Antithrombotic Therapy in Hereditary Hemorrhagic Telangiectasia: Real-World Data from the Gemelli Hospital HHT Registry

 ,
,  , , ,
, , , 
 and
and
Abstract
1. Introduction
2. Materials and Methods
Statistical Analysis
3. Results
3.1. Characteristics of the Study Population
3.2. Antithrombotic Therapy (AT): Indications, Type, Dosage, Duration, and Reasons for Therapy Discontinuation
3.3. AT: Effectiveness and Safety
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Shovlin, C.L.; Buscarini, E.; Kjeldsen, A.D.; Mager, H.J.; Sabba, C.; Droege, F.; Geisthoff, U.; Ugolini, S.; Dupuis-Girod, S. European reference network for rare vascular diseases (VASCERN) outcome measures for hereditary haemorrhagic telangiectasia (HHT). Orphanet J. Rare Dis. 2018, 13, 136. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.; Bayrak-Toydemir, P.; Pyeritz, E.R. Hereditary hemorrhagic telangiectasia: An overview of diagnosis, management, and pathogenesis. Genet. Med. 2011, 13, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Sharathkumar, A.A.; Shapiro, A. Hereditary haemorrhagic telangiectasia. Haemophilia 2008, 14, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, N.L.; Govani, F.S.; Jackson, J.E.; Begbie, M.E.; Shovlin, C.L. Elevated factor VIII in hereditary haemorrhagic telangiectasia (HHT): Association with venous thromboembolism. Thromb. Haemost. 2007, 98, 1031–1039. [Google Scholar] [CrossRef]
- Livesey, J.A.; Manning, R.A.; Meek, J.H.; Jackson, J.E.; Kulinskaya, E.; Laffan, M.A.; Shovlin, C.L. Low serum iron levels are associated with elevated plasma levels of coagulation factor VIII and pulmonary emboli/deep venous thromboses in replicate cohorts of patients with hereditary haemorrhagic telangiectasia. Thorax 2011, 67, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Buscarini, E.; Leandro, G.; Conte, D.; Danesino, C.; Daina, E.; Manfredi, G.; Lupinacci, G.; Brambilla, G.; Menozzi, F.; de Grazia, F.; et al. Natural history and outcome of hepatic vascular malformations in a large cohort of patients with hereditary hemorrhagic teleangiectasia. Dig. Dis. Sci. 2011, 56, 2166–2178. [Google Scholar] [CrossRef]
- Ginon, I.; Decullier, E.; Finet, G.; Cordier, J.-F.; Marion, D.; Saurin, J.-C.; Dupuis-Girod, S. Hereditary hemorrhagic telangiectasia, liver vascular malformations and cardiac consequences. Eur. J. Intern. Med. 2013, 24, e35–e39. [Google Scholar] [CrossRef]
- Shovlin, C.L.; Awan, I.; Cahilog, Z.; Abdulla, F.N.; Guttmacher, A.E. Reported cardiac phenotypes in hereditary hemorrhagic telangiectasia emphasize burdens from arrhythmias, anemia and its treatments, but suggest reduced rates of myocardial infarction. Int J. Cardiol. 2016, 215, 179–185. [Google Scholar] [CrossRef]
- Schünemann, H.J.; Cushman, M.; Burnett, A.E.; Kahn, S.R.; Beyer-Westendorf, J.; Spencer, F.A.; Rezende, S.M.; Zakai, N.A.; Bauer, K.A.; Dentali, F.; et al. American society of hematology 2018 guidelines for management of venous thromboembolism: Prophylaxis for hospitalized and nonhospitalized medical patients. Blood Adv. 2018, 2, 3198–3225. [Google Scholar] [CrossRef]
- Anderson, D.R.; Morgano, G.P.; Bennett, C.; Dentali, F.; Francis, C.W.; Garcia, D.A.; Kahn, S.R.; Rahman, M.; Rajasekhar, A.; Rogers, F.B.; et al. American society of hematology 2019 guidelines for management of venous thromboembolism: Prevention of venous thromboembolism in surgical hospitalized patients. Blood Adv. 2019, 3, 3898–3944. [Google Scholar] [CrossRef]
- Shovlin, C.L.; Millar, C.M.; Droege, F.; Kjeldsen, A.; Manfredi, G.; Suppressa, P.; Ugolini, S.; Coote, N.; Fialla, A.D.; Geisthoff, U.; et al. Safety of direct oral anticoagulants in patients with hereditary hemorrhagic telangiectasia. Orphanet J. Rare Dis. 2019, 14, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Riera-Mestre, A.; Mora-Luján, J.M.; Trujillo-Santos, J.; Toro, J.D.; Nieto, J.A.; Pedrajas, J.M.; López-Reyes, R.; Soler, S.; Ballaz, A.; Cerdà, P.; et al. Natural history of patients with venous thromboembolism and hereditary hemorrhagic telangiectasia. Findings from the RIETE registry. Orphanet J. Rare Dis. 2019, 14, 196. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, E.; Agostini, F.; Porfidia, A.; Giarretta, I.; Feliciani, D.; Martino, L.D.; Tortora, A.; Gasbarrini, A.; Pola, R.; Passali, G.; et al. Safety of antithrombotic therapy in subjects with hereditary hemorrhagic telangiectasia: Prospective data from a multidisciplinary working group. Orphanet J. Rare Dis. 2019, 14, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Guttmacher, A.E.; Buscarini, E.; Faughnan, M.E.; Hyland, R.H.; Westermann, C.J.J.; Kjeldsen, A.D.; Plauchu, H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am. J. Med. Genet. 2000, 91, 66–67. [Google Scholar] [CrossRef]
- Schulman, S.; Kearon, C. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J. Thromb. Haemost. 2005, 3, 692–694. [Google Scholar] [CrossRef]
- Kaatz, S.; Ahmad, D.; Spyropoulos, A.C.; Schulman, S.; Anticoagulation, F.T.S.O.C.O. Definition of clinically relevant non-major bleeding in studies of anticoagulants in atrial fibrillation and venous thromboembolic disease in non-surgical patients: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2015, 13, 2119–2126. [Google Scholar] [CrossRef]
- Lesca, G.; Olivieri, C.; Burnichon, N.; Pagella, F.; Carette, M.-F.; Gilbert-Dussardier, B.; Goizet, C.; Roume, J.; Rabilloud, M.; Saurin, J.-C.; et al. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia: Data from the French-Italian HHT network. Genet. Med. 2007, 9, 14–22. [Google Scholar] [CrossRef]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.-C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur. J. Cardiothorac. Surg. 2016, 50, e1–e88. [Google Scholar] [CrossRef]
- Steffel, J.; Verhamme, P.; Potpara, T.S.; Albaladejo, P.; Antz, M.; Desteghe, L.; Haeusler, K.G.; Oldgren, J.; Reinecke, H.; Roldán-Schilling, V.; et al. The 2018 European heart rhythm association practical guide on the use of non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation. Eur. Hear. J. 2018, 39, 1330–1393. [Google Scholar] [CrossRef]
- Yao, X.; Shah, N.D.; Sangaralingham, L.R.; Gersh, B.J.; Noseworthy, P.A. Non–Vitamin K antagonist oral anticoagulant dosing in patients with atrial fibrillation and renal dysfunction. J. Am. Coll. Cardiol. 2017, 69, 2779–2790. [Google Scholar] [CrossRef]
- Shovlin, C.L.; Condliffe, R.; Donaldson, J.W.; Kiely, D.G.; Wort, S.J. British thoracic society clinical statement on pulmonary arteriovenous malformations. Thorax 2017, 72, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.P.; Shehata, N.; Faughnan, M.E. Hereditary hemorrhagic telangiectasia patients can tolerate anticoagulation. Ann. Hematol. 2012, 91, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Devlin, H.L.; Hosman, A.E.; Shovlin, C.L. Antiplatelet and anticoagulant agents in hereditary hemorrhagic telangiectasia. New Engl. J. Med. 2013, 368, 876–878. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Mean age (years ± SD) | 59.3 ± 17.0 |
| Gender (male/female ratio) | 14/12 |
| HHT1/HHT2/Clinical diagnosis (n) | 9/15/2 |
| Epistaxis (n/total) | 26/26 |
| Mucocutaneous telangiectases (n/total) | 23/26 |
| Family history of HHT (n/total) | 24/26 |
| Pulmonary AVMs (n/screened) | 10/22 |
| Hepatic AVMs (n/screened) | 5/24 |
| Cerebral AVMs (n/screened) | 2/20 |
| Gastrointestinal AVMs (n/screened) | 10/15 |
| Previous gastrointestinal bleeding (n/total) | 10/26 |
| Anticoagulant drug | Number of therapeutic courses (n = 19) |
| enoxaparin | 6 |
| fondaparinux | 4 |
| VKA | 4 |
| DOAC | 5 |
| Antiplatelet drug | Number of therapeutic courses (n = 11) |
| ASA (number of courses) | 9 |
| clopidogrel (number of courses) | 1 |
| indobufen (number of courses) | 1 |
| Reasons why anticoagulant therapy was prescribed | |
| AF | 6 |
| Heart valve bioprosthesis | 2 |
| PE | 1 |
| CVT | 1 |
| SVT | 3 |
| RAO in a patient with Horton’s disease | 1 |
| Secondary prevention after stroke/TIA in a patient with pAVMs | 1 |
| Surgical VTE prophylaxis | 4 |
| Reasons why antiplatelet therapy was prescribed | |
| Secondary prevention after stroke in patients with pAVMs | 3 |
| Secondary prevention after stroke/TIA | 3 |
| Secondary prevention after MI | 2 |
| Horton’s disease | 1 |
| Primary CV prevention | 2 |
| Drug | Genetic Mutation | Indication for Treatment | Dosage Prescribed | Treatment Duration | Ongoing Treatment at the Time of Study | Reason for Therapy Cessation | Therapy Cessation Decided by Doctor or Patient |
|---|---|---|---|---|---|---|---|
| warfarin | ACVRL1 (HHT2) | AF | variable, based on INR | 6 months | No | hematuria | doctor |
| rivaroxaban | ACVRL1 (HHT2) | AF | 15 mg o.d. | 6 months | No | GI bleeding | doctor |
| rivaroxaban | ACVRL1 (HHT2) | AF | 15 mg o.d. | 27 months | No | GI bleeding | doctor |
| rivaroxaban | ACVRL1 (HHT2) | AF | 15 mg o.d. | 13 months | Yes | --- | --- |
| dabigatran | ACVRL1 (HHT2) | AF | 110 mg b.i.d. | 21 months | Yes | --- | --- |
| apixaban | ACVRL1 (HHT2) | AF | 2.5 mg b.i.d. | 20 months | Yes | --- | --- |
| acenocoumarol | ENG (HHT1) | stroke in pAVM | variable, based on INR | 46 months | Yes | --- | --- |
| enoxaparin | ENG (HHT1) | SVT | 4000 U b.i.d. | 1 month | No | completion of treatment | doctor |
| enoxaparin | ACVRL1 (HHT2) | SVT | 4000 U b.i.d. | 1 month | No | completion of treatment | doctor |
| fondaparinux | ACVRL1 (HHT2) | SVT | 2.5 mg o.d. | 1 month | No | completion of treatment | doctor |
| warfarin | ENG (HHT1) | heart valve bioprosthesis | variable, based on INR | 3 months | No | completion of treatment | doctor |
| fondaparinux | Clinical diagnosis | heart valve bioprosthesis | 2.5 mg o.d. | 3 months | No | completion of treatment | doctor |
| warfarin | ACVRL1 (HHT2) | PE | variable, based on INR | 1 month | No | shift to DOAC | doctor |
| enoxaparin | ENG (HHT1) | RAO in Horton’s disease | 6000 U b.i.d | 5 months | No | completion of treatment | doctor |
| fondaparinux | ENG (HHT1) | CVT | 5 mg o.d. | 6 months | No | completion of treatment | doctor |
| enoxaparin | ACVRL1 (HHT2) | surgical VTE prophylaxis | 4000 U o.d. | 1 month | No | completion of treatment | doctor |
| enoxaparin | ACVRL1 (HHT2) | surgical VTE prophylaxis | 4000 U o.d. | 3 months | No | completion of treatment | doctor |
| enoxaparin | ACVRL1 (HHT2) | surgical VTE prophylaxis | 4000 U o.d. | 4 months | No | completion of treatment | doctor |
| enoxaparin | ACVRL1 (HHT2) | surgical VTE prophylaxis | 4000 U o.d. | 1 month | No | completion of treatment | doctor |
| ASA | ENG (HHT1) | stroke in pAVM | 100 mg o.d. | 43 months | No | worsening of epistaxis | patient |
| ASA | ENG (HHT1) | stroke in pAVM | 100 mg o.d. | 72 months | No | pAVM embolization | doctor |
| ASA | ENG (HHT1) | stroke in pAVM | 100 mg o.d. | 14 months | Yes | --- | --- |
| ASA | Clinical diagnosis | Stroke | 100 mg o.d. | 30 months | No | worsening of epistaxis | patient |
| ASA | ACVRL1 (HHT2) | Stroke | 100 mg o.d. | 5 months | Yes | --- | --- |
| indobufen | ENG (HHT1) | TIA | 200 mg o.d. | 21 months | Yes | --- | --- |
| ASA | ACVRL1 (HHT2) | secondary prevention after MI | 100 mg o.d. | 94 months | Yes | --- | --- |
| ASA | ACVRL1 (HHT2) | secondary prevention after MI | 100 mg o.d. | 50 months | Yes | --- | --- |
| ASA | ACVRL1 (HHT2) | primary CV prevention | 100 mg o.d. | 24 months | No | worsening of epistaxis | patient |
| ASA | ACVRL1 (HHT2) | primary CV prevention | 100 mg o.d. | 18 months | No | medical decision | doctor |
| clopidogrel | ENG (HHT1) | Horton’s disease | 75 mg o.d. | 14 months | Yes | --- | --- |
| Minor bleedings different from epistaxis (n/total of AT courses) | 0/30 |
| CRNM bleedings (n/total of AT courses) | 0/30 |
| Major bleedings (n/total of AT courses) | 3/30 |
| - in subjects taking anticoagulants (n/total of anticoagulant courses) | 3/19 |
| - in subjects taking antiplatelets (n/total of antiplatelet courses) | 0/11 |
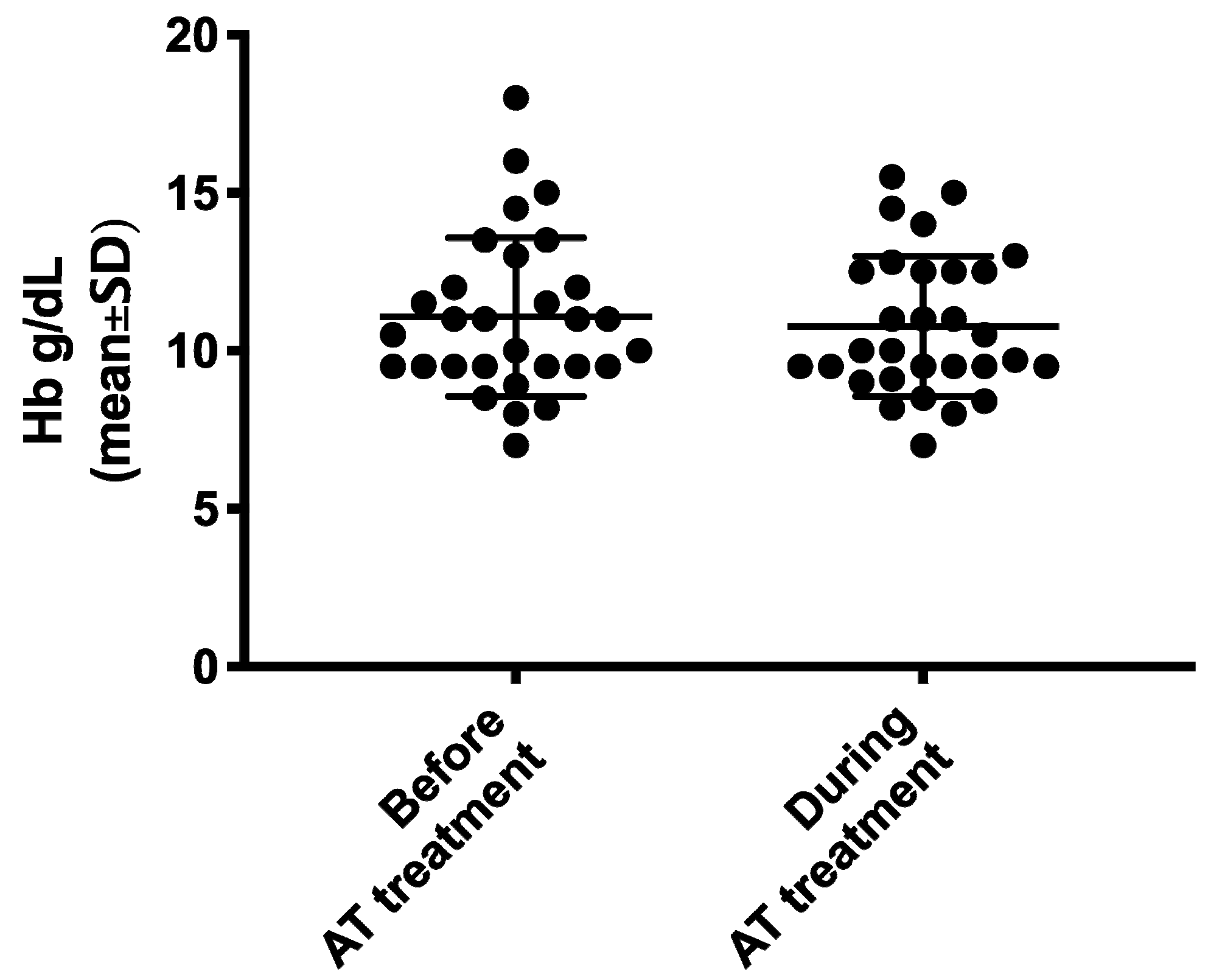
| Hb levels (g/dL) during AT versus prior to AT (mean ± SD) | 10.8 ± 2.2 vs. 11.1 ± 2.5 (95% CI −0.90–0.31) p = 0.3256 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaetani, E.; Agostini, F.; Giarretta, I.; Porfidia, A.; Di Martino, L.; Gasbarrini, A.; Pola, R.; on behalf of the Multidisciplinary Gemelli Hospital Group for HHT. Antithrombotic Therapy in Hereditary Hemorrhagic Telangiectasia: Real-World Data from the Gemelli Hospital HHT Registry. J. Clin. Med. 2020, 9, 1699. https://doi.org/10.3390/jcm9061699
Gaetani E, Agostini F, Giarretta I, Porfidia A, Di Martino L, Gasbarrini A, Pola R, on behalf of the Multidisciplinary Gemelli Hospital Group for HHT. Antithrombotic Therapy in Hereditary Hemorrhagic Telangiectasia: Real-World Data from the Gemelli Hospital HHT Registry. Journal of Clinical Medicine. 2020; 9(6):1699. https://doi.org/10.3390/jcm9061699
Chicago/Turabian StyleGaetani, Eleonora, Fabiana Agostini, Igor Giarretta, Angelo Porfidia, Luigi Di Martino, Antonio Gasbarrini, Roberto Pola, and on behalf of the Multidisciplinary Gemelli Hospital Group for HHT. 2020. "Antithrombotic Therapy in Hereditary Hemorrhagic Telangiectasia: Real-World Data from the Gemelli Hospital HHT Registry" Journal of Clinical Medicine 9, no. 6: 1699. https://doi.org/10.3390/jcm9061699
APA StyleGaetani, E., Agostini, F., Giarretta, I., Porfidia, A., Di Martino, L., Gasbarrini, A., Pola, R., & on behalf of the Multidisciplinary Gemelli Hospital Group for HHT. (2020). Antithrombotic Therapy in Hereditary Hemorrhagic Telangiectasia: Real-World Data from the Gemelli Hospital HHT Registry. Journal of Clinical Medicine, 9(6), 1699. https://doi.org/10.3390/jcm9061699