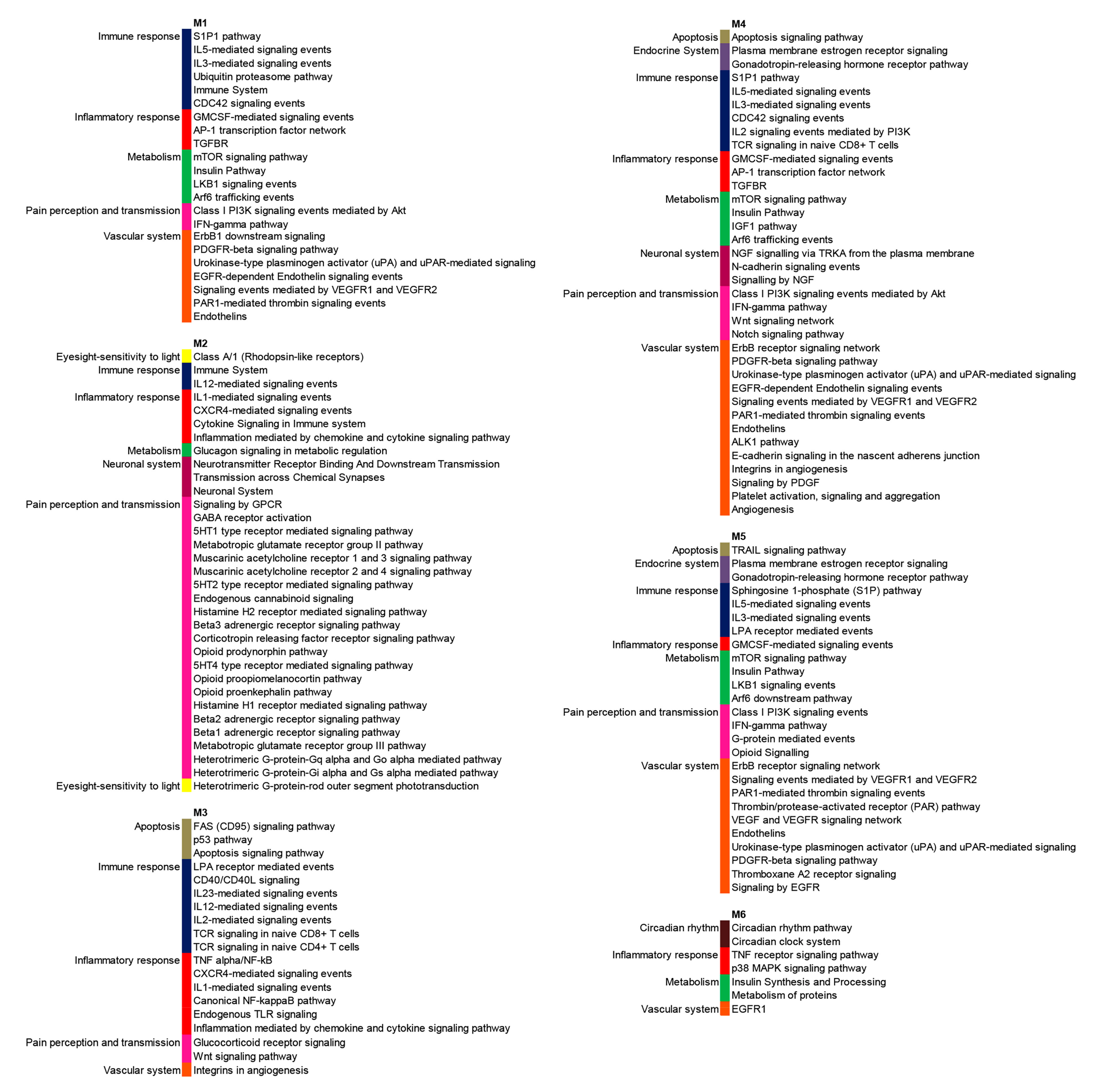
3.2. PPI Network and Modular Analysis of Genes Modulated in FM
3.3. High-Throughput Long Non-Coding RNA Expression Profiling in Peripheral Blood Mononuclear Cells of Patients with FM
3.4. Frequency of IL-17 Positive CD4+ T Cells in PBMCs from Patients with FM
3.5. Detection of Soluble Mediators in FM Sera
3.6. Autoantibodies Detection in FM Patients’ Sera
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Boerma, T.; Harrison, J.; Jakob, R.; Mathers, C.; Schmider, A.; Weber, S. Revising the ICD: Explaining the WHO approach. Lancet 2016, 388, 2476–2477. [Google Scholar] [CrossRef]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.; Goldenberg, D.L.; Katz, R.S.; Mease, P.; Russell, A.S.; Russell, I.J.; Winfield, J.B.; Yunus, M.B. The American College of Rheumatology Preliminary Diagnostic Criteria for Fibromyalgia and Measurement of Symptom Severity. Arthritis Care Res. (Hoboken). 2010, 62, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Sarzi-Puttini, P.; Atzeni, F.; Masala, I.F.; Sala, F.; Chapman, J.; Choy, E. Are the ACR 2010 diagnostic criteria for fibromyalgia better than the 1990 criteria? Autoimmun. Rev. 2018, 17, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Fitzcharles, M.A.; Ste-Marie, P.A.; Goldenberg, D.L.; Pereira, J.X.; Abbey, S.; Choinière, M. 2012 Canadian Guidelines for the Diagnosis and Management of Fibromyalgia Syndrome: Executive Summary. Pain Res. Manag. 2013, 18, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.M.; Bennett, R.M.; Croord, L.J.; Dean, L.E.; Clauw, D.J.; Goldenberg, D.L. AAPT Diagnostic Criteria for Fibromyalgia. J. Pain 2019, 20, 611–628. [Google Scholar] [CrossRef] [PubMed]
- Salaffi, F.; Di Carlo, M.; Farah, S.; Atzeni, F.; Buskila, D.; Ablin, J.N.; Häuser, W.; Sarzi-Puttini, P. Diagnosis of fibromyalgia: Comparison of the 2011/2016 ACR and AAPT criteria and validation of the modified Fibromyalgia Assessment Status. Rheumatology (Oxford) 2020, 24, keaa061. [Google Scholar] [CrossRef] [PubMed]
- Ryabkova, V.A.; Churilov, L.P.; Shoenfeld, Y. Neuroimmunology: What Role for Autoimmunity, Neuroinflammation, and Small Fiber Neuropathy in Fibromyalgia, Chronic Fatigue Syndrome, and Adverse Events after Human Papillomavirus Vaccination? Int. J. Mol. Sci. 2019, 20, 5164. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, D.S.; Forsberg, A.; Sandström, A.; Bergan, C.; Kadetoff, D.; Protsenko, E.; Lampa, J.; Lee, Y.C.; Höglund, C.O.; Catana, C.; et al. Brain glial activation in fibromyalgia—A multi-site positron emission tomography investigation. Brain Behav. Immun. 2019, 75, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Littlejohn, G.; Guymer, E. Neurogenic inflammation in fibromyalgia. Semin Immunopathol. 2018, 40, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, A.; Tiosano, S.; Amital, H. The complexities of fibromyalgia and its comorbidities. Curr. Opin. Rheumatol. 2018, 30, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Fitzcharles, M.A.; Perrot, S.; Häuser, W. Comorbid fibromyalgia: A qualitative review of prevalence and importance. Eur. J. Pain. 2018, 22, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Bazzichi, L.; Giacomelli, C.; Consensi, A.; Giorgi, V.; Batticciotto, A.; Di Franco, M.; Sarzi-Puttini, P. One year in review 2020: Fibromyalgia. Clin. Exp. Rheumatol. 2020, 38, S3–S8. [Google Scholar]
- Bair, M.J.; Krebs, E.E. Fibromyalgia. Ann. Intern. Med. 2020, 172, ITC33–ITC48. [Google Scholar] [CrossRef] [PubMed]
- Clauw, D.J. Fibromyalgia and related conditions. Mayo Clin. Proc. 2015, 90, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.S.; Correa, H.; Silva, G.C.; Campos, F.S.; Baião, F.R.; Ribeiro, L.S.; Faria, A.M.; d’Avila Reis, D. May genetic factors in fibromyalgia help to identify patients with differentially altered frequencies of immune cells? Clin. Exp. Immunol. 2008, 154, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Banfi, G.; Diani, M.; Pigatto, P.D.; Reali, E.T. Cell Subpopulations in the Physiopathology of Fibromyalgia: Evidence and Perspectives. Int. J. Mol. Sci. 2020, 21, 1186. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Zhang, T.J.; Meng, L.B.; Cheng, X.T.; Hua, Z. Bioinformatics analysis of gene and microRNA targets for fibromyalgia. Clin. Exp. Rheumatol. 2020. Epub ahead of print. [Google Scholar]
- Dolcino, M.; Tinazzi, E.; Puccetti, A.; Lunardi, C. In Systemic Sclerosis, a Unique Long Non Coding RNA Regulates Genes and Pathways Involved in the Three Main Features of the Disease (Vasculopathy, Fibrosis and Autoimmunity) and in Carcinogenesis. J. Clin. Med. 2019, 8, E320. [Google Scholar] [CrossRef] [PubMed]
- Dolcino, M.; Pelosi, A.; Fiore, P.F.; Patuzzo, G.; Tinazzi, E.; Lunardi, C.; Puccetti, A. Long Non-Coding RNAs Play a Role in the Pathogenesis of Psoriatic Arthritis by Regulating MicroRNAs and Genes Involved in Inflammation and Metabolic Syndrome. Front. Immunol. 2018, 9, 1533. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, A.; Pelosi, A.; Fiore, P.F.; Patuzzo, G.; Lunardi, C.; Dolcino, M. MicroRNA Expression Profiling in Behçet’s Disease. J. Immunol. Res. 2018, 2018, 2405150. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Chen, X.; Xue, H.; Tang, Y.; Zhang, P.; Kang, Q.; Hao, Y.; Chen, R.; Zhao, Y.; He, S. NPInter v4.0: An integrated database of ncRNA interactions. Nucleic Acids Res. 2020, 48, D160–D165. [Google Scholar] [CrossRef] [PubMed]
- Pathan, M.; Keerthikumar, S.; Ang, C.S.; Gangoda, L.; Quek, C.Y.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. Funrich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. String 8-a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of biological networks and gene expression data using cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Dolcino, M.; Tinazzi, E.; Puccetti, A.; Lunardi, C. Long Non-Coding RNAs Target Pathogenetically Relevant Genes and Pathways in Rheumatoid Arthritis. Cells 2019, 8, E816. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, A.; Fiore, P.F.; Pelosi, A.; Tinazzi, E.; Patuzzo, G.; Argentino, G.; Moretta, F.; Lunardi, C.; Dolcino, M. Gene Expression Profiling in Behcet’s Disease Indicates an Autoimmune Component in the Pathogenesis of the Disease and Opens New Avenues for Targeted Therapy. J. Immunol. Res. 2018, 2018, 4246965. [Google Scholar] [CrossRef] [PubMed]
- Brkic, Z.; Corneth, O.B.; van Helden-Meeuwsen, C.G.; Dolhain, R.J.; Maria, N.I.; Paulissen, S.M.; Davelaar, N.; van Hamburg, J.P.; van Daele, P.L.; Dalm, V.A.; et al. T helper 17 cell cytokines and interferon type I: Partners in crime in systemic lupus erythematosus? Arthritis. Res. Ther. 2014, 16, R62. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, A.; Espinosa, A.; Wahren-Herlenius, M. IL-17: A new actor in IFN-driven systemic autoimmune diseases. Eur. J. Immunol. 2012, 42, 2274–2284. [Google Scholar] [CrossRef] [PubMed]
- Axtell, R.C.; de Jong, B.A.; Boniface, K.; van der Voort, L.F.; Bhat, R.; De Sarno, P.; Naves, R.; Han, M.; Zhong, F.; Castellanos, J.G.; et al. T helper type 1 and 17 cells determine efficacy of interferon-β in multiple sclerosis and experimental encephalomyelitis. Nat. Med. 2010, 16, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Dolcino, M.; Ottria, A.; Barbieri, A.; Patuzzo, G.; Tinazzi, E.; Argentino, G.; Beri, R.; Lunardi, C.; Puccetti, A. Gene Expression Profiling in Peripheral Blood Cells and Synovial Membranes of Patients with Psoriatic Arthritis. PLoS ONE 2015, 10, e0128262. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | p-Value | Gene Symbol | Description |
|---|---|---|---|
| Th-17 Related Genes | |||
| TC0100010686.hg.1 | 0.005 | CACYBP | calcyclin binding protein |
| TC0200010980.hg.1 | <0.001 | CCL20 | chemokine (C-C motif) ligand 20 |
| TC1700007558.hg.1 | 0.005 | CCL7 | chemokine (C-C motif) ligand 7 |
| TC0 × 00008540.hg.1 | 0.004 | CD40LG | CD40 ligand |
| TC2000007682.hg.1 | 0.001 | CEBPB | CCAAT/enhancer binding protein (C/EBP). beta |
| TC1200011177.hg.1 | 0.008 | IFNG | interferon. Gamma |
| TC0100017107.hg.1 | 0.001 | IL10 | interleukin 10 |
| TC0200013916.hg.1 | 0.001 | IL1B | interleukin 1 beta |
| TC0700006890.hg.1 | <0.001 | IL6 | interleukin 6 |
| TC2000007514.hg.1 | 0.002 | MMP9 | matrix metallopeptidase 9 |
| TC0400008271.hg.1 | 0.002 | NFKB1 | nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 |
| TC0100009241.hg.1 | 0.005 | S1PR1 | sphingosine-1-phosphate receptor 1 |
| TC1600009395.hg.1 | 0.006 | SOCS1 | suppressor of cytokine signaling 1 |
| TC0600007596.hg.1 | <0.001 | TNF | tumor necrosis factor |
| Type I Interferon Related Genes | |||
| TC0600007495.hg.1 | 0.003 | HLA-A | major histocompatibility complex. class I. A |
| TC0200014772.hg.1 | 0.008 | IFIH1 | interferon induced. with helicase C domain 1 |
| TC0100017107.hg.1 | 0.001 | IL10 | interleukin 10 |
| TC0700006890.hg.1 | <0.001 | IL6 | interleukin 6 |
| TC0400012606.hg.1 | 0.002 | IRF2 | interferon regulatory factor 2 |
| TC1400010584.hg.1 | 0.005 | IRF9 | interferon regulatory factor 9 |
| TC0700008875.hg.1 | <0.001 | MET | MET proto-oncogene. receptor tyrosine kinase |
| TC1500007833.hg.1 | <0.001 | PML | promyelocytic leukemia |
| TC0900012231.hg.1 | 0.0004 | SHB | Src homology 2 domain containing adaptor protein B |
| TC1600009395.hg.1 | 0.006 | SOCS1 | suppressor of cytokine signaling 1 |
| TC0 × 00007149.hg.1 | 0.006 | TIMP1 | TIMP metallopeptidase inhibitor 1 |
| TC0 × 00006625.hg.1 | <0.001 | TLR7 | toll-like receptor 7 |
| TC1400008362.hg.1 | 0.008 | TRAF3 | TNF receptor-associated factor 3 |
| TC1900009627.hg.1 | 0.005 | TYK2 | tyrosine kinase 2 |
| TC0600008109.hg.1 | 0.003 | VEGFA | vascular endothelial growth factor A |
| LncRNAs | FC | Targeted miRNAs | Targeted FM-DEGs | Targeted Module-Associated FM-DEGs | Targeted Modules |
|---|---|---|---|---|---|
| CTD-2651B20.6 | 5.23 | 2 | 12 | 4 | M3 M5 M6 |
| RP1-151F17.1 | 4.65 | 15 | 75 | 16 | M2 M3 M4 M5 M6 |
| AC009299.3 | −4.52 | 29 | 183 | 38 | M1 M2 M3 M4 M5 M6 |
| RP11-283I3.6 | −5.05 | 1 | none | none | none |
| RP11-747H7.3 | −9 | 8 | 57 | 12 | M1 M2 M3 M4 M5 M6 |
| Module Associated Gene | Module | Diseases (GeneCards) |
|---|---|---|
| SOCS1 | 1 | coronary heart disease 1, myeloproliferative neoplasm, b-cell lymphoma, placental insufficiency, multiple sclerosis |
| ASB6 | 1 | None |
| LMO7 | 1 | cardiomyopathy, dilated, 1dd |
| UBE2H | 1 | pontocerebellar hypoplasia, Type 7 |
| TNFSF11 | 2 | bone disease, depersonalization disorder, psoriatic arthritis |
| TNFRSF1B | 2 | juvenile rheumatoid arthritis, psoriatic arthritis, Guillain-Barre syndrome |
| CCL3 | 2 | aseptic meningitis, bone disease, rheumatoid arthritis, arthritis, demyelinating disease, immune deficiency disease |
| GNG5 | 2 | echolalia |
| S1PR1 | 2 | autoimmune disease of central nervous system, multiple sclerosis |
| IL10 | 3 | rheumatoid arthritis, arthritis, ulcerative colitis, rheumatic heart disease, chronic fatigue syndrome |
| THBS1 | 3 | thrombotic thrombocytopenic purpura, thrombocytopenia |
| TRAF3 | 3 | encephalitis, lymphoma, Hodgkin, classic, myeloma, multiple |
| KIF2A | 3 | cortical dysplasia, complex, with other brain malformations 3, Walker-Warburg syndrome |
| CCL7 | 3 | nephrogenic systemic fibrosis, demyelinating disease, inflammatory bowel disease, multiple sclerosis |
| RCC2 | 3 | spermatogenic failure 5, renal cell carcinoma, nonpapillary |
| BORA | 3 | branchiootic syndrome |
| CEP97 | 3 | spinocerebellar ataxia 11, nephronophthisis, bardet-biedl syndrome |
| NHLRC2 | 3 | fibrosis, neurodegeneration, and cerebral angiomatosis |
| HSPA5 | 4 | rheumatoid arthritis, wolfram syndrome 1 |
| CDKN1A | 4 | Tumors |
| BCL2L11 | 4 | autoimmune lymphoproliferative syndrome, leukemia |
| PPP2R2A | 4 | syndromic intellectual disability |
| GRIA1 | 4 | depression |
| ZEB1 | 4 | congenital hypothyroidism, corneal endothelial dystrophy |
| BET1 | 4 | leukodystrophy, hypomyelinating |
| CWC22 | 4 | bruxism |
| CLP1 | 4 | pontocerebellar hypoplasia |
| SEC23IP | 4 | Warrensburg’s syndrome |
| CSTF1 | 4 | None |
| PPIL1 | 4 | None |
| TFRC | 5 | immunodeficiency 46, iron deficiency anemia |
| EXOSC10 | 5 | systemic scleroderma, collagen disease |
| FAS | 5 | lymphoproliferative syndrome, Sjogren syndrome |
| CCNH | 5 | capillary malformation-arteriovenous malformation 1, weber syndrome, xeroderma pigmentosum |
| WASL | 5 | Wiskott–Aldrich syndrome, x-linked recessive disease |
| CTSB | 5 | acute pancreatitis |
| GNA13 | 5 | Burkitt lymphoma, retinitis pigmentosa 14 |
| SURF4 | 5 | spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 2, macular degeneration, age-related |
| HBEGF | 5 | perinatal necrotizing enterocolitis, interstitial cystitis, bladder disease, urinary tract obstruction |
| MEIS1 | 5 | myeloid leukemia, restless legs syndrome |
| SDC2 | 5 | autism, rheumatic disease, fibrodysplasia ossificans progressiva |
| RPS3 | 6 | Warrensburg syndrome |
| CEBPB | 6 | alk-positive anaplastic large cell lymphoma, myxoid liposarcoma, retinoblastoma, acute promyelocytic leukemia, |
| DDIT3 | 6 | liposarcoma, fatty liver disease, lipomatosis, multiple |
| CTPS1 | 6 | nemaline myopathy, immunodeficiency, sleeping sickness |
| CRY2 | 6 | major depressive disorder, advanced sleep phase syndrome, nephronophthisis, delayed sleep phase disorder |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolcino, M.; Tinazzi, E.; Puccetti, A.; Lunardi, C. Gene Expression Profiling in Fibromyalgia Indicates an Autoimmune Origin of the Disease and Opens New Avenues for Targeted Therapy. J. Clin. Med. 2020, 9, 1814. https://doi.org/10.3390/jcm9061814
Dolcino M, Tinazzi E, Puccetti A, Lunardi C. Gene Expression Profiling in Fibromyalgia Indicates an Autoimmune Origin of the Disease and Opens New Avenues for Targeted Therapy. Journal of Clinical Medicine. 2020; 9(6):1814. https://doi.org/10.3390/jcm9061814
Chicago/Turabian StyleDolcino, Marzia, Elisa Tinazzi, Antonio Puccetti, and Claudio Lunardi. 2020. "Gene Expression Profiling in Fibromyalgia Indicates an Autoimmune Origin of the Disease and Opens New Avenues for Targeted Therapy" Journal of Clinical Medicine 9, no. 6: 1814. https://doi.org/10.3390/jcm9061814
APA StyleDolcino, M., Tinazzi, E., Puccetti, A., & Lunardi, C. (2020). Gene Expression Profiling in Fibromyalgia Indicates an Autoimmune Origin of the Disease and Opens New Avenues for Targeted Therapy. Journal of Clinical Medicine, 9(6), 1814. https://doi.org/10.3390/jcm9061814