Anti-VEGF Drugs in the Treatment of Multiple Myeloma Patients


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Angiogenesis in MM Progression
2.1. The Bone Marrow Microenvironment
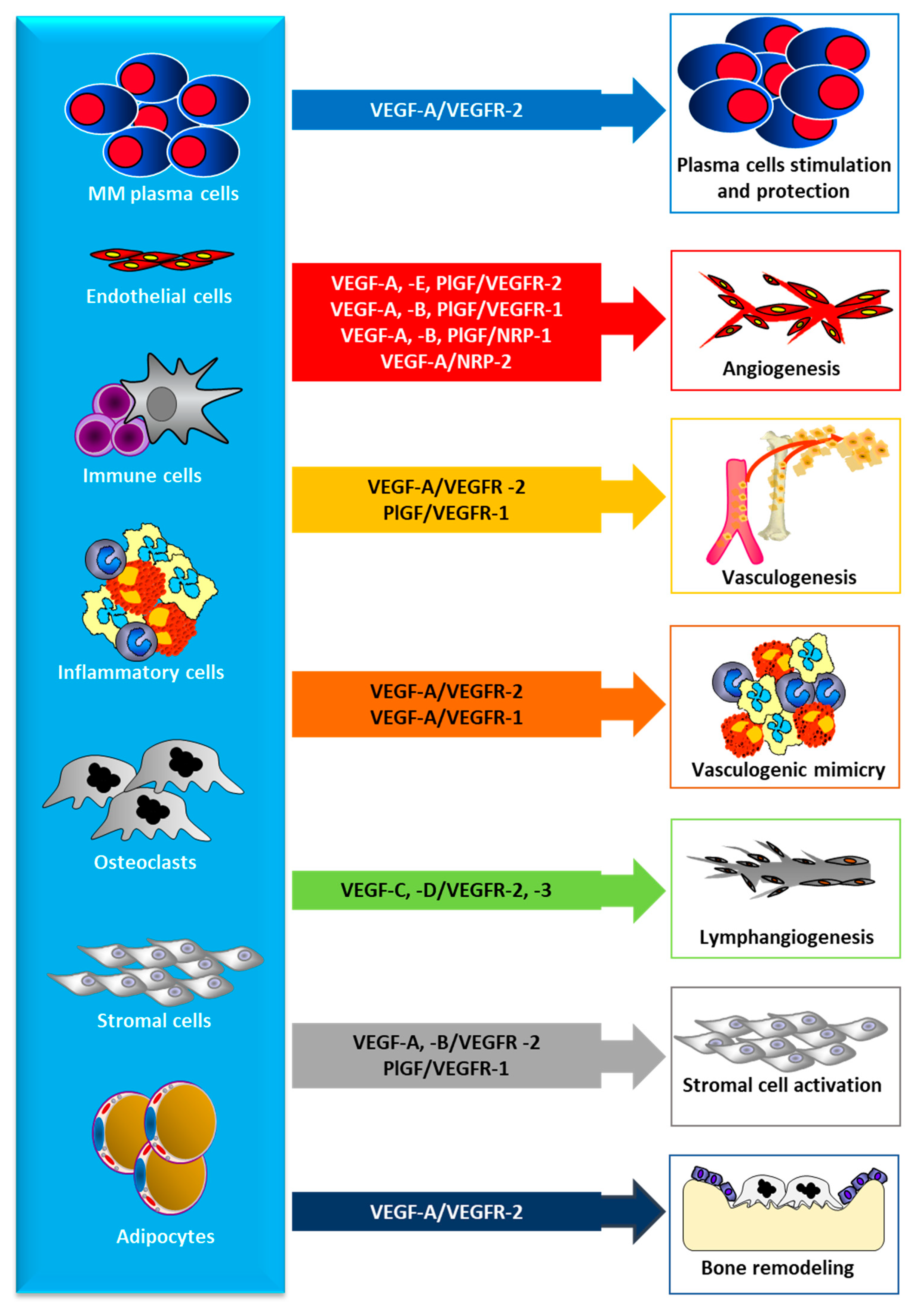
2.2. Angiogenesis in MM
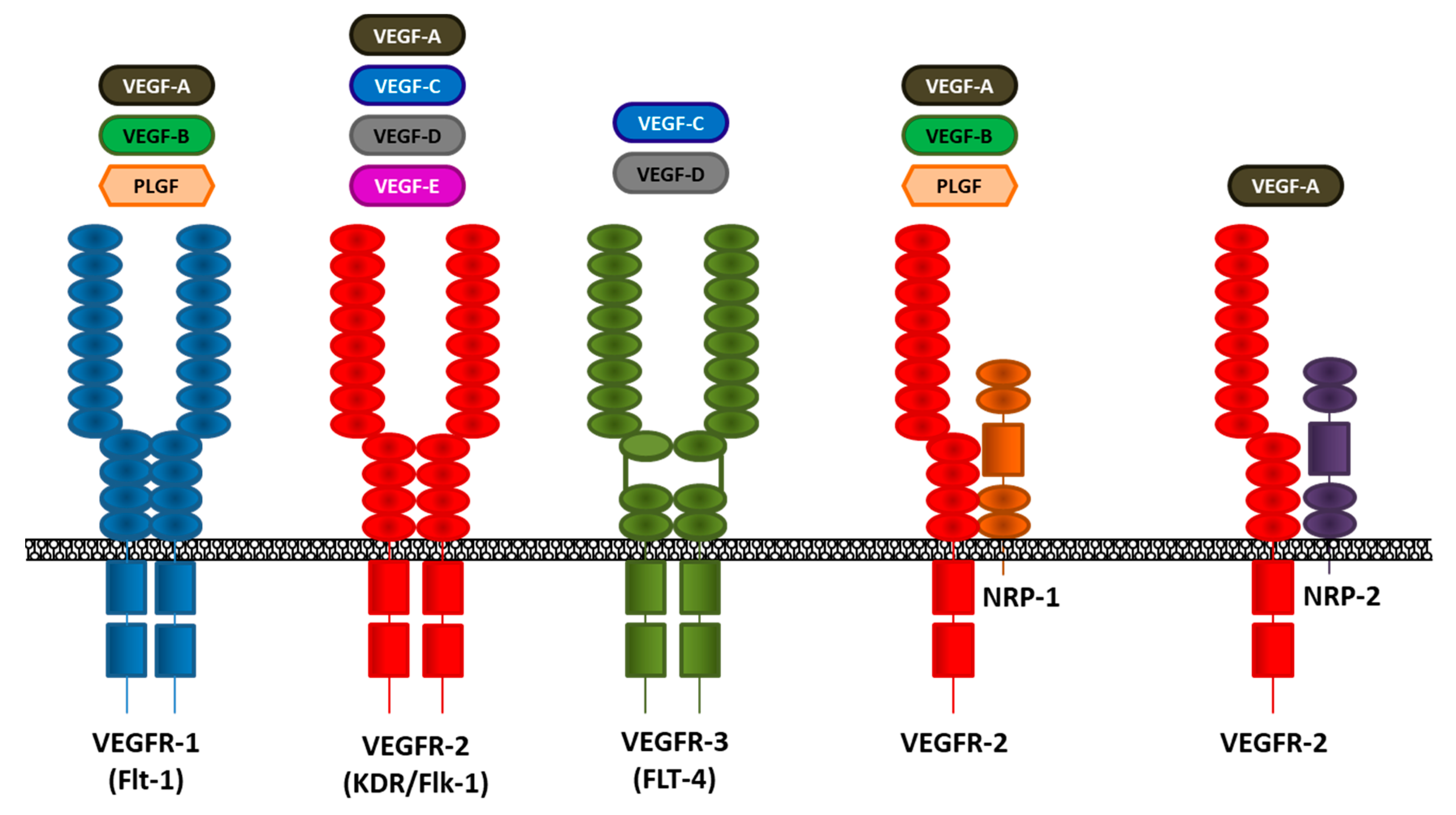
3. The VEGF/VEGFR Pathway
4. VEGF/VEGFR Inhibition in MM
4.1. Preclinical Evidence on the Anti-Angiogenic Effect of Currently Used Therapy
4.1.1. Proteasome Inhibitors
4.1.2. Immunomodulators (IMIDs)
4.1.3. Bisphosphonates
4.2. Preclinical Evidence for Novel Anti-Angiogenic Inhibitors
4.2.1. Monoclonal Antibodies
4.2.2. Other Molecules
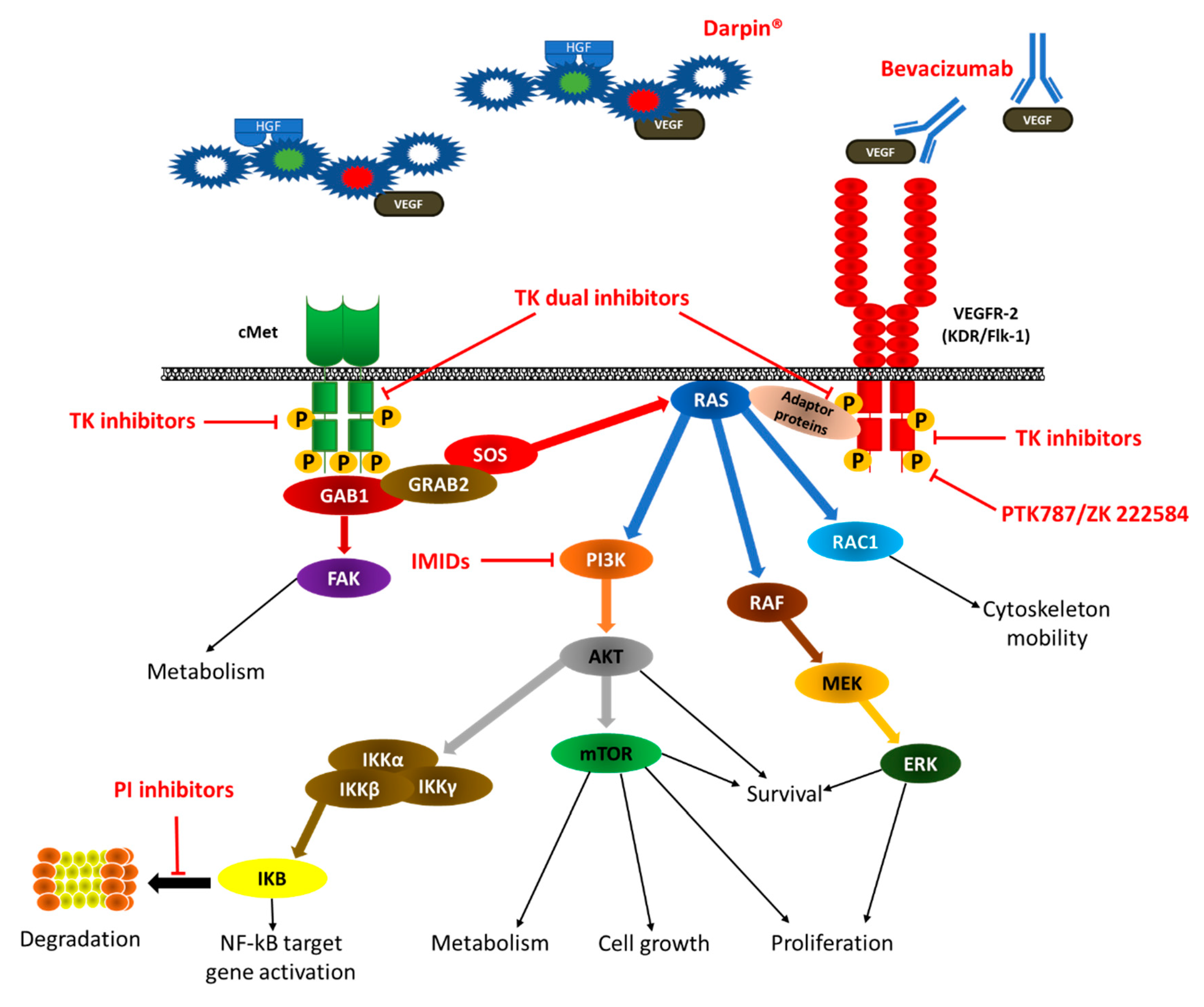
4.2.3. Dual Inhibition of VEGF/cMET
4.3. Clinical Experiences
4.3.1. Proteasome Inhibitors
4.3.2. Immunomodulators (IMIDs)
4.3.3. Anti-VEGF Monoclonal Antibodies
4.3.4. Dual Inhibition of VEGF/cMET
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 17046. [Google Scholar] [CrossRef]
- Rajkumar, S.; Kumar, S. Multiple myeloma: Diagnosis and treatment. Mayo Clin. Proc. 2016, 91, 101–119. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: Every year a new standard? Hematol. Oncol. 2019, 37 (Suppl. 1), 62–65. [Google Scholar] [CrossRef] [PubMed]
- Eslick, R.; Talaulikar, D. Multiple myeloma: From diagnosis to treatment. Aust. Fam. Phys. 2013, 42, 684–688. [Google Scholar] [PubMed]
- Ria, R.; Reale, A.; Mangialardi, G.; Dammacco, F.; Ribatti, D.; Vacca, A. The Bone Marrow Microenvironment in Multiple Myeloma: Cellular and Molecular Basis of Disease Progression. In Cell Respiration and Cell Survival. Process, Type and Effects; Osterhoudt, G., Barhydt, J., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2011; pp. 93–124. [Google Scholar]
- Langley, R.R.; Fidler, I.J. Tumor Cell-Organ Microenvironment Interactions in the Pathogenesis of Cancer Metastasis. Endocr. Rev. 2007, 28, 297–321. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Vacca, A. Multiple myeloma as a model for the role of bone marrow niches in the control of angiogenesis. Int. Rev. Cell. Mol. Biol. 2015, 314, 259–282. [Google Scholar]
- Glavey, S.V.; Manier, S.; Sacco, A.; Salem, K.; Kawano, Y.; Bouyssou, J.; Ghobrial, I.M.; Roccaro, A.M. Epigenetics in Multiple Myeloma. Cancer Treat. Res. 2016, 169, 35–49. [Google Scholar]
- Ria, R.; Solimando, A.; Melaccio, A.; Sportelli, A.; Vacca, A. Angiogenesis and Antiangiogenesis in Multiple Myeloma. In Update on Multiple Myeloma; Ahmed Al-Anazi, K., Ed.; IntechOpen Pub.: London, UK, 2018; pp. 97–123. [Google Scholar]
- Patel, U.; Luthra, R.; Medeiros, L.J.; Patel, K.P. Diagnostic, Prognostic, and Predictive Utility of Recurrent Somatic Mutations in Myeloid Neoplasms. Clin. Lymphoma Myeloma Leuk. 2017, 17S, S62–S74. [Google Scholar] [CrossRef]
- Alzrigat, M.; Párraga, A.A.; Jernberg-Wiklund, H. Epigenetics in multiple myeloma: From mechanisms to therapy. Semin. Cancer. Biol. 2017, 51, 101–115. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ria, R.; Vacca, A. Bone Marrow Stromal Cells-Induced Drug Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2020, 21, 613. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. BM niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ribatti, D.; Roncali, L.; Ranieri, G.; Serio, G.; Silvestris, F.; Dammacco, F. Bone marrow angiogenesis and progression in multiple myeloma. Br. J. Haematol. 1994, 87, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Scavelli, C.; Nico, B.; Cirulli, T.; Ria, R.; Di Pietro, G.; Mangieri, D.; Bacigalupo, A.; Mangialardi, G.; Coluccia, A.M.; Caravita, T.; et al. Vasculogenic mimicry by bone marrow macrophages in patients with multiple myeloma. Oncogene 2008, 27, 663–674. [Google Scholar] [CrossRef]
- Ria, R.; Piccoli, C.; Cirulli, T.; Falzetti, F.; Mangialardi, G.; Guidolin, D.; Tabilio, A.; Di Renzo, N.; Guarini, A.; Ribatti, D.; et al. Endothelial differentiation of hematopoietic stem and progenitor cells from patients with multiple myeloma. Clin. Cancer Res. 2008, 14, 1678–1685. [Google Scholar] [CrossRef]
- Kastrinakis, N.G.; Gorgoulis, V.G.; Foukas, P.G.; Dimopoulos, M.A.; Kittas, C. Molecular aspects of multiple myeloma. Ann. Oncol. 2000, 11, 1217–1228. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Chesi, M.V. Molecular classification and risk stratification of myeloma. Hematol. Oncol. 2013, 31 (Suppl. 1), 38–41. [Google Scholar] [CrossRef]
- Mondello, P.; Cuzzocrea, S.; Navarra, M.; Mian, M. Bone marrow micro-environment is a crucial player for myelomagenesis and disease progression. Oncotarget 2017, 8, 20394–20409. [Google Scholar] [CrossRef]
- Bhutani, M.; Landgren, O.; Usmani, S.Z. Multiple myeloma: Is it time for biomarker-driven therapy? Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e493–e503. [Google Scholar] [CrossRef]
- Moschetta, M.; Kawano, Y.; Podar, K. Targeting the Bone Marrow Microenvironment. Cancer Treat. Res. 2016, 169, 63–102. [Google Scholar]
- Robak, P.; Drozdz, I.; Szemraj, J.; Robak, T. Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Ponzoni, M.; Ria, R.; Grioni, M.; Cattaneo, E.; Villa, I.; Sabrina Bertilaccio, M.T.; Chesi, M.; Rubinacci, A.; Tonon, G.; et al. Modifications of the mouse bone marrow microenvironment favour angiogenesis and correlate with disease progression from asymptomatic to symptomatic multiple myeloma. Oncoimmunology 2015, 4, e1008850. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ria, R.; Semeraro, F.; Merchionne, F.; Coluccia, M.; Boccarelli, A.; Scavelli, C.; Nico, B.; Gernone, A.; Battelli, F.; et al. Endothelial cells in the bone marrow of patients with multiple myeloma. Blood 2003, 102, 3340–3348. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Todoerti, K.; Berardi, S.; Coluccia, A.M.; De Luisi, A.; Mattioli, M.; Ronchetti, D.; Morabito, F.; Guarini, A.; Petrucci, M.T.; et al. Gene expression profiling of bone marrow endothelial cells in patients with multiple myeloma. Clin. Cancer Res. 2009, 15, 5369–5378. [Google Scholar] [CrossRef] [PubMed]
- Risau, W.; Flamme, I. Vasculogenesis. Annu. Rev. Cell Dev. Biol. 1995, 11, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Postnatal vasculogenesis. Mech. Dev. 2001, 100, 157–163. [Google Scholar] [CrossRef]
- Reale, A.; Melaccio, A.; Lamanuzzi, A.; Saltarella, I.; Dammacco, F.; Vacca, A.; Ria, R. Functional and Biological Role of Endothelial Precursor Cells in Tumour Progression: A New Potential Therapeutic Target in Haematological Malignancies. Stem Cells Int. 2016, 2016, 7954580. [Google Scholar] [CrossRef]
- Moschetta, M.; Mishima, Y.; Sahin, I.; Manier, S.; Glavey, S.; Vacca, A.; Roccaro, A.M.; Ghobrial, I.M. Role of endothelial progenitor cells in cancer progression. Biochim. Biophys. Acta 2014, 1846, 26–39. [Google Scholar] [CrossRef]
- Berardi, S.; Caivano, A.; Ria, R.; Nico, B.; Savino, R.; Terracciano, R.; De Tullio, G.; Ferrucci, A.; De Luisi, A.; Moschetta, M.; et al. Four proteins governing overangiogenic endothelial cell phenotype in patients with multiple myeloma are plausible therapeutic targets. Oncogene 2012, 31, 2258–2269. [Google Scholar] [CrossRef]
- Vacca, A.; Ria, R.; Reale, A.; Ribatti, D. Angiogenesis in multiple myeloma. Chem. Immunol. Allergy 2014, 99, 180–196. [Google Scholar]
- Ria, R.; Catacchio, I.; Berardi, S.; De Luisi, A.; Caivano, A.; Piccoli, C.; Ruggieri, V.; Frassanito, M.A.; Ribatti, D.; Nico, B.; et al. HIF-1α of bone marrow endothelial cells implies relapse and drug resistance in patients with multiple myeloma and may act as a therapeutic target. Clin. Cancer Res. 2014, 20, 847–858. [Google Scholar] [CrossRef]
- Döme, B.; Hendrix, M.J.; Paku, S.; Tóvári, J.; Tímár, J. Alternative vascularisation mechanisms in cancer: Pathology and therapeutic implications. Am. J. Pathol. 2007, 170, 1–15. [Google Scholar] [CrossRef]
- Pinto, M.P.; Sotomayor, P.; Carrasco-Avino, G.; Corvalan, A.H.; Owen, G.I. Escaping Antiangiogenic Therapy: Strategies Employed by Cancer Cells. Int. J. Mol. Sci. 2016, 17, 1489. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ribatti, D. Angiogenesis and vasculogenesis in multiple myeloma: Role of inflammatory cells. Recent Res. Cancer Res. 2011, 183, 187–195. [Google Scholar]
- Rehman, J.; Li, J.; Orschell, C.M.; March, K.L. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation 2003, 107, 1164–1169. [Google Scholar] [CrossRef]
- Nico, B.; Mangieri, D.; Crivellato, E.; Vacca, A.; Ribatti, D. Mast Cells contribute to vasculogenic mimicry in multiple myeloma. Stem Cells Dev. 2008, 17, 19–22. [Google Scholar] [CrossRef]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone marrow microenvironment in multiple myeloma progression. J. Biomed. Biotechnol. 2012, 2012, 157496. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.; Micke, P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin. Cancer Biol. 2014, 25, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A. Role of Endothelial Cells and Fibroblasts in Multiple Myeloma Angiogenic Switch. Cancer Treat. Res. 2016, 169, 51–61. [Google Scholar] [PubMed]
- Asosingh, K.; De Raeve, H.; Menu, E.; Van Riet, I.; Van Marck, E.; Van Camp, B.; Vanderkerken, K. Angiogenic switch during 5T2MM murine myeloma tumorigenesis: Role of CD45 heterogeneity. Blood 2004, 103, 3131–3177. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Rao, L.; Moschetta, M.; Ria, R.; Di Marzo, L.; De Luisi, A.; Racanelli, V.; Catacchio, I.; Berardi, S.; Basile, A.; et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: In vitro and in vivo studies. Leukemia 2014, 28, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Zavidij, O.; Haradhvala, N.J.; Mouhieddine, T.H.; Sklavenitis-Pistofidis, R.; Cai, S.; Reidy, M.; Rahmat, M.; Flaifel, A.; Ferland, B.; Su, N.K.; et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat. Cancer 2020, 1, 493–506. [Google Scholar] [CrossRef]
- Ribatti, D.; Vacca, A.; Nico, B.; Quondamatteo, F.; Ria, R.; Minischetti, M.; Marzullo, A.; Herken, R.; Roncali, L.; Dammacco, F. Bone marrow angiogenesis and mast cell density increase simultaneously with progression of human multiple myeloma. Br. J. Cancer 1999, 79, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A. The role of inflammatory cells in angiogenesis in multiple myeloma. Adv. Exp. Med. Biol. 2014, 816, 361–376. [Google Scholar]
- Jurczyszyn, A.; Czepiel, J.; Gdula-Argasińska, J.; Paśko, P.; Czapkiewicz, A.; Librowski, T.; Perucki, W.; Butrym, A.; Castillo, J.J.; Skotnicki, A.B. The Analysis of the Relationship between Multiple Myeloma Cells and Their Microenvironment. J. Cancer 2015, 6, 160–168. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Horenstein, A.L.; Quarona, V.; Toscani, D.; Costa, F.; Chillemi, A.; Pistoia, V.; Giuliani, N.; Malavasi, F. Adenosine Generated in the Bone Marrow Niche Through a CD38-Mediated Pathway Correlates with Progression of Human Myeloma. Mol. Med. 2016, 22, 694–704. [Google Scholar] [CrossRef]
- Costa, F.; Toscani, D.; Chillemi, A.; Bolzoni, M.; Marchica, V.; Vescovini, R.; Mancini, C.; Martella, E.; Campanini, N.; Schifano, C.; et al. Expression of CD38 in myeloma bone niche: A rational basis for the use of anti-CD38 immunotherapy to inhibit osteoclast formation. Oncotarget 2017, 8, 56598–56611. [Google Scholar] [CrossRef]
- Tamma, R.; Ribatti, D. Bone Niches, Hematopoietic Stem Cells, and Vessel Formation. Int. J. Mol. Sci. 2017, 18, 151. [Google Scholar] [CrossRef]
- Zhang, H.; Vakil, V.; Braunstein, M.; Smith, E.L.; Maroney, J.; Chen, L.; Dai, K.; Berenson, J.R.; Hussain, M.M.; Klueppelberg, U.; et al. Circulating endothelial progenitor cells in multiple myeloma: Implications and significance. Blood 2005, 105, 3286–3294. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Zhang, H.M.; Zhang, W.J.; Wang, H.X. Changes in circulating endothelial progenitor cells predict responses of multiple myeloma patients to treatment with bortezomib and dexamethasone. Braz. J. Med. Biol. Res. 2015, 48, 736–742. [Google Scholar] [CrossRef]
- Rigolin, G.M.; Fraulini, C.; Ciccone, M.; Mauro, E.; Bugli, A.M.; De Angeli, C.; Negrini, M.; Cuneo, A.; Castoldi, G. Neoplastic circulating endothelial cells in multiple myeloma 13q14 deletion. Blood 2006, 107, 2531–2535. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; De Veirman, K.; De Becker, A.; Vanderkerken, K.; Van Riet, I. Mesenchymal stem cells in multiple myeloma: A therapeutical tool or target? Leukemia 2018, 32, 155–1514. [Google Scholar] [CrossRef] [PubMed]
- Reagan, M.R.; Ghobrial, I.M. Multiple myeloma mesenchymal stem cells: Characterization, origin, and tumor-promoting effects. Clin. Cancer Res. 2012, 18, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Herrero, C.; Pérez-Simón, J.A. Immunomodulatory effect of mesenchymal stem cells. Braz. J. Med. Biol. Res. 2010, 43, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Amadori, S. The role of angiogenesis in hematologic Malignancies. J. Hematother. Stem Cell Res. 2002, 11, 49–68. [Google Scholar] [CrossRef]
- Vacca, A.; Ribatti, D.; Roccaro, A.M.; Frigeri, A.; Dammacco, F. Bone marrow angiogenesis in patients with active multiple myeloma. Semin. Oncol. 2001, 28, 543–550. [Google Scholar] [CrossRef]
- Yang, Z.; Yao, H.; Fei, F.; Li, Y.; Qu, J.; Li, C.; Zhang, S. Generation of erythroid cells from polyploid giant cancer cells: Re-thinking about tumor blood supply. J. Cancer Res. Clin. Oncol. 2018, 144, 617–627. [Google Scholar] [CrossRef]
- Medinger, M.; Passweg, J. Role of tumour angiogenesis in haematological malignancies. Swiss Med. Wkly 2014, 144, w14050. [Google Scholar] [CrossRef]
- Ria, R.; Roccaro, A.M.; Merchionne, F.; Vacca, A.; Dammacco, F.; Ribatti, D. Vascular endothelial growth factor and its receptors in multiple myeloma. Leukemia 2003, 17, 1961–1966. [Google Scholar] [CrossRef]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cell secrete a novel heparin-binding growth factor specific for vascular endothelial cell. Biochem. Biophys. Res. Commun. 2012, 161, 8511989. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Positive and negative modulation of angiogenesis by VEGFR1 ligands. Sci. Signal 2009, 2, re12009. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, F.; Han, W.; Shen, B.; Luo, J.; Shibuya, M.; He, Y. VEGFR-3 ligand-binding and kinase activity are required for lymphangiogenesis but not for angiogenesis. Cell Res. 2010, 20, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hooper, A.T.; Zhong, Z.; Witte, L.; Bohlen, P.; Rafii, S.; Hicklin, D.J. The vascular endothelial growth factor receptor (VEGFR-1) supports growth and survival of human breast carcinoma. Int. J. Cancer 2006, 119, 1519–1529. [Google Scholar] [CrossRef]
- Autiero, M.; Luttun, A.; Tjwa, M.; Carmeliet, P. Placental growth factor and its receptor, vascular endothelial growth factor receptor-1: Novel targets for stimulation of ischemic tissue revascularization and inhibition of angiogenic and inflammatory disorders. J. Thromb. Haemost. 2003, 1, 1356–1370. [Google Scholar] [CrossRef]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell Signal 2007, 19, 2003–2012. [Google Scholar] [CrossRef]
- Brekken, R.A.; Overholser, J.P.; Stastny, V.A.; Waltenberger, J.; Minna, J.D.; Thorpe, P.E. Selective inhibition of vascular endothelial growth factor (VEGF) receptor 2 (KDR/Flk-1) activity by a monoclonal anti-VEGF antibody blocks tumor growth in mice. Cancer Res. 2000, 60, 5117–5124. [Google Scholar]
- Katoh, O.; Tauchi, H.; Kawaishi, K.; Kimura, A.; Satow, Y. Expression of the vascular endothelial growth factor (VEGF) receptor gene, KDR, in hematopoietic cells and inhibitory effect of VEGF on apoptotic cell death caused by ionizing radiation. Cancer Res. 1995, 55, 5687–5692. [Google Scholar]
- Cursiefen, C.; Chen, L.; Borges, L.P.; Jackson, D.; Cao, J.; Radziejewski, C.; D’Amore, P.A.; Dana, M.R.; Wiegand, S.J.; Streilein, J.W. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J. Clin. Invest. 2004, 113, 1040–1050. [Google Scholar] [CrossRef]
- Vacca, A.; Ria, R.; Ribatti, D.; Semeraro, F.; Djonov, V.; Di Raimondo, F.; Dammacco, F. A Paracrine Loop in The Vascular Endothelial Growth Factor Pathway Triggers Tumor Angiogenesis And Growth in Multiple Myeloma. Haematologica 2003, 88, 176–185. [Google Scholar]
- Ria, R.; Vacca, A.; Russo, F.; Cirulli, T.; Massaia, M.; Tosi, P.; Cavo, M.; Guidolin, D.; Ribatti, D.; Dammacco, F. A VEGF-dependent autocrine loop mediates proliferation and capillarogenesis in bone marrow endothelial cells of patients with multiple myeloma. Thromb. Haemost. 2004, 92, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Carder, P.J.; Downey, S.; Forbes, M.A.; MacLennan, K.; Allgar, V.; Kaufman, S.; Hallam, S.; Bicknell, R.; Walker, J.J.; et al. Vascular endothelial growth factor (VEGF) in breast cancer: Comparison of plasma, serum and tissue VEGF and microvessel density and effects of tamoxifen. Cancer Res. 2000, 60, 2898–2905. [Google Scholar] [PubMed]
- Kay, N.E.; Bone, N.D.; Tschumper, R.C.; Howell, K.H.; Geyer, S.M.; Dewald, G.W.; Hanson, C.A.; Jelinek, D.F. B-CLL cells are capable of synthesis and secretion of both pro- and anti-angiogenic molecules. Leukemia 2002, 16, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Lyden, D.; Benezra, R.; Hattori, K.; Heissig, B. Vascular and haematopoietic stem cells: Novel targets for anti-angiogenesis therapy? Nat. Rev. Cancer. 2002, 2, 826–835. [Google Scholar] [CrossRef]
- Moschetta, M.; Basile, A.; Ferrucci, A.; Frassanito, M.A.; Rao, L.; Ria, R.; Solimando, A.G.; Giuliani, N.; Boccarelli, A.; Fumarola, F.; et al. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin. Cancer Res. 2013, 19, 4371–4382. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef]
- Dor, Y.; Porat, R.; Keshet, E. Vascular endothelial growth factor and vascular adjustments to perturbations in oxygen homeostasis. Am. J. Physiol. Cell Physiol. 2001, 280, C1367–C1374. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The von Hippel-Lindau tumor suppressor protein and clear cell renal carcinoma. Clin. Cancer Res. 2007, 13, 680s–682s. [Google Scholar] [CrossRef]
- Cong, X.L.; Li, B.; Yang, R.C.; Feng, S.Z.; Chen, S.J.; Han, Z.C. Enhanced growth suppression of Philadephia1 leukemia cells by targeting bcr3/abl2 and VEGF through antisense strategy. Leukemia 2005, 19, 1517–1524. [Google Scholar] [CrossRef][Green Version]
- Kumar, S.A.; Hu, X.; Brown, M.; Kuschak, B.; Hernandez, T.A.; Johnston, J.B.; Gibson, S.B. Lysophosphatidic acid receptor expression in chronic lymphocytic leukemia leads to cell survival mediated though vascular endothelial growth factor expression. Leuk. Lymphoma 2009, 50, 2038–2048. [Google Scholar] [CrossRef]
- Aguayo, A.; Kantarjian, H.; Manshouri, T.; Gidel, C.; Estey, E.; Thomas, D.; Koller, C.; Estrov, Z.; O’Brien, S.; Keating, M.; et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000, 96, 2240–2245. [Google Scholar] [CrossRef] [PubMed]
- Bellamy, W.T.; Richter, L.; Sirjani, D.; Roxas, C.; Glinsmann-Gibson, B.; Frutiger, Y.; Grogan, T.M.; List, A.F. Vascular endothelial cell growth factor is an autocrine promoter of abnormal localized immature myeloid precursors and leukemia progenitor formation in myelodysplastic syndromes. Blood 2001, 97, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Carvalho, G.; Fabre, C.; Grosjean, J.; Fenaux, P.; Kroemer, G. Targeting NF-kappaB in hematologic malignancies. Cell Death Differ. 2006, 13, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Sujobert, P.; Bardet, V.; Cornillet-Lefebvre, P.; Hayflick, J.S.; Prie, N.; Verdier, F.; Vanhaesebroeck, B.; Muller, O.; Pesce, F.; Ifrah, N.; et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 2005, 106, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Le, X.; Abbruzzese, J.L.; Peng, Z.; Qian, C.N.; Tang, H.; Xiong, Q.; Wang, B.; Li, X.C.; Xie, K. Constitutive Sp1 activity is essential for differential constitutive expression of vascular endothelial growth factor in human pancreatic adenocarcinoma. Cancer Res. 2001, 61, 4143–4154. [Google Scholar]
- Hsu, T.C.; Young, M.R.; Cmarik, J.; Colburn, N.H. Activator protein 1 (AP-1) and nuclear factor kappaB (NF-kappa B)-dependent transcriptional events in carcinogenesis. Free Radic. Biol. Med. 2000, 28, 1338–1348. [Google Scholar] [CrossRef]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Pollmann, C.; Huang, X.; Mall, J.; Bech-Otschir, D.; Naumann, M.; Dubiel, W. The constitutive photomorphogenesis 9 signalosome directs vascular endothelial growth factor production in tumor cells. Cancer Res. 2001, 61, 8416–8421. [Google Scholar]
- Poulaki, V.; Mitsiades, C.S.; McMullan, C.; Sykoutri, D.; Fanourakis, G.; Kotoula, V.; Tseleni-Balafouta, S.; Koutras, D.A.; Mitsiades, N. Regulation of vascular endothelial growth factor expression by insulin-like growth factor I in thyroid carcinomas. J. Clin. Endocrinol. Metab. 2003, 88, 5392–5398. [Google Scholar] [CrossRef]
- Chuang, C.H.; Huang, C.S.; Hu, M.L. Vitamin E and rutin synergistically inhibit expression of vascular endothelial growth factor through down-regulation of binding activity of activator protein-1 in human promyelocytic leukemia (HL-60) cells. Chem. Biol. Interact. 2010, 183, 434–441. [Google Scholar] [CrossRef]
- Venables, J.P. Unbalanced alternative splicing and its significance in cancer. Bioessays 2006, 28, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Tai, Y.T.; Davies, F.E. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood 2001, 98, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Hussong, J.W.; Rodgers, G.M.; Shami, P.J. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood 2000, 95, 309–313. [Google Scholar] [CrossRef]
- Padró, T.; Ruiz, S.; Bieker, R.; Bürger, H.; Steins, M.; Kienast, J.; Büchner, T.; Berdel, W.E.; Mesters, R.M. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood 2000, 95, 2637–2644. [Google Scholar] [CrossRef] [PubMed]
- Gora-Tybor, J.; Blonski, J.Z.; Robak, T. Circulating vascular endothelial growth factor (VEGF) and its soluble receptors in patients with chronic lymphocytic leukemia. Eur. Cytokine Netw. 2005, 16, 41–46. [Google Scholar]
- Vacca, A.; Ribatti, D.; Presta, M.; Minischetti, M.; Iurlaro, M.; Ria, R.; Albini, A.; Bussolino, F.; Dammacco, F. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood 1999, 93, 3064–3073. [Google Scholar] [CrossRef]
- Ugarte-Berzal, E.; Redondo-Muñoz, J.; Eroles, P.; Del Cerro, M.H.; García-Marco, J.A.; Terol, M.J.; García-Pardo, A. VEGF/VEGFR2 interaction down-regulates matrix metalloproteinase-9 via STAT1 activation and inhibits B chronic lymphocytic leukemia cell migration. Blood 2010, 115, 846–849. [Google Scholar] [CrossRef]
- Hata, A.N.; Breyer, R.M. Pharmacology and signaling of prostaglandin receptors: Multiple roles in inflammation and immune modulation. Pharmacol. Ther. 2004, 103, 147–166. [Google Scholar] [CrossRef]
- Wu, G.; Luo, J.; Rana, J.S.; Laham, R.; Sellke, F.W.; Li, J. Involvement of COX-2 in VEGF-induced angiogenesis via P38 and JNK pathways in vascular endothelial cells. Cardiovasc. Res. 2006, 69, 512–519. [Google Scholar] [CrossRef]
- Su, J.L.; Shih, J.Y.; Yen, M.L.; Jeng, Y.M.; Chang, C.C.; Hsieh, C.Y.; Wei, L.H.; Yang, P.C.; Kuo, M.L. Cyclooxygenase-2 induces EP1- and HER-2/Neu-dependent vascular endothelial growth factor-C up-regulation: A novel mechanism of lymphangiogenesis in lung adenocarcinoma. Cancer Res. 2004, 64, 554–564. [Google Scholar] [CrossRef]
- Ria, R.; Reale, A.; Vacca, A. Novel agents and new therapeutic approaches for treatment of multiple myeloma. World J. Methodol. 2014, 4, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Barchnicka, A.; Olejniczak-Nowakowska, M.; Krupa-Kotara, K.; Grosicki, S. The importance of antiangiogenic effect in multiple myeloma treatment. Adv. Clin. Exp. Med. 2018, 27, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Emerging options in multiple myeloma: Targeted, immune, and epigenetic therapies. Hematology. Am. Soc. Hematol. Ed. Prog. 2017, 2017, 518–524. [Google Scholar] [CrossRef]
- Podar, K.; Anderson, K.C. The pathophysiologic role of VEGF in hematologic malignancies: Therapeutic implications. Blood 2005, 105, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Terpos, E.; Kastritis, E.; Dimopoulos, M.A. Emerging antibodies for the treatment of multiple myeloma. Exp. Opin. Emerging Drugs 2016, 21, 225–237. [Google Scholar] [CrossRef]
- Cook, K.M.; Figg, W.D. Angiogenesis inhibitors: Current strategies and future prospects. CA A Cancer J. Clin. 2010, 60, 222–243. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.N.; Liu, P. Targeting MET in cancer therapy. Chronic Dis. Transl. Med. 2017, 3, 148–153. [Google Scholar] [CrossRef]
- Lopuch, S.; Kawalec, P.; Wiśniewska, N. Effectiveness of targeted therapy as monotherapy or combined therapy in patients with relapsed or refractory multiple myeloma: A systematic review and meta-analysis. Hematology 2015, 20, 1–10. [Google Scholar] [CrossRef]
- Hideshima, T.; Chauhan, D.; Hayashi, T.; Akiyama, M.; Mitsiades, N.; Mitsiades, C.; Podar, K.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Proteasome inhibitor PS-341 abrogates IL-6 triggered signaling cascades via caspase-dependent downregulation of gp130 in multiple myeloma. Oncogene 2003, 22, 8386–8393. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Hideshima, T.; Raje, N.; Kumar, S.; Ishitsuka, K.; Yasui, H.; Shiraishi, N.; Ribatti, D.; Nico, B.; Vacca, A.; et al. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006, 66, 184–191. [Google Scholar] [CrossRef]
- Williams, S.; Pettaway, C.; Song, R.; Papandreou, C.; Logothetis, C.; McConkey, D.J. Differential effects of the proteasome inhibitor bortezomib on apoptosis and angiogenesis in human prostate tumor xenografts. Mol. Cancer Ther. 2003, 2, 835–843. [Google Scholar] [PubMed]
- Sunwoo, J.B.; Chen, Z.; Dong, G.; Yeh, N.; Crowl Bancroft, C.; Sausville, E.; Adams, J.; Elliott, P.; Van Waes, C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin. Cancer Res. 2001, 7, 1419–1428. [Google Scholar]
- Shono, T.; Ono, M.; Izumi, H.; Jimi, S.I.; Matsushima, K.; Okamoto, T.; Kohno, K.; Kuwano, M. Involvement of the transcription factor NF-kB in tubular morphogenesis of human microvascular endothelial cells by oxidative stress. Mol. Cell Biol. 1996, 16, 4231–4239. [Google Scholar] [CrossRef] [PubMed]
- Scatena, M.; Almeida, M.; Chaisson, M.L.; Fausto, N.; Nicosia, R.F.; Giachelli, C.M. NF-kB mediates avb3 indegrin-induced endothelial cell survival. J. Cell. Biol. 1998, 141, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D.J.; Zhu, K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist. Updat. 2008, 11, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Karl, E.; Warner, K.; Zeitlin, B.; Kaneko, T.; Wurtzel, L.; Jin, T.; Chang, J.; Wang, S.; Wang, C.Y.; Strieter, R.M.; et al. Bcl-2 acts in a proangiogenic signaling pathway through nuclear factor-kB and CXC chemokines. Cancer Res. 2005, 65, 5063–5069. [Google Scholar] [CrossRef]
- Oikawa, T.; Sasaki, T.; Nakamura, M.; Shimamura, M.; Tanahashi, N.; Omra, S.; Tanaka, K. The proteasome is involved in angiogenesis. Biochem. Biophys. Res. Commun. 1998, 8, 243–248. [Google Scholar] [CrossRef]
- Drexler, H.C.A.; Risau, W.; Konerding, M.A. Inhibition of proteasome function induces programmed cell death in proliferating endothelial cells. FASEB J. 2000, 14, 65–77. [Google Scholar] [CrossRef]
- LeBlanc, R.; Catley, L.P.; Hideshima, T.; Lentzsch, S.; Mitsiades, C.S.; Mitsiades, N.; Neuberg, D.; Goloubeva, O.; Pien, C.S.; Adams, J.; et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002, 62, 4996–5000. [Google Scholar]
- Anargyrou, K.; Terpos, E.; Vassilakopoulos, T.P.; Pouli, A.; Sachanas, S.; Tzenou, T.; Masouridis, S.; Christoulas, D.; Angelopoulou, M.K.; Dimitriadou, E.M.; et al. Greek Myeloma Study Group. Normalization of the serum angiopoietin-1 to angiopoietin-2 ratio reflects response in refractory/resistant multiple myeloma patients treated with bortezomib. Haematologica 2008, 93, 451–454. [Google Scholar] [CrossRef]
- Politou, M.; Naresh, K.; Terpos, E.; Crawley, D.; Lampert, I.; Apperley, J.F.; Rahemtulla, A. Anti-angiogenic effect of bortezomib in patients with multiple myeloma. Acta Haematol. 2005, 114, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Saltarella, I.; Morabito, F.; Giuliani, N.; Terragna, C.; Omedè, P.; Palumbo, A.; Bringhen, S.; De Paoli, L.; Martino, E.; Larocca, A.; et al. Prognostic or predictive value of circulating cytokines and angiogenic factors for initial treatment of multiple myeloma in the GIMEMA MM0305 randomized controlled trial. J. Hematol. Oncol. 2019, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.S.; Kirma, N.B.; Santhamma, B.; Tekmal, R.R.; Agyin, J.K. Effects of a novel proteasome inhibitor BU-32 on multiple myeloma cells. Cancer Chemother. Pharmacol. 2014, 73, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Tian, Z.; Zhou, B.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin. Cancer Res. 2011, 17, 5311–5321. [Google Scholar] [CrossRef]
- Price, D.K.; Ando, Y.; Kruger, E.A.; Weiss, M.; Figg, W.D. 5’-OH-thalidomide, a metabolite of thalidomide, inhibits angiogenesis. Ther. Drug Monit. 2002, 24, 104–110. [Google Scholar] [CrossRef]
- Ng, S.S.; Gutschow, M.; Weiss, M.; Hauschildt, S.; Teubert, U.; Hecker, T.K.; Luzzio, F.A.; Kruger, E.A.; Eger, K.; Figg, W.D. Antiangiogenic activity of N-substituted and tetrafluorinated thalidomide analogues. Cancer Res. 2003, 63, 3189–3194. [Google Scholar]
- Franks, M.E.; Macpherson, G.R.; Figg, W.D. Thalidomide. Lancet 2004, 363, 1802–1811. [Google Scholar] [CrossRef]
- Therapontos, C.; Erskine, L.; Gardner, E.; Figg, W.D.; Vargesson, N. Thalidomide induces limb defects by preventing angiogenic outgrowth during early limb formation. Proc. Natl. Acad. Sci. USA 2009, 106, 8573–8578. [Google Scholar] [CrossRef]
- Rashid, A.; Kuppa, A.; Kunwar, A.; Panda, D. Thalidomide (5HPP-33) suppresses microtubule dynamics and depolymerizes the microtubule network by binding at the vinblastine binding site on tubulin. Biochemistry 2015, 54, 2149–2159. [Google Scholar] [CrossRef]
- Inatsuki, S.; Noguchi, T.; Miyachi, H.; Oda, S.; Iguchi, T.; Kizaki, M.; Hashimoto, Y.; Kobayashi, H. Tubulin-polymerization inhibitors derived from thalidomide. Bioorg. Med. Chem. Lett. 2005, 15, 321–325. [Google Scholar] [CrossRef]
- Hansen, J.M.; Gong, S.G.; Philbert, M.; Harris, C. Misregulation of gene expression in the redox-sensitive NF-kappab-dependent limb outgrowth pathway by thalidomide. Dev. Dyn. 2002, 225, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, J.; Jungck, D.; Koch, A. Apoptosis induction by thalidomide: Critical for limb teratogenicity but therapeutic potential in idiopathic pulmonary fibrosis? Curr. Mol. Pharmacol. 2011, 4, 26–61. [Google Scholar] [CrossRef] [PubMed]
- Tamilarasan, K.P.; Kolluru, G.K.; Rajaram, M.; Indhumathy, M.; Saranya, R.; Chatterjee, S. Thalidomide attenuates nitric oxide mediated angiogenesis by blocking migration of endothelial cells. BMC Cell. Biol. 2006, 7, 17. [Google Scholar] [CrossRef]
- Majumder, S.; Rajaram, M.; Muley, A.; Reddy, H.S.; Tamilarasan, K.P.; Kolluru, G.K.; Sinha, S.; Siamwala, J.H.; Gupta, R.; Ilavarasan, R.; et al. Thalidomide attenuates nitric oxide-driven angiogenesis by interacting with soluble guanylyl cyclase. Br. J. Pharmacol. 2009, 158, 1720–1734. [Google Scholar] [CrossRef]
- Siamwala, J.H.; Veeriah, V.; Priya, M.K.; Rajendran, S.; Saran, U.; Sinha, S.; Nagarajan, S.; Pradeep, T.; Chatterjee, S. Nitric oxide rescues thalidomide mediated teratogenicity. Sci. Rep. 2012, 2, 679. [Google Scholar] [CrossRef]
- Yabu, T.; Tomimoto, H.; Taguchi, Y.; Yamaoka, S.; Igarashi, Y.; Okazaki, T. Thalidomide-induced antiangiogenic action is mediated by ceramide through depletion of VEGF receptors, and is antagonized by sphingosine-1-phosphate. Blood 2005, 106, 125–134. [Google Scholar] [CrossRef]
- D’Amato, R.J.; Loughnan, M.S.; Flynn, E.; Folkman, J. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef]
- Kenyon, B.M.; Browne, F.; D’Amato, R.J. Effects of thalidomide and related metabolites in a mouse corneal model of neovascularisation. Exp. Eye Res. 1997, 64, 971–978. [Google Scholar] [CrossRef]
- Stephens, T.D.; Bunde, C.J.; Fillmore, B.J. Mechanism of action in thalidomide teratogenesis. Biochem. Pharmacol. 2000, 59, 1489–1499. [Google Scholar] [CrossRef]
- Vargesson, N. Thalidomide-induced limb defects: Resolving a 50-year-old puzzle. BioEssays 2009, 31, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Vargesson, N. Thalidomide embryopathy: An enigmatic challenge. ISRN Dev. Biol. 2013, 2013, 241016. [Google Scholar] [CrossRef]
- Feng, Q.; Tan, H.H.; Ge, Z.Z.; Gao, Y.J.; Chen, H.M.; Xiao, S.D. Thalidomide-induced angiopoietin 2, Notch1 and Dll4 downregulation under hypoxic condition in tissues with gastrointestinal vascular malformation and human umbilical vein endothelial cells. J. Dig. Dis. 2014, 15, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fu, S.; Chen, H.; Feng, Q.; Gao, Y.; Xue, H.; Ge, Z.; Fang, J.; Xiao, S. Inhibition of endothelial Slit2/Robo1 signalling by thalidomide restrains angiogenesis by blocking the PI3K/Akt pathway. Dig. Dis. Sci. 2014, 59, 2958–2966. [Google Scholar] [CrossRef]
- Vacca, A.; Scavelli, C.; Montefusco, V.; Di Pietro, G.; Neri, A.; Mattioli, M.; Bicciato, S.; Nico, B.; Ribatti, D.; Dammacco, F.; et al. Thalidomide downregulates angiogenic genes in bone marrow endothelial cells of patients with active multiple myeloma. J. Clin. Oncol. 2005, 23, 5334–5346. [Google Scholar] [CrossRef]
- Lu, L.; Payvandi, F.; Wu, L.; Zhang, L.-H.; Hariri, R.J.; Man, H.-W.; Chen, R.S.; Muller, G.W.; Hughes, C.C.W.; Stirling, D.I.; et al. The anti-cancer drug lenalidomide inhibits angiogenesis andmetastasis viamultiple inhibitory effects on endothelial cell function in normoxic and hypoxic conditions. Microvasc. Res. 2009, 77, 78–86. [Google Scholar] [CrossRef]
- Mitsiades, N. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: Therapeutic implications. Blood 2002, 99, 4525–4530. [Google Scholar] [CrossRef]
- Chang, D.H. Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: Therapeutic implications. Blood 2006, 108, 618–621. [Google Scholar] [CrossRef]
- Dredge, K.; Horsfall, R.; Robinson, S.P.; Zhang, L.-H.; Lu, L.; Tang, Y.; Shirley, M.A.; Muller, G.; Schafer, P.; Stirling, D.; et al. Orally administered lenalidomide (CC-5013) is anti-angiogenic in vivo and inhibits endothelial cell migration and Akt phosphorylation in vitro. Microvasc. Res. 2005, 69, 56–63. [Google Scholar] [CrossRef]
- Gorgun, G.; Calabrese, E.; Soydan, E.; Hideshima, T.; Perrone, G.; Bandi, M.; Cirstea, D.; Santo, L.; Hu, Y.; Tai, Y.T.; et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood 2010, 116, 3227–3237. [Google Scholar] [CrossRef]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef] [PubMed]
- De Luisi, A.; Ferrucci, A.; Coluccia, A.M.L.; Ria, R.; Moschetta, M.; de Luca, E.; Pieroni, L.; Maffia, M.; Urbani, A.; Di Pietro, G.; et al. Lenalidomide restrains motility and overangiogenic potential of bone marrow endothelial cells in patients with active multiple myeloma. Clin. Cancer Res. 2011, 17, 1935–1946. [Google Scholar] [CrossRef] [PubMed]
- Cibeira, M.T.; Rozman, M.; Segarra, M.; Lozano, E.; Rosiñol, L.; Cid, M.C.; Filella, X.; Bladé, J. Bone marrow angiogenesis and angiogenic factors in multiple myeloma treated with novel agents. Cytokine 2008, 41, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Chanan-Khan, A.A.; Swaika, A.; Paulus, A.; Kumar, S.K.; Mikhael, J.R.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q. Pomalidomide: The new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer J. 2013, 3, e143. [Google Scholar] [CrossRef] [PubMed]
- Scavelli, C.; Di Pietro, G.; Cirulli, T.; Coluccia, M.; Boccarelli, A.; Giannini, T.; Mangialardi, G.; Bertieri, R.; Coluccia, A.M.L.; Ribatti, D.; et al. Zoledronic acid affects over-angiogenic phenotype of endothelial cells in patients with multiple myeloma. Mol. Cancer Ther. 2007, 6, 3256–3262. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Di Pietro, G.; Ria, R.; Gnoni, A.; Mangialardi, G.; Guarini, A.; Ditonno, P.; Musto, P.; D’Auria, F.; Ricciardi, M.R.; et al. Bortezomib and zoledronic acid on angiogenic and vasculogenic activities of bone marrow macrophages in patients with multiple myeloma. Eur. J. Cancer 2010, 46, 420–429. [Google Scholar] [CrossRef]
- Kim, J.; Denu, R.A.; Dollar, B.A.; Escalante, L.E.; Kuether, J.P.; Callander, N.S.; Asimakopoulos, F.; Hematti, P. Macrophages and mesenchymal stromal cells support survival and proliferation of multiple myeloma cells. Br. J. Haematol. 2012, 158, 336–346. [Google Scholar] [CrossRef]
- Lin, B.; Podar, K.; Gupta, D.; Tai, Y.T.; Li, S.; Weller, E.; Hideshima, T.; Lentzsch, S.; Davies, F.; Li, C.; et al. The vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584 inhibits growth and migration of multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2002, 62, 5019–5026. [Google Scholar]
- Borjan, B.; Steiner, N.; Karbon, S.; Kern, J.; Francesch, A.; Hermann, M.; Willenbacher, W.; Gunsilius, E.; Untergasser, G. The Aplidin analogs PM01215 and PM02781 inhibit angiogenesis in vitro and in vivo. BMC Cancer 2015, 15, 738. [Google Scholar] [CrossRef]
- Gentile, M.; Martino, M.; Recchia, A.G.; Vigna, E.; Morabito, L.; Morabito, F. Sorafenib for the treatment of multiple myeloma. Expert Opin. Investig. Drugs 2016, 25, 743–749. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; Timm, M.; Haug, J.L.; Kimlinger, T.K.; Wellik, L.E.; Witzig, T.E.; Rajkumar, S.V.; Adjei, A.A.; Kumar, S. Sorafenib, a dual Raf kinase/vascular endothelial growth factor receptor inhibitor has significant anti-myeloma activity and synergizes with common anti-myeloma drugs. Oncogene 2010, 29, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.A.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.G.; Vergara, D.; et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clin. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, X.; Jiang, Y.; Guo, M.; Zhang, S.; Li, J.; He, J.; Liu, J.; Wang, J.; Ouyang, L. Recent advances in the development of dual VEGFR and c-Met small molecule inhibitors as anticancer drugs. Eur. J. Med. Chem. 2016, 108, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Tonon, G.; Sattler, M.; Tai, Y.T.; Legouill, S.; Yasui, H.; Ishitsuka, K.; Kumar, S.; Kumar, R.; Pandite, L.N.; et al. The small-molecule VEGF receptor inhibitor pazopanib (GW786034B) targets both tumor and endothelial cells in multiple myeloma. Proc. Natl. Acad. Sci. USA 2006, 103, 19478–19483. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [PubMed]
- Hennequin, L.F.; Stokes, E.S.; Thomas, A.P.; Johnstone, C.; Plé, P.A.; Ogilvie, D.J.; Dukes, M.; Wedge, S.R.; Kendrew, J.; Curwen, J.O. Novel 4-anilinoquinazolines with C-7 basic side chains: Design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors. J. Med. Chem. 2002, 45, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; De Veirman, K.; Giannico, D.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Prete, M.; Harstrick, A.; et al. Targeting angiogenesis in multiple myeloma by the VEGF and HGF blocking DARPin ® protein MP0250: A preclinical study. Oncotarget 2018, 9, 13366–13381. [Google Scholar] [CrossRef]
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (Velcade trade mark) in the Treatment of Multiple Myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef]
- Ribatti, D.; Vacca, A. Therapeutic renaissance of thalidomide in the treatment of haematological malignancies. Leukemia 2005, 19, 1525–1531. [Google Scholar] [CrossRef]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef]
- Palumbo, A.; Giaccone, L.; Bertola, A.; Pregno, P.; Bringhen, S.; Rus, C.; Triolo, S.; Gallo, E.; Pileri, A.; Boccadoro, M. Low-dose thalidomide plus dexamethasone is an effective salvage therapy for advanced myeloma. Haematologica 2001, 86, 399–403. [Google Scholar] [PubMed]
- Dimopoulos, M.A.; Zervas, K.; Kouvatseas, G.; Galani, E.; Grigoraki, V.; Kiamouris, C.; Vervessou, E.; Samantas, E.; Papadimitriou, C.; Economou, O.; et al. Thalidomide and dexamethasone combination for refractory multiple myeloma. Ann. Oncol. 2001, 12, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Hayman, S.; Gertz, M.A.; Dispenzieri, A.; Lacy, M.Q.; Greipp, P.R.; Geyer, S.; Iturria, N.; Fonseca, R.; Lust, J.A.; et al. Combination therapy with thalidomide plus dexamethasone for newly diagnosed myeloma. J. Clin. Oncol. 2002, 20, 4319–4323. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.; Rankin, K.; Gavino, M.; Delasalle, K.; Alexanian, R. Thalidomide alone or with dexamethasone for previously untreated multiple myeloma. J. Clin. Oncol. 2003, 21, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Cavo, M.; Zamagni, E.; Tosi, P.; Cellini, C.; Cangini, D.; Tacchetti, P.; Testoni, N.; Tonelli, M.; de Vivo, A.; Palareti, G.; et al. First-line therapy with thalidomide and dexamethasone in preparation for autologous stem cell transplantation for multiple myeloma. Haematologica 2004, 89, 826–831. [Google Scholar]
- McCarthy, P.L.; Palumbo, A. Maintenance therapy for multiple myeloma. Hematol. Oncol. Clin. N. Am. 2014, 28, 839–859. [Google Scholar] [CrossRef]
- Palumbo, A.; Avonto, I.; Bruno, B.; Ambrosini, M.T.; Bringhen, S.; Cavallo, F.; Falco, P.; Boccadoro, M. Intravenous melphalan, thalidomide and prednisone in refractory and relapsed multiple myeloma. Eur. J. Haematol. 2006, 76, 273–277. [Google Scholar] [CrossRef]
- Garcia-Sanz, R.; Gonzalez-Porras, J.R.; Hernandez, J.M.; Polo-Zarzuela, M.; Sureda, A.; Barrenetxea, C.; Palomera, L.; López, R.; Grande-García, C.; Alegre, A.; et al. The oral combination of thalidomide, cyclophosphamide and dexamethasone (ThaCyDex) is effective in relapsed/refractory multiple myeloma. Leukemia 2004, 18, 856–863. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Hamilos, G.; Zomas, A.; Gika, D.; Efstathiou, E.; Grigoraki, V.; Poziopoulos, C.; Xilouri, I.; Zorzou, M.P.; Anagnostopoulos, N.; et al. Pulsed cyclophosphamide, thalidomide and dexamethasone: An oral regimen for previously treated patients with multiple myeloma. Hematol. J. 2004, 5, 112–117. [Google Scholar] [CrossRef]
- Hovenga, S.; Daenen, S.M.; de Wolf, J.T.; van Imhoff, G.W.; Kluin-Nelemans, H.C.; Sluiter, W.J.; Vellenga, E. Combined thalidomide and cyclophosphamide treatment for refractory or relapsed multiple myeloma patients: A prospective phase II study. Ann. Hematol. 2005, 84, 311–316. [Google Scholar] [CrossRef]
- Kyriakou, C.; Thomson, K.; D’Sa, S.; Flory, A.; Hanslip, J.; Goldstone, A.H.; Yong, K.L. Low-dose thalidomide in combination with oral weekly cyclophosphamide and pulsed dexamethasone is a well tolerated and effective regimen in patients with relapsed and refractory multiple myeloma. Br. J. Haematol. 2005, 129, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Offidani, M.; Corvatta, L.; Marconi, M.; Visani, G.; Alesiani, F.; Brunori, M.; Galieni, P.; Catarini, M.; Burattini, M.; Centurioni, R.; et al. Low-dose thalidomide with pegylated liposomal doxorubicin and high-dose dexamethasone for relapsed/refractory multiple myeloma: A prospective, multicenter, phase II study. Haematologica 2006, 91, 133–136. [Google Scholar] [PubMed]
- Offidani, M.; Corvatta, L.; Piersantelli, M.N.; Visani, G.; Alesiani, F.; Brunori, M.; Galieni, P.; Catarini, M.; Burattini, M.; Centurioni, R.; et al. Thalidomide, dexamethasone, and pegylated liposomal doxorubicin (ThaDD) for patients older than 65 years with newly diagnosed multiple myeloma. Blood 2006, 108, 2159–2164. [Google Scholar] [CrossRef] [PubMed]
- Zervas, K.; Dimopoulos, M.A.; Hatzicharissi, E.; Anagnostopoulos, A.; Papaioannou, M.; Mitsouli, C.; Panagiotidis, P.; Korantzis, J.; Tzilianos, M.; Maniatis, A. Greek Myeloma Study Group. Primary treatment of multiple myeloma with thalidomide, vincristine, liposomal doxorubicin and dexamethasone (T-VAD doxil): A phase II multicenter study. Ann. Oncol. 2004, 15, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.A.; Baz, R.; Srkalovic, G.; Agrawal, N.; Suppiah, R.; His, E.; Andresen, S.; Karam, M.A.; Reed, J.; Faiman, B.; et al. Phase 2 study of pegylated liposomal doxorubicin, vincristine, decreased-frequency dexamethasone, and thalidomide in newly diagnosed and relapsed-refractory multiple myeloma. Mayo Clin. Proc. 2006, 81, 889–895. [Google Scholar] [CrossRef]
- Ciolli, S.; Leoni, F.; Gigli, F.; Rigacci, L.; Bosi, A. Low dose Velcade, thalidomide and dexamethasone (LD-VTD): An effective regimen for relapsed and refractory multiple myeloma patients. Leuk. Lymphoma 2006, 47, 171–173. [Google Scholar] [CrossRef]
- Palumbo, A.; Ambrosini, M.T.; Benevolo, G.; Pregno, P.; Pescosta, N.; Callea, V.; Cangialosi, C.; Caravita, T.; Morabito, F.; Musto, P.; et al. Bortezomib, melphalan, prednisone, and thalidomide for relapsed multiple myeloma. Blood 2007, 109, 2767–2772. [Google Scholar] [CrossRef][Green Version]
- Wang, M.; Giralt, S.; Delasalle, K.; Handy, B.; Alexanian, R. Bortezomib in combination with thalidomide-dexamethasone for previously untreated multiple myeloma. Hematology 2007, 12, 235–239. [Google Scholar] [CrossRef]
- Pineda-Roman, M.; Zangari, M.; van Rhee, F.; Anaissie, E.; Szymonifka, J.; Hoering, A.; Petty, N.; Crowley, J.; Shaughnessy, J.; Epstein, J.; et al. VTD combination therapy with bortezomib-thalidomide-dexamethasone is highly effective in advanced and refractory multiple myeloma. Leukemia 2008, 22, 1419–1427. [Google Scholar] [CrossRef]
- Sonneveld, P.; Asselbergs, E.; Zweegman, S.; van der Holt, B.; Kersten, M.J.; Vellenga, E.; van Marwijk-Kooy, M.; Broyl, A.; de Weerdt, O.; Lonergan, S.; et al. Phase 2 study of carfilzomib, thalidomide, and dexamethasone as induction/consolidation therapy for newly diagnosed multiple myeloma. Blood 2015, 125, 449–456. [Google Scholar] [CrossRef]
- Mikhael, J.R.; Reeder, C.B.; Libby, E.N.; Costa, L.J.; Bergsagel, P.L.; Buadi, F.; Mayo, A.; Nagi Reddy, S.K.; Gano, K.; Dueck, A.C.; et al. Phase Ib/II trial of CYKLONE (cyclophosphamide, carfilzomib, thalidomide and dexamethasone) for newly diagnosed myeloma. Br. J. Haematol. 2015, 169, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Granell, M.; Oriol, A.; Martinez-Lopez, J.; Blade, J.; Hernandez, M.T.; Martín, J.; Gironella, M.; Lynch, M.; Bleickardt, E.; et al. Elotuzumab in combination with thalidomide and low-dose dexamethasone: A phase 2 single-arm safety study in patients with relapsed/refractory multiple myeloma. Br. J. Haematol. 2016, 175, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Barlogie, B.; Munshi, N.; Zangari, M.; Fassas, A.; Jacobson, J.; van Rhee, F.; Cottler-Fox, M.; Muwalla, F.; Tricot, G. DTPACE: An effective, novel combination chemotherapy with thalidomide for previously treated patients with myeloma. J. Clin. Oncol. 2003, 21, 2732–2739. [Google Scholar] [CrossRef] [PubMed]
- Kropff, M.H.; Lang, N.; Bisping, G.; Domine, N.; Innig, G.; Hentrich, M.; Mitterer, M.; Südhoff, T.; Fenk, R.; Straka, C.; et al. Hyperfractionated cyclophosphamide in combination with pulsed dexamethasone and thalidomide (HyperCDT) in primary refractory or relapsed multiple myeloma. Br. J. Haematol. 2003, 122, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Facon, T.; Mary, J.Y.; Hulin, C.; Benboubker, L.; Attal, M.; Pegourie, B.; Renaud, M.; Harousseau, J.L.; Guillerm, G.; Chaleteix, C.; et al. Melphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99–06): A randomised trial. Lancet 2007, 370, 1209–1218. [Google Scholar] [CrossRef]
- Palumbo, A.; Bringhen, S.; Caravita, T.; Merla, E.; Capparella, V.; Callea, V.; Cangialosi, C.; Grasso, M.; Rossini, F.; Galli, M.; et al. Oral melphalan and prednisone chemotherapy plus thalidomide compared with melphalan and prednisone alone in elderly patients with multiple myeloma: Randomised controlled trial. Lancet 2006, 367, 825–831. [Google Scholar] [CrossRef]
- Cavo, M.; Pantani, L.; Petrucci, M.T.; Patriarca, F.; Zamagni, E.; Donnarumma, D.; Crippa, C.; Boccadoro, M.; Perrone, G.; Falcone, A.; et al. Bortezomib-thalidomide-dexamethasone is superior to thalidomide-dexamethasone as consolidation therapy after autologous hematopoietic stem cell transplantation in patients with newly diagnosed multiple myeloma. Blood 2012, 120, 9–19. [Google Scholar] [CrossRef]
- Richardson, P.G.; Schlossman, R.L.; Weller, E.; Hideshima, T.; Mitsiades, C.; Davies, F.; LeBlanc, R.; Catley, L.P.; Doss, D.; Kelly, K.; et al. Immunomodulatory drug CC-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma. Blood 2002, 100, 3063–3067. [Google Scholar] [CrossRef]
- Weber, D.M.; Chen, C.; Niesvizky, R.; Wang, M.; Belch, A.; Stadtmauer, E.A.; Siegel, D.; Borrello, I.; Rajkumar, S.V.; Chanan-Khan, A.A.; et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N. Engl. J. Med. 2007, 357, 2133–2142. [Google Scholar] [CrossRef]
- Hou, J.; Du, X.; Jin, J.; Cai, Z.; Chen, F.; Zhou, D.B.; Yu, L.; Ke, X.; Li, X.; Wu, D.; et al. A multicenter, open-label, phase 2 study of lenalidomide plus low-dose dexamethasone in Chinese patients with relapsed/refractory multiple myeloma: The MM-021 trial. J. Hematol. Oncol. 2013, 6, 41. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Spencer, A.; Attal, M.; Prince, H.M.; Harousseau, J.L.; Dmoszynska, A.; San Miguel, J.; Hellmann, A.; Facon, T.; Foà, R.; et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2007, 357, 2123–2132. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Hayman, S.R.; Lacy, M.Q.; Dispenzieri, A.; Geyer, S.M.; Kabat, B.; Zeldenrust, S.R.; Kumar, S.; Greipp, P.R.; Fonseca, R.; et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood 2005, 106, 4050–4053. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Jacobus, S.; Callander, N.S.; Fonseca, R.; Vesole, D.H.; Williams, M.E.; Abonour, R.; Siegel, D.S.; Katz, M.; Greipp, P.R. Eastern Cooperative Oncology Group. Lenalidomide plus high-dose dexamethasone versus lenalidomide plus low-dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: An open-label randomised controlled trial. Lancet Oncol. 2010, 11, 29–37. [Google Scholar] [CrossRef]
- Richardson, P.G.; Weller, E.; Lonial, S.; Jakubowiak, A.J.; Jagannath, S.; Raje, N.S.; Avigan, D.E.; Xie, W.; Ghobrial, I.M.; Schlossman, R.L.; et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood 2010, 116, 679–686. [Google Scholar] [CrossRef]
- McCarthy, P.L.; Owzar, K.; Hofmeister, C.C.; Hurd, D.D.; Hassoun, H.; Richardson, P.G.; Giralt, S.; Stadtmauer, E.A.; Weisdorf, D.J.; Vij, R.; et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N. Engl. J. Med. 2012, 366, 1770–1781. [Google Scholar] [CrossRef]
- Attal, M.; Lauwers-Cances, V.; Marit, G.; Caillot, D.; Moreau, P.; Facon, T.; Stoppa, A.M.; Hulin, C.; Benboubker, L.; Garderet, L.; et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N. Engl. J. Med. 2012, 366, 1782–1791. [Google Scholar] [CrossRef]
- Palumbo, A.; Cavallo, F.; Gay, F.; Di Raimondo, F.; Ben Yehuda, D.; Petrucci, M.T.; Pezzatti, S.; Caravita, T.; Cerrato, C.; Ribakovsky, E.; et al. Autologous transplantation and maintenance therapy in multiple myeloma. N. Engl. J. Med. 2014, 371, 895–905. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Franssen, L.E.; Levin, M.D.; Bos, G.M.J.; Broijl, A.; Klein, S.K.; Koene, H.R.; Bloem, A.C.; Beeker, A.; Faber, L.M.; et al. Phase 1/2 study of lenalidomide combined with low-dose cyclophosphamide and prednisone in lenalidomide-refractory multiple myeloma. Blood 2016, 128, 2297–2306. [Google Scholar] [CrossRef]
- Schey, S.A.; Morgan, G.J.; Ramasamy, K.; Hazel, B.; Ladon, D.; Corderoy, S.; Jenner, M.; Phekoo, K.; Boyd, K.; Davies, F.E. The addition of cyclophosphamide to lenalidomide and dexamethasone in multiply relapsed/refractory myeloma patients; a phase I/II study. Br. J. Haematol. 2010, 150, 326–333. [Google Scholar] [CrossRef]
- Kumar, S.K.; Lacy, M.Q.; Hayman, S.R.; Stewart, K.; Buadi, F.K.; Allred, J.; Laumann, K.; Greipp, P.R.; Lust, J.A.; Gertz, M.A.; et al. Lenalidomide, cyclophosphamide and dexamethasone (CRd) for newly diagnosed multiple myeloma: Results from a phase 2 trial. Am. J. Hematol. 2011, 86, 640–645. [Google Scholar] [CrossRef]
- Lentzsch, S.; O’Sullivan, A.; Kennedy, R.C.; Abbas, M.; Dai, L.; Pregja, S.L.; Burt, S.; Boyiadzis, M.; Roodman, G.D.; Mapara, M.Y.; et al. Combination of bendamustine, lenalidomide, and dexamethasone (BLD) in patients with relapsed or refractory multiple myeloma is feasible and highly effective: Results of phase 1/2 open-label, dose escalation study. Blood 2012, 119, 4608–4613. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Krishnan, A.; LaPlant, B.; Laumann, K.; Roy, V.; Zimmerman, T.; Gertz, M.A.; Buadi, F.K.; Stockerl Goldstein, K.; Birgin, A.; et al. Bendamustine, lenalidomide, and dexamethasone (BRD) is highly effective with durable responses in relapsed multiple myeloma. Am. J. Hematol. 2015, 90, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Falco, P.; Corradini, P.; Falcone, A.; Di Raimondo, F.; Giuliani, N.; Crippa, C.; Ciccone, G.; Omedè, P.; Ambrosini, M.T.; et al. Melphalan, prednisone, and lenalidomide treatment for newly diagnosed myeloma: A report from the GIMEMA--Italian Multiple Myeloma Network. J. Clin. Oncol. 2007, 25, 4459–4465. [Google Scholar] [CrossRef] [PubMed]
- Baz, R.; Walker, E.; Karam, M.A.; Choueiri, T.K.; Jawde, R.A.; Bruening, K.; Reed, J.; Faiman, B.; Ellis, Y.; Brand, C.; et al. Lenalidomide and pegylated liposomal doxorubicin-based chemotherapy for relapsed or refractory multiple myeloma: Safety and efficacy. Ann. Oncol. 2006, 17, 1766–1771. [Google Scholar] [CrossRef]
- Baz, R.C.; Shain, K.H.; Hussein, M.A.; Lee, J.H.; Sullivan, D.M.; Oliver, E.F.; Nardelli, L.A.; Nodzon, L.A.; Zhao, X.; Ochoa-Bayona, J.L.; et al. Phase II study of pegylated liposomal doxorubicin, low-dose dexamethasone, and lenalidomide in patients with newly diagnosed multiple myeloma. Am. J. Hematol. 2014, 89, 62–67. [Google Scholar] [CrossRef]
- Richardson, P.G.; Weller, E.; Jagannath, S.; Avigan, D.E.; Alsina, M.; Schlossman, R.L.; Mazumder, A.; Munshi, N.C.; Ghobrial, I.M.; Doss, D.; et al. Multicenter, phase I, dose-escalation trial of lenalidomide plus bortezomib for relapsed and relapsed/refractory multiple myeloma. J. Clin. Oncol. 2009, 27, 5713–5719. [Google Scholar] [CrossRef]
- Richardson, P.G.; Xie, W.; Jagannath, S.; Jakubowiak, A.; Lonial, S.; Raje, N.S.; Alsina, M.; Ghobrial, I.M.; Schlossman, R.L.; Munshi, N.C.; et al. A phase 2 trial of lenalidomide, bortezomib, and dexamethasone in patients with relapsed and relapsed/refractory myeloma. Blood 2014, 123, 1461–1469. [Google Scholar] [CrossRef]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Spicka, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef]
- Jakubowiak, A.J.; Dytfeld, D.; Griffith, K.A.; Lebovic, D.; Vesole, D.H.; Jagannath, S.; Al-Zoubi, A.; Anderson, T.; Nordgren, B.; Detweiler-Short, K.; et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood 2012, 120, 1801–1809. [Google Scholar] [CrossRef]
- Niesvizky, R.; Martin, T.G., 3rd; Bensinger, W.I.; Alsina, M.; Siegel, D.S.; Kunkel, L.A.; Wong, A.F.; Lee, S.; Orlowski, R.Z.; Wang, M. Phase Ib dose-escalation study (PX-171–006) of carfilzomib, lenalidomide, and low-dose dexamethasone in relapsed or progressive multiple myeloma. Clin. Cancer Res. 2013, 19, 2248–2256. [Google Scholar] [CrossRef]
- Wang, M.; Martin, T.; Bensinger, W.; Alsina, M.; Siegel, D.S.; Kavalerchik, E.; Huang, M.; Orlowski, R.Z.; Niesvizky, R. Phase 2 dose-expansion study (PX-171–006) of carfilzomib, lenalidomide, and low-dose dexamethasone in relapsed or progressive multiple myeloma. Blood 2013, 122, 3122–3128. [Google Scholar] [CrossRef]
- Kumar, S.K.; Berdeja, J.G.; Niesvizky, R.; Lonial, S.; Laubach, J.P.; Hamadani, M.; Stewart, A.K.; Hari, P.; Roy, V.; Vescio, R.; et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: An open-label phase 1/2 study. Lancet Oncol. 2014, 15, 1503–1512. [Google Scholar] [CrossRef]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Cho, H.J.; Dhadwal, A.; Morgan, G.; La, L.; Zarychta, K.; Catamero, D.; Florendo, E.; Stevens, N.; Verina, D.; et al. A Phase II Study of Panobinostat with Lenalidomide and Weekly Dexamethasone in Myeloma. Blood Adv. 2017, 1, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef]
- Lonial, S.; Vij, R.; Harousseau, J.L.; Facon, T.; Moreau, P.; Mazumder, A.; Kaufman, J.L.; Leleu, X.; Tsao, L.C.; Westland, C.; et al. Elotuzumab in combination with lenalidomide and low-dose dexamethasone in relapsed or refractory multiple myeloma. J. Clin. Oncol. 2012, 30, 1953–1959. [Google Scholar] [CrossRef]
- Plesner, T.; Arkenau, H.-T.; Gimsing, P.; Krejcik, J.; Lemech, C.; Minnema, M.C.; Lassen, U.; Laubach, J.P.; Palumbo, A.; Lisby, S.; et al. Phase 1/2 study of daratumumab, lenalidomide, and dexamethasone for relapsed multiple myeloma. Blood 2016, 128, 1821–1828. [Google Scholar] [CrossRef]
- San Miguel, J.; Mateos, M.V.; Shah, J.J.; Ocio, E.M.; Rodriguez-Otero, P.; Reece, D.; Munshi, N.C.; Avigan, D.; Ge, Y.; Balakumaran, A.; et al. Pembrolizumab in Combination with Lenalidomide and Low-Dose Dexamethasone for Relapsed/Refractory Multiple Myeloma (RRMM): Keynote-023. Blood 2015, 126, 505. [Google Scholar] [CrossRef]
- Richardson, P.G.; Siegel, D.; Baz, R.; Kelley, S.L.; Munshi, N.C.; Laubach, J.; Sullivan, D.; Alsina, M.; Schlossman, R.; Ghobrial, I.M.; et al. Phase 1 study of pomalidomide MTD, safety, and efficacy in patients with refractory multiple myeloma who have received lenalidomide and bortezomib. Blood 2013, 121, 1961–1967. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Palumbo, A.; Corradini, P.; Cavo, M.; Delforge, M.; Di Raimondo, F.; Weisel, K.C.; Oriol, A.; Hansson, M.; Vacca, A.; et al. Safety and efficacy of pomalidomide plus low-dose dexamethasone in STRATUS (MM-010): A phase 3b study in refractory multiple myeloma. Blood 2016, 128, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Lacy, M.Q.; Hayman, S.R.; Gertz, M.A.; Dispenzieri, A.; Buadi, F.; Kumar, S.; Greipp, P.R.; Lust, J.A.; Russell, S.J.; Dingli, D.; et al. Pomalidomide (CC4047) plus low-dose dexamethasone as therapy for relapsed multiple myeloma. J. Clin. Oncol. 2009, 27, 5008–5014. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Siegel, D.S.; Vij, R.; Hofmeister, C.C.; Baz, R.; Jagannath, S.; Chen, C.; Lonial, S.; Jakubowiak, A.; Bahlis, N.; et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: A randomized phase 2 study. Blood 2014, 123, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Leleu, X.; Attal, M.; Arnulf, B.; Moreau, P.; Traulle, C.; Marit, G.; Mathiot, C.; Petillon, M.O.; Macro, M.; Roussel, M.; et al. Pomalidomide plus low-dose dexamethasone is active and well tolerated in bortezomib and lenalidomide-refractory multiple myeloma: Intergroupe Francophone du Myelome 2009–02. Blood 2013, 121, 1968–1975. [Google Scholar] [CrossRef]
- Paludo, J.; Mikhael, J.R.; LaPlant, B.R.; Halvorson, A.E.; Kumar, S.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; Dispenzieri, A.; Lust, J.A.; et al. Pomalidomide, Bortezomib and Dexamethasone (PVD) for Patients with Relapsed Lenalidomide Refractory Multiple Myeloma (MM). Blood 2017, 130, 1198–1204. [Google Scholar] [CrossRef]
- Richardson, P.G.; Hofmeister, C.; Raje, N.S.; Siegel, D.; Lonial, S.; Laubach, J.P.; Efebera, Y.A.; Vesole, D.H.; Nooka, A.K.; Rosenblatt, J.; et al. A Phase 1, Multicenter Study of Pomalidomide, Bortezomib, and Low-Dose Dexamethasone in Patients with Proteasome Inhibitor Exposed and Lenalidomide-Refractory Myeloma (Trial MM-005). Blood 2015, 126, 3036. [Google Scholar] [CrossRef]
- Shah, J.J.; Stadtmauer, E.A.; Abonour, R.; Cohen, A.D.; Bensinger, W.I.; Gasparetto, C.; Kaufman, J.L.; Lentzsch, S.; Vogl, D.T.; Gomes, C.L.; et al. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood 2015, 126, 2284–2290. [Google Scholar] [CrossRef]
- Krishnan, A.; Kapoor, P.; Palmer, J.M.; Tsai, N.C.; Kumar, S.; Lonial, S.; Htut, M.; Karanes, C.; Nathwani, N.; Rosenzweig, M.; et al. Phase I/II trial of the oral regimen ixazomib, pomalidomide, and dexamethasone in relapsed/refractory multiple myeloma. Leukemia 2018, 32, 1567–1574. [Google Scholar] [CrossRef]
- Larocca, A.; Montefusco, V.; Bringhen, S.; Rossi, D.; Crippa, C.; Mina, R.; Galli, M.; Marcatti, M.; La Verde, G.; Giuliani, N.; et al. Pomalidomide, cyclophosphamide, and prednisone for relapsed/refractory multiple myeloma: A multicenter phase 1/2 open-label study. Blood 2013, 122, 2799–2806. [Google Scholar] [CrossRef]
- Baz, R.C.; Martin, T.G., 3rd; Lin, H.Y.; Zhao, X.; Shain, K.H.; Cho, H.J.; Wolf, J.L.; Mahindra, A.; Chari, A.; Sullivan, D.M.; et al. Randomized multicenter phase 2 study of pomalidomide, cyclophosphamide, and dexamethasone in relapsed refractory myeloma. Blood 2016, 127, 2561–2568. [Google Scholar] [CrossRef]
- Hussain, M.J.; Robinson, M.M.; Hamadeh, I.; Arnall, J.; Bhutani, M.; Atrash, S.; Friend, R.; Pineda-Roman, M.; Symanowski, J.T.; Usmani, S.Z.; et al. Daratumumab, pomalidomide and dexamethasone combination therapy in daratumumab and/or pomalidomide refractory multiple myeloma. Br. J. Haematol. 2019, 186, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Assoun, S.; Brosseau, S.; Steinmetz, C.; Gounant, V.; Zalcman, G. Bevacizumab in advanced lung cancer: State of the art. Future Oncol. 2017, 13, 2515–2535. [Google Scholar] [CrossRef] [PubMed]
- Somlo, G.; Lashkari, A.; Bellamy, W.; Zimmerman, T.M.; Tuscano, J.M.; O’Donnell, M.R.; Mohrbacher, A.F.; Forman, S.J.; Frankel, P.; Chen, H.X.; et al. Phase II randomized trial of bevacizumab versus bevacizumab and thalidomide for relapsed/refractory multiple myeloma: A California Cancer Consortium trial. Br. J. Haematol. 2011, 154, 533–535. [Google Scholar] [CrossRef]
- White, D.; Kassim, A.; Bhaskar, B.; Yi, J.; Wamstad, K.; Paton, V.E. Results from AMBER, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer 2013, 119, 339–347. [Google Scholar] [CrossRef]
- Yordanova, A.; Hose, D.; Neben, K.; Witzens-Harig, M.; Gütgemann, I.; Raab, M.S.; Moehler, T.; Goldschmidt, H.; Schmidt-Wolf, I.G. Sorafenib in patients with refractory or recurrent multiple myeloma. Hematol. Oncol. 2013, 31, 197–200. [Google Scholar] [CrossRef]
- Srkalovic, G.; Hussein, M.A.; Hoering, A.; Zonder, J.A.; Popplewell, L.L.; Trivedi, H.; Mazzoni, S.; Sexton, R.; Orlowski, R.Z.; Barlogie, B. A phase II trial of BAY 43-9006 (sorafenib) (NSC-724772) in patients with relapsing and resistant multiple myeloma: SWOG S0434. Cancer Med. 2014, 3, 1275–1283. [Google Scholar] [CrossRef]
- Kumar, S.; Porrata, L.F.; Ansell, S.M.; Colgan, J.P.; LaPlant, B.; Gertz, M.A.; Inwards, D.J.; Johnston, P.B.; Haberman, T.; Micallef, I.N.; et al. Phase 1 Trial of Sorafenib and Everolimus In Patients with Lymphoma or Multiple Myeloma. Blood 2010, 116, 2802. [Google Scholar] [CrossRef]
- Carlomagno, F.; Vitagliano, D.; Guida, T.; Ciardiello, F.; Tortora, G.; Vecchio, G.; Ryan, A.J.; Fontanini, G.; Fusco, A.; Santoro, M. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002, 62, 7284–7290. [Google Scholar]
- Kovacs, M.J.; Reece, D.E.; Marcellus, D.; Meyer, R.M.; Mathews, S.; Dong, R.P.; Eisenhauer, E. A phase II study of ZD6474 (Zactima, a selective inhibitor of VEGFR and EGFR tyrosine kinase in patients with relapsed multiple myeloma--NCIC CTG IND.145. Invest. New Drugs 2006, 24, 529–535. [Google Scholar]
- Grząśko, N.; Knop, S.; Goldschmidt, H.; Raab, M.S.; Dürig, J.; Bringhen, S.; D’Agostino, M.; Gamberi, B.; Rivolti, E.; Vacca, A.; et al. The MP0250-CP201 Mirror Study: A Phase 2 Study Update of MP0250 Plus Bortezomib and Dexamethasone in Relapse/Refractory Multiple Myeloma (RRMM) Patients Previously Exposed to Proteasome Inhibitors and Immunomodulatory Drugs. Blood 2019, 134 (Suppl. 1), 1899. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ria, R.; Melaccio, A.; Racanelli, V.; Vacca, A. Anti-VEGF Drugs in the Treatment of Multiple Myeloma Patients. J. Clin. Med. 2020, 9, 1765. https://doi.org/10.3390/jcm9061765
Ria R, Melaccio A, Racanelli V, Vacca A. Anti-VEGF Drugs in the Treatment of Multiple Myeloma Patients. Journal of Clinical Medicine. 2020; 9(6):1765. https://doi.org/10.3390/jcm9061765
Chicago/Turabian StyleRia, Roberto, Assunta Melaccio, Vito Racanelli, and Angelo Vacca. 2020. "Anti-VEGF Drugs in the Treatment of Multiple Myeloma Patients" Journal of Clinical Medicine 9, no. 6: 1765. https://doi.org/10.3390/jcm9061765
APA StyleRia, R., Melaccio, A., Racanelli, V., & Vacca, A. (2020). Anti-VEGF Drugs in the Treatment of Multiple Myeloma Patients. Journal of Clinical Medicine, 9(6), 1765. https://doi.org/10.3390/jcm9061765