Theranostic Designed Near-Infrared Fluorescent Poly (Lactic-co-Glycolic Acid) Nanoparticles and Preliminary Studies with Functionalized VEGF-Nanoparticles

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. PLGA-NPs
2.2. PLGA-NPs Functionalization with VEGF
2.3. Calculation of Average Size and Zeta Potential
2.4. In Vitro Binding of VEGF-PLGA-NPs and PLGA-NPs to KDR-Fc
2.5. In Vivo Studies
2.5.1. Mouse Model
2.5.2. Pharmacokinetic of PLGA-NPs
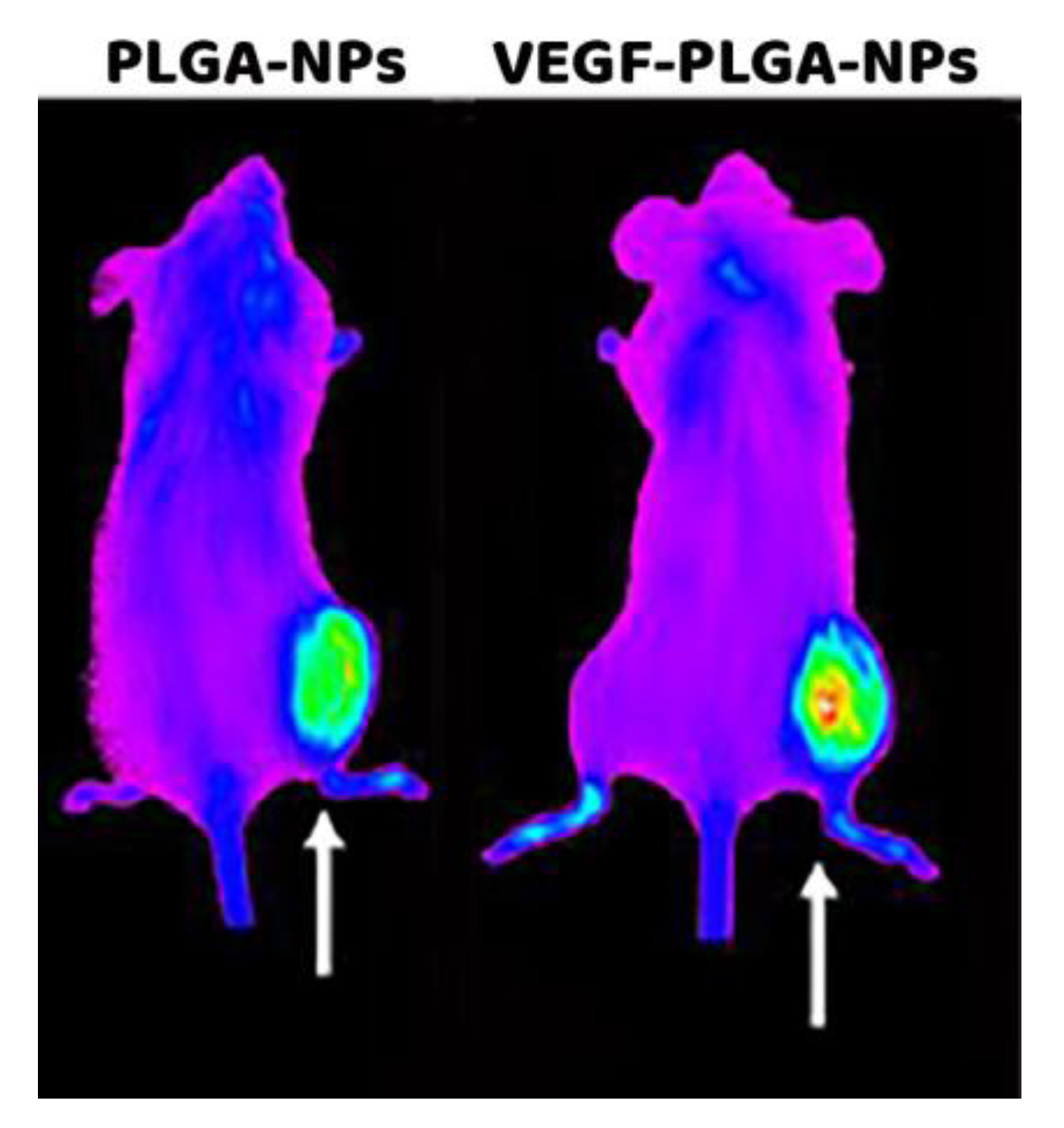
2.5.3. Tumor Targeting of VEGF-PLGA-NPs and of PLGA-NPs
2.6. Statistical Analysis
3. Results
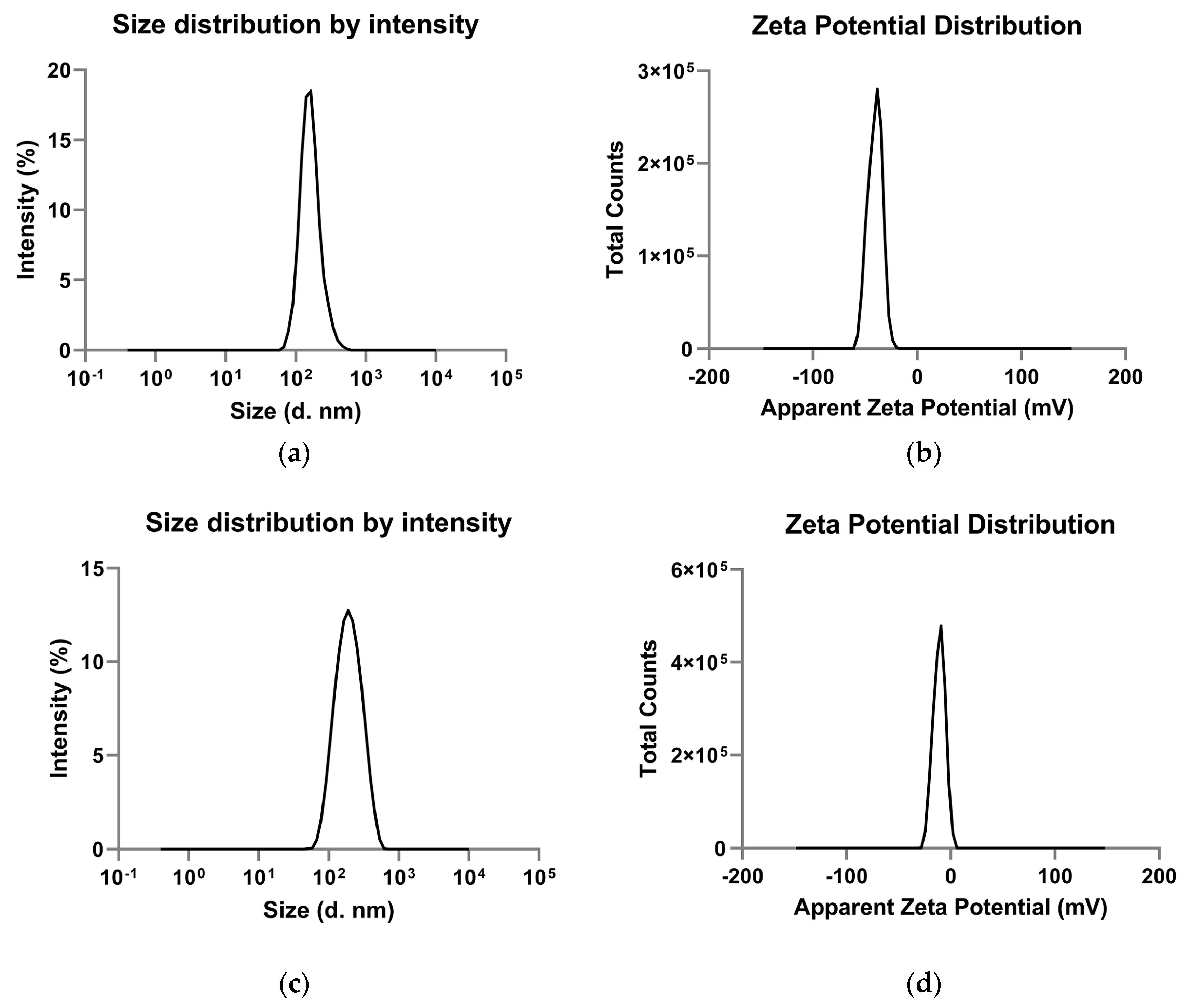
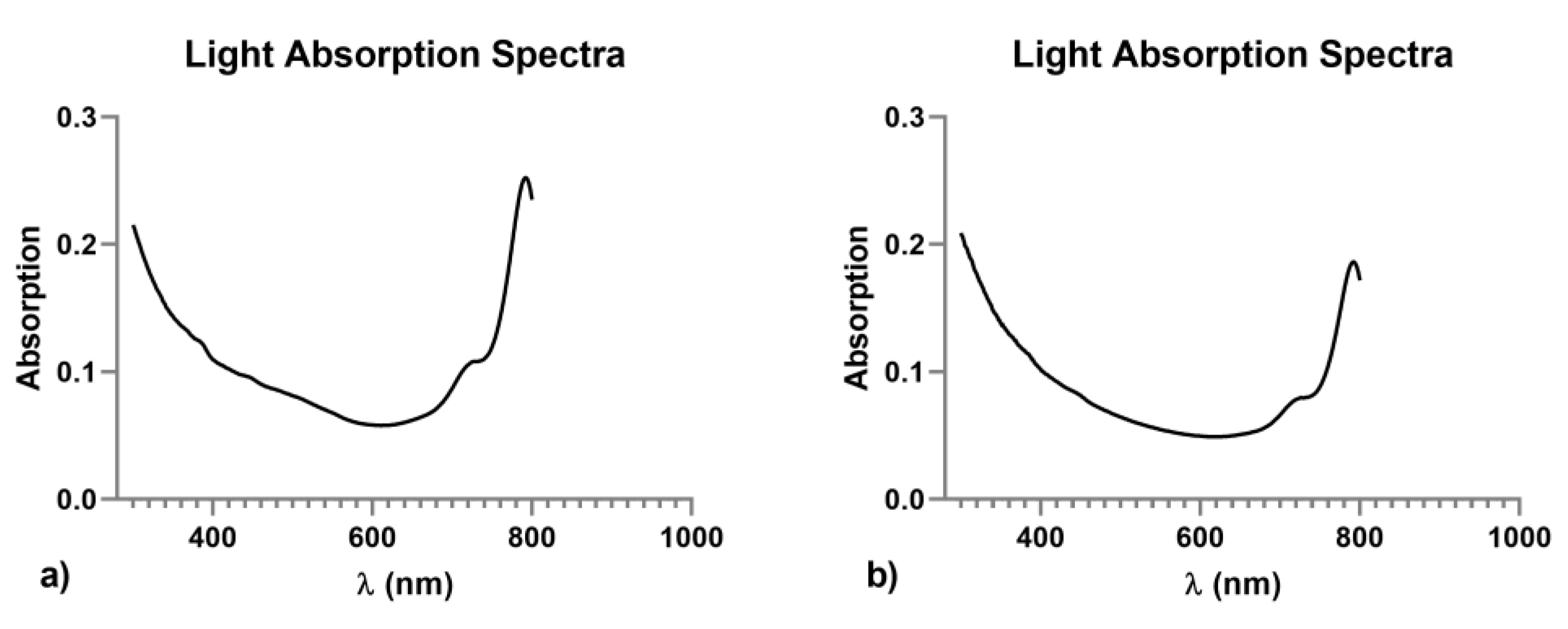
3.1. Characterization of Native and VEGF Functionalized PLGA-NPs
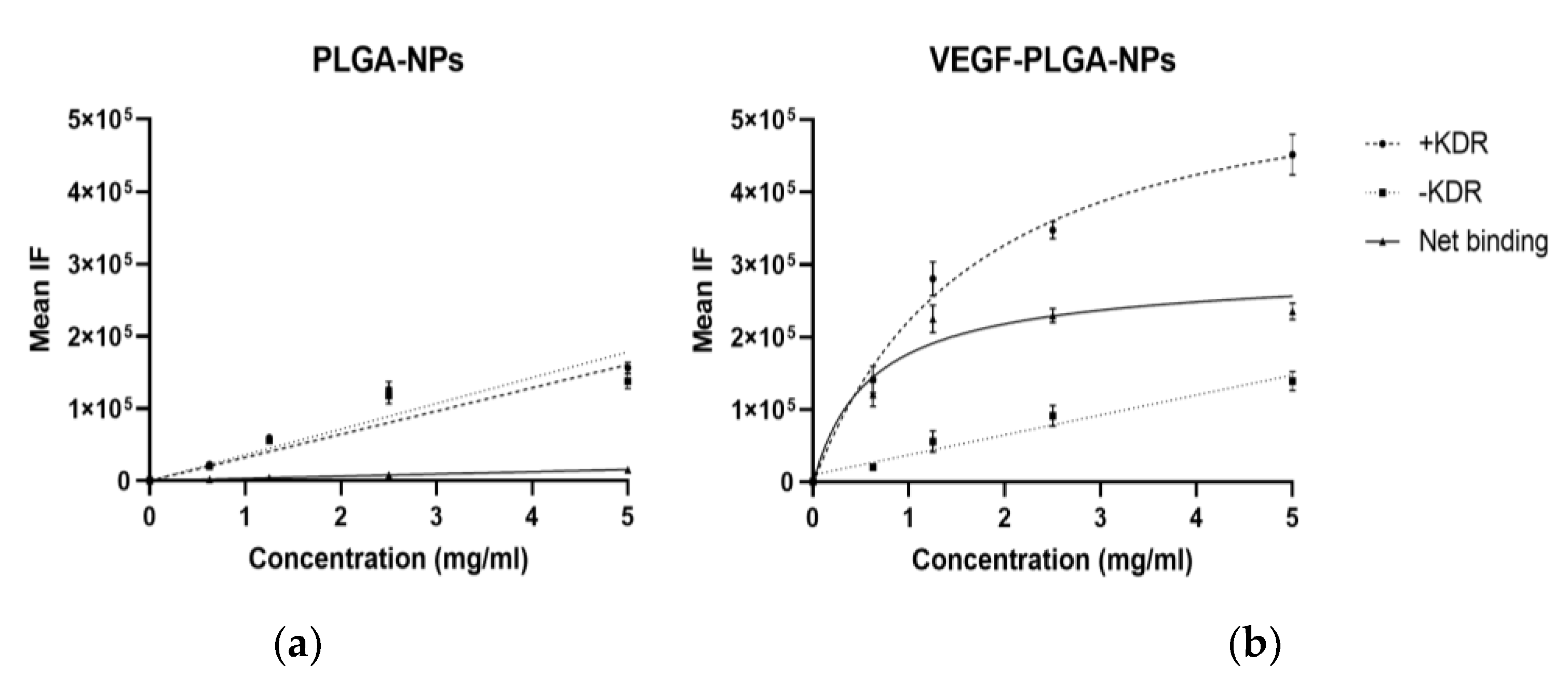
3.2. In Vitro Binding of PLGA-NPs and VEGF-PLGA-NPs to KDR-Fc
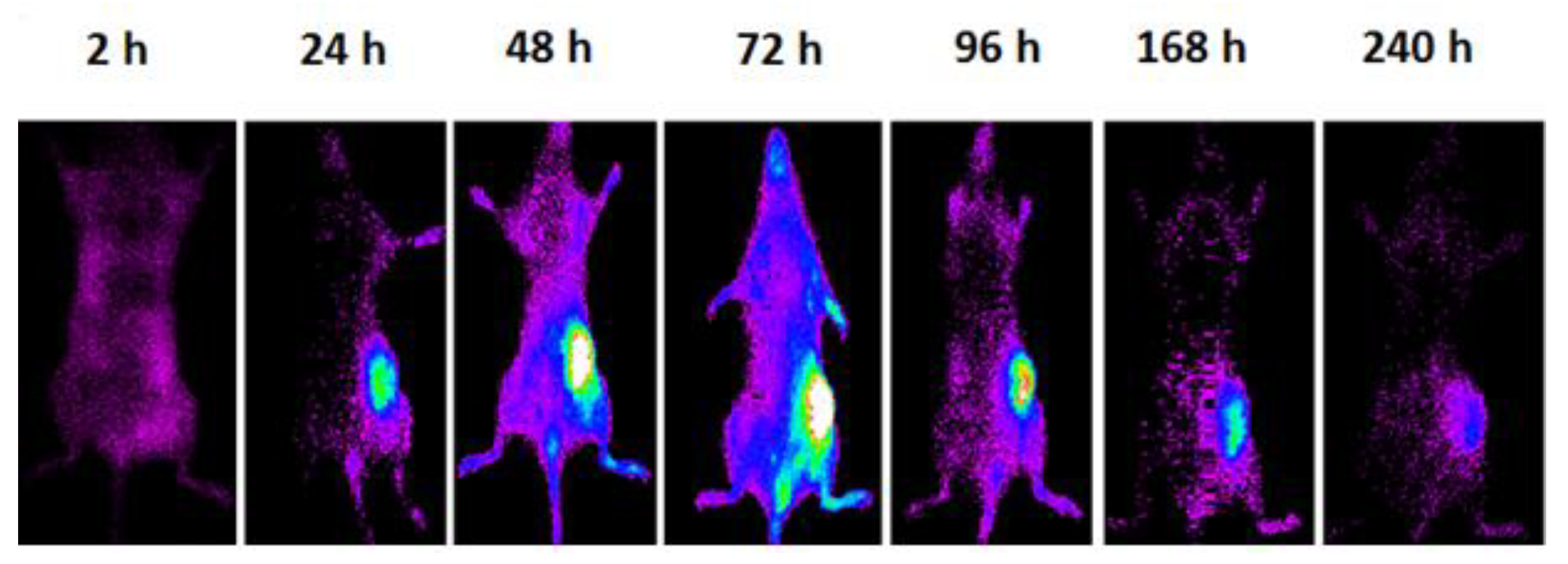
3.3. In Vivo Studies
Pharmacokinetic and Tumor Targeting of PLGA-NPs
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yohan, D.; Chithrani, B.D. Applications of nanoparticles in nanomedicine. J. Biomed. Nanotechnol. 2014, 10, 2371–2392. [Google Scholar] [CrossRef] [PubMed]
- ASTM E2456-06 (2012), Standard Terminology Relating to Nanotechnology. ASTM International. Available online: www.astm.org (accessed on 1 May 2012).
- Chu, K.S.; Hasan, W.; Rawal, S.; Walsh, M.D.; Enlow, E.M.; Luft, J.C.; Bridges, A.S.; Kuijer, J.L.; Napier, M.E.; Zamboni, W.C.; et al. Plasma, tumor and tissue pharmacokinetics of Docetaxel delivered via nanoparticles of different sizes and shapes in mice bearing SKOV-3 human ovarian carcinoma xenograft. Nanomedicine 2013, 9, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Zeng, X.; Liu, T.; Wang, Z.; Xiong, Q.; Ouyang, C.; Huang, L.; Mei, L. Docetaxel-loaded nanoparticles based on star-shaped mannitol-core PLGA-TPGS diblock copolymer for breast cancer therapy. Acta Biomater. 2013, 9, 8910–8920. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Valencia, P.M.; Zhang, L.; Langer, R.; Farokhzad, O.C. Polymeric nanoparticles for drug delivery. Methods Mol. Biol. 2010, 624, 163–175. [Google Scholar] [PubMed]
- Dinarvand, R.; Sepehri, N.; Manoochehri, S.; Rouhani, H.; Atyabi, F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int. J. Nanomed. 2011, 6, 877–895. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.; Islam, J.; Thurecht, J.K.; Corrie, S.R. Nanoparticle-based medicines: A review of FDA-approved materials and clinical trials to date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef]
- Kulkarni, S.A.; Feng, S.S. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm. Res. 2013, 30, 2512–2522. [Google Scholar] [CrossRef]
- Greish, K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol. Biol. 2010, 624, 25–37. [Google Scholar]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Shields, A.F.; Siegel, B.A.; Miller, K.D.; Krop, I.; Ma, C.X.; LoRusso, P.M.; Munster, P.N.; Campbell, K.; Gaddy, D.F.; et al. 64Cu-MM-302 Positron Emission Tomography Quantifies Variability of Enhanced Permeability and Retention of Nanoparticles in Relation to Treatment Response in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 4190–4202. [Google Scholar] [CrossRef] [PubMed]
- Baetke, S.C.; Lammers, T.; Kiessling, F. Applications of nanoparticles for diagnosis and therapy of cancer. Br. J. Radiol. 2015, 88, 20150207. [Google Scholar] [CrossRef]
- Bi, Y.; Hao, F.; Yan, G.; Teng, L.; Lee, R.J.; Xie, J. Actively Targeted Nanoparticles for Drug Delivery to Tumor. Curr. Drug Metab. 2016, 17, 763–782. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Taurone, S.; Galli, F.; Signore, A.; Agostinelli, E.; Dierckx, R.A.J.O.; Minni, A.; Pucci, M.; Artico, M. VEGF in nuclear medicine: Clinical application in cancer and future perspectives. Int. J. Oncol. 2016, 49, 437–447. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ntziachristos, V.; Bremer, C.; Weissleder, R. Fluorescence imaging with near-infrared light: New technological advances that enable in vivo molecular imaging. Eur. Radiol. 2003, 13, 195–208. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine, Basic Local Alignment Search (BLAST®). Entrez Gene IDs: 7422 (Human); 22339 (Mouse). Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 3 April 2020).
- Byrne, A.M.; Bouchier-Hayes, D.J.; Harmey, J.H. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF). J. Cell Mol. Med. 2005, 9, 777–794. [Google Scholar] [CrossRef]
- Tian, J.; Min, Y.; Rodgers, Z.; Au, K.M.; Hagan, C.T.; Zhang, M.; Roche, K.; Yang, F.; Wagner, K.; Wang, A.Z. Co-delivery of paclitaxel and cisplatin with biocompatible PLGA-PEG nanoparticles enhances chemoradiotherapy in non-small cell lung cancer models. J. Mater. Chem. B 2017, 5, 6049–6057. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Mita, M.M.; Ramanathan, R.K.; Weiss, G.J.; Mita, A.C.; Lo Russo, P.M.; Burris, H.A.; Hart, L.L.; Low, S.C.; Parsons, D.M.; et al. Phase I Study of PSMA-Targeted Docetaxel-Containing Nanoparticle BIND-014 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 3157–3163. [Google Scholar] [CrossRef]
- Varani, M.; Galli, F.; Auletta, S.; Signore, A. Radiolabelled nanoparticles for cancer diagnosis. Clin. Transl. Imaging 2018, 6, 271–292. [Google Scholar] [CrossRef]
- Pillai, G.J.; Greeshma, M.M.; Menon, D. Impact of poly(lactic-co-glycolic acid) nanoparticle surface charge on protein, cellular and haematological interactions. Colloids Surf. B Biointerfaces 2015, 136, 1058–1066. [Google Scholar] [CrossRef]
- Fornaguera, C.; Calderó, G.; Mitjans, M.; Vinardell, M.P.; Solans, C.; Vauthier, C. Interactions of PLGA nanoparticles with blood components: Protein adsorption, coagulation, activation of the complement system and hemolysis studies. Nanoscale 2015, 7, 6045–6058. [Google Scholar] [CrossRef]
- Karra, N.; Nassar, T.; Ripin, A.N.; Schwob, O.; Borlak, J.; Benita, S. Antibody conjugated PLGA nanoparticles for targeted delivery of paclitaxel palmitate: Efficacy and biofate in a lung cancer mouse model. Small 2013, 9, 4221–4236. [Google Scholar] [CrossRef] [PubMed]
- Jahan, S.T.; Sadat, S.M.A.; Walliser, M.; Haddadi, A. Targeted Therapeutic Nanoparticles: An Immense Promise to Fight against Cancer. J. Drug Deliv. 2017, 2017, 9090325. [Google Scholar] [CrossRef] [PubMed]
- Weidner, N.; Carroll, P.R.; Flax, J.; Blumenfeld, W.; Folkman, J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am. J. Pathol. 1993, 143, 401–409. [Google Scholar] [PubMed]
- Fearnley, G.W.; Smith, G.A.; Abdul-Zani, I.; Yuldasheva, N.; Mughal, N.A.; Homer-Vanniasinkam, S.; Kearney, M.T.; Zachary, I.C.; Tomlinson, D.C.; Harrison, M.A.; et al. VEGF-A isoforms program differential VEGFR2 signal transduction, trafficking and proteolysis. Biol. Open 2016, 5, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Rydén, L.; Linderholm, B.; Nielsen, N.H.; Emdin, S.; Jönsson, P.E.; Landberg, G. Tumor specific VEGF-A and VEGFR2/KDR protein are co-expressed in breast cancer. Breast Cancer Res. Treat. 2003, 82, 147–154. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Novotny, W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 2005, 333, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lupo, G.; Caporarello, N.; Olivieri, M.; Cristaldi, M.; Motta, C.; Bramanti, V.; Avola, R.; Salmeri, M.; Nicoletti, F.; Anfuso, C.D. Anti-angiogenic Therapy in Cancer: Downsides and New Pivots for Precision Medicine. Front Pharm. 2017, 7, 519. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Sela, E.; Chorny, M.; Koroukhov, N.; Danenberg, H.D.; Golomb, G. A new double emulsion solvent diffusion technique for encapsulating hydrophilic molecules in PLGA nanoparticles. J. Control. Release 2009, 133, 90–95. [Google Scholar] [CrossRef]
- Xie, H.; Smith, J.W. Fabrication of PLGA nanoparticles with a fluidic nanoprecipitation system. J. Nanobiotechnol. 2010, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Karal-Yılmaz, O.; Serhatlı, M.; Baysal, K.; Baysal, B.M. Preparation and in vitro characterization of vascular endothelial growth factor (VEGF)-loaded poly (D,L-lactic-co-glycolic acid) microspheres using a double emulsion/solvent evaporation technique. J. Microencapsul. 2011, 28, 46–54. [Google Scholar] [CrossRef]
- Rui, J.; Dadsetan, M.; Runge, M.B.; Spinner, R.J.; Yaszemski, M.J.; Windebank, A.J.; Wang, H. Controlled release of vascular endothelial growth factor using poly-lactic-co-glycolic acid microspheres: In vitro characterization and application in polycaprolactone fumarate nerve conduits. Acta Biomater. 2012, 8, 511–518. [Google Scholar] [CrossRef]
- Jiang, X.; Lin, H.; Jiang, D.; Xu, G.; Fang, X.; He, L.; Xu, M.; Tang, B.; Wang, Z.; Cui, D.; et al. Co-delivery of VEGF and bFGF via a PLGA nanoparticle-modified BAM for effective contracture inhibition of regenerated bladder tissue in rabbits. Sci. Rep. 2016, 6, 20784. [Google Scholar] [CrossRef]
- Shi, Y.; Zhou, M.; Zhang, J.; Lu, W. Preparation and cellular targeting study of VEGF-conjugated PLGA nanoparticles. J. Microencapsul. 2015, 32, 699–704. [Google Scholar] [CrossRef]
- Kumar, A.; Dixit, C.K. Methods for characterization of nanoparticles. Advances in Nanomedicine for the Delivery of Therapeutic Nucleic Acids; Woodhead Publishing: Sawston, UK; Cambridge, UK, 2017; pp. 43–58. [Google Scholar]
- Coll, J.L. Cancer optical imaging using fluorescent nanoparticles. Nanomedicine 2011, 6, 7–10. [Google Scholar] [CrossRef]
- Weissleder, R.; Pittet, M.J. Imaging in the era of molecular oncology. Nature 2008, 452, 580–589. [Google Scholar] [CrossRef]
- Raja, C.; Graham, P.; Rizvi, S.; Song, E.; Goldsmith, H.; Thompson, J.; Bosserhoff, A.; Morgenstern, A.; Apostolidis, C.; Kearsley, J.; et al. Interim analysis of toxicity and response in phase 1 trial of systemic targeted alpha therapy for metastatic melanoma. Cancer Biol. Ther. 2007, 6, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Artico, M.; Taurone, S.; Manni, S.; Bianchi, E.; Piaggio, G.; Weintraub, B.D.; Szkudlinski, M.W.; Agostinelli, E.; Dierckx, R.A.J.O.; et al. Radiolabeling of VEGF165 with 99mTc to evaluate VEGFR expression in tumor angiogenesis. Int. J. Oncol. 2017, 50, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Das, M.; Swarnakar, N.K.; Jain, S. Engineered PLGA nanoparticles: An emerging delivery tool in cancer therapeutics. Crit. Rev. Ther. Drug Carrier Syst. 2011, 28, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.M.; Wang, X.; Marin-Muller, C.; Wang, H.; Lin, P.H.; Yao, Q.; Chen, C. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev. Mol. Diagn. 2009, 9, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Pratiwi, F.W.; Kuo, C.W.; Chen, B.C.; Chen, P. Recent advances in the use of fluorescent nanoparticles for bioimaging. Nanomedicine 2019, 14, 1759–1769. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLGA-NPs Mean ± SD | VEGF-PLGA-NPs Mean ± SD | t Test (p) | |
|---|---|---|---|
| Zeta average (nm) | 180.2 ± 17.08 | 173.03 ± 7.39 | n.s. |
| Polydispersity index | 0.25 ± 0.02 | 0.17 ± 0.01 | 0.01 |
| Mean intensity (nm) | 169.73 ± 15.10 | 208.60 ± 4.97 | 0.03 |
| Zeta potential (mV) | −37.6 ± 0.67 | −9.43 ± 0.25 | 0.0001 |
| Parameter | 2 h | 24 h | 48 h | 72 h | p |
|---|---|---|---|---|---|
| Mean ± SD (95% CI) | Mean ± SD (95% CI) | Mean ± SD (95% CI) | Mean ± SD (95% CI) | ||
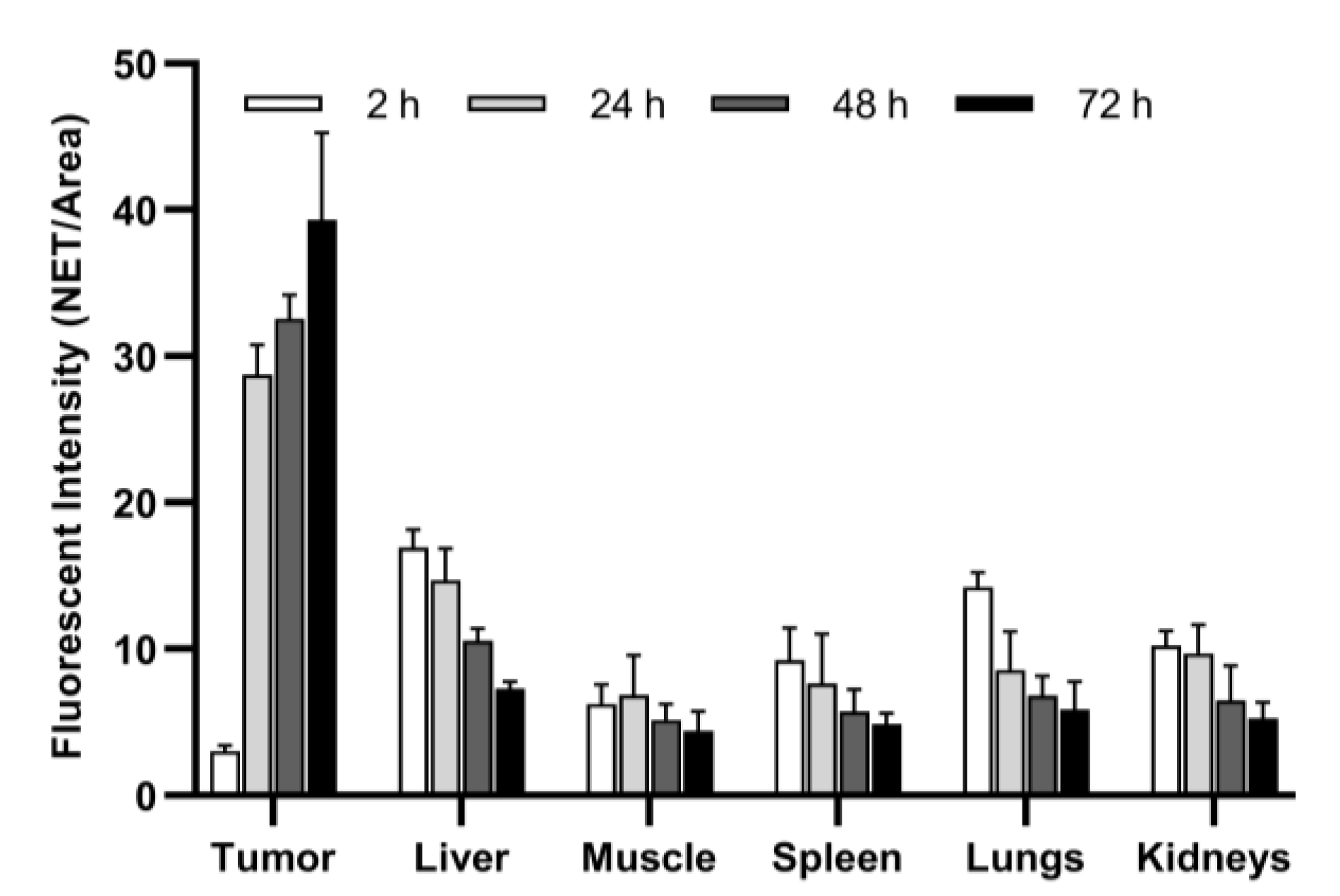
| Tumor * | 3.03 ± 0.37 (2.58 to 3.49) | 28.75 ± 2.02 (26.23 to 31.26) | 32.58 ± 1.62 (30.57 to 34.59) | 39.32 ± 5.95 (31.92 to 46.71) | <0.0001 |
| Liver ** | 16.94 ± 1.19 (15.47 to 18.41) | 14.67 ± 2.19 (11.94 to 17.39) | 10.56 ± 0.85 (9.50 to 11.61) | 7.26 ± 0.51 (6.62 to 7.89) | <0.0001 |
| Muscle ** | 6.23 ± 1.34 (4.57 to 7.89) | 6.83 ± 2.72 (3.46 to 10.21) | 5.16 ± 1.05 (3.86 to 6.46) | 4.39 ± 1.34 (2.73 to 6.05) | n.s. |
| Spleen | 9.25 ± 2.19 (6.53 to 11.96) | 7.64 ± 3.38 (3.44 to 11.84) | 5.73 ± 1.52 (3.84 to 7.62) | 4.84 ± 0.75 (3.90 to 5.77) | 0.02 |
| Lungs | 14.22 ± 1.01 (12.96 to 15.48) | 8.56 ± 2.64 (5.28 to 11.85) | 6.83 ± 1.29 (5.22 to 8.44) | 5.86 ± 1.92 (3.47 to 8.25) | <0.0001 |
| Kidneys | 10.21 ± 1.01 (8.96 to 11.46) | 9.70 ± 1.96 (7.26 to 12.14) | 6.49 ± 2.35 (3.58 to 9.40) | 5.28 ± 1.08 (3.93 to 6.62) | 0.0006 |
| Parameter | PLGA-NPs | VEGF-PLGA-NPs | t Test (p) |
|---|---|---|---|
| Mean ± SD (95% CI) | Mean ± SD (95% CI) | ||
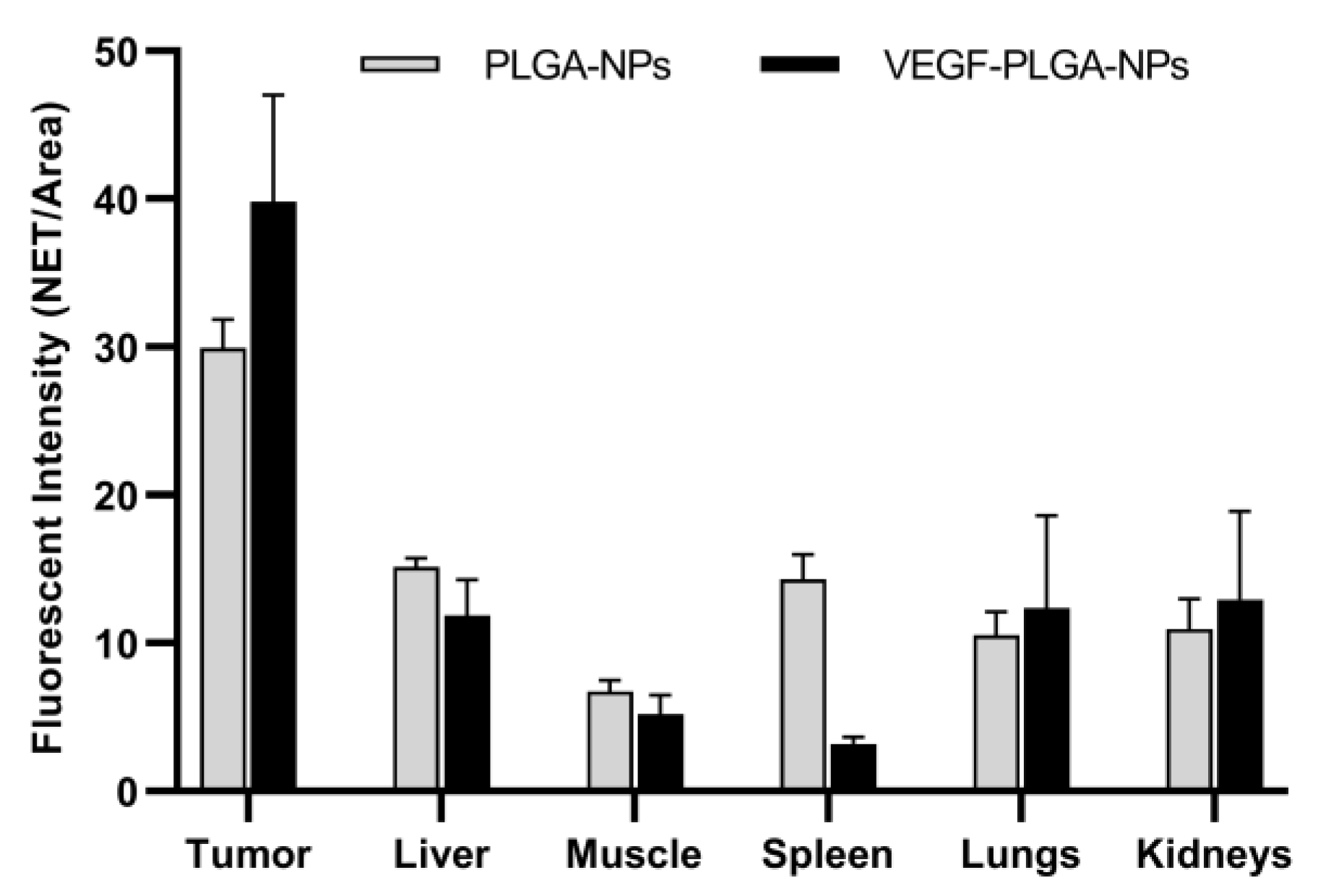
| Tumor ° | 29.95 ± 1.92 (27.56 to 32.33) | 39.83 ± 7.17 (30.92 to 48.74) | 0.03 |
| Liver ° | 15.17 ± 0.55 (14.48 to 15.85) | 11.86 ± 2.42 (5.84 to 17.88) | n.s. |
| Muscle | 6.73 ± 0.73 (5.82 to 7.65) | 5.18 ± 1.31 (3.56 to 6.80) | n.s. |
| Spleen ° | 14.29 ± 1.71 (12.18 to 16.41) | 3.20 ± 0.44 (2.50 to 3.89) | <0.0001 |
| Lungs ° | 10.53 ± 1.59 (8.55 to 12.50) | 12.37 ± 6.24 (4.62 to 20.12) | n.s. |
| Kidneys | 10.96 ± 2.03 (8.43 to 13.49) | 12.92 ± 5.99 (5.48 to 20.36) | n.s. |
| T/M * | 4.49 ± 0.54 (3.81 to 5.17) | 7.90 ± 1.61 (5.90 to 9.90) | 0.0003 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varani, M.; Galli, F.; Capriotti, G.; Mattei, M.; Cicconi, R.; Campagna, G.; Panzuto, F.; Signore, A. Theranostic Designed Near-Infrared Fluorescent Poly (Lactic-co-Glycolic Acid) Nanoparticles and Preliminary Studies with Functionalized VEGF-Nanoparticles. J. Clin. Med. 2020, 9, 1750. https://doi.org/10.3390/jcm9061750
Varani M, Galli F, Capriotti G, Mattei M, Cicconi R, Campagna G, Panzuto F, Signore A. Theranostic Designed Near-Infrared Fluorescent Poly (Lactic-co-Glycolic Acid) Nanoparticles and Preliminary Studies with Functionalized VEGF-Nanoparticles. Journal of Clinical Medicine. 2020; 9(6):1750. https://doi.org/10.3390/jcm9061750
Chicago/Turabian StyleVarani, Michela, Filippo Galli, Gabriela Capriotti, Maurizio Mattei, Rosella Cicconi, Giuseppe Campagna, Francesco Panzuto, and Alberto Signore. 2020. "Theranostic Designed Near-Infrared Fluorescent Poly (Lactic-co-Glycolic Acid) Nanoparticles and Preliminary Studies with Functionalized VEGF-Nanoparticles" Journal of Clinical Medicine 9, no. 6: 1750. https://doi.org/10.3390/jcm9061750
APA StyleVarani, M., Galli, F., Capriotti, G., Mattei, M., Cicconi, R., Campagna, G., Panzuto, F., & Signore, A. (2020). Theranostic Designed Near-Infrared Fluorescent Poly (Lactic-co-Glycolic Acid) Nanoparticles and Preliminary Studies with Functionalized VEGF-Nanoparticles. Journal of Clinical Medicine, 9(6), 1750. https://doi.org/10.3390/jcm9061750