Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site

 ,
,  , , , ,
, , , ,  , ,
, , 
Abstract

1. Introduction
2. Material and Methods
2.1. Bioinformatics Analysis of the SARS-CoV-2 Spike Variability
2.2. Structural Bioinformatics, Molecular Dynamics
2.3. In Silico Structure Based Virtual Screening
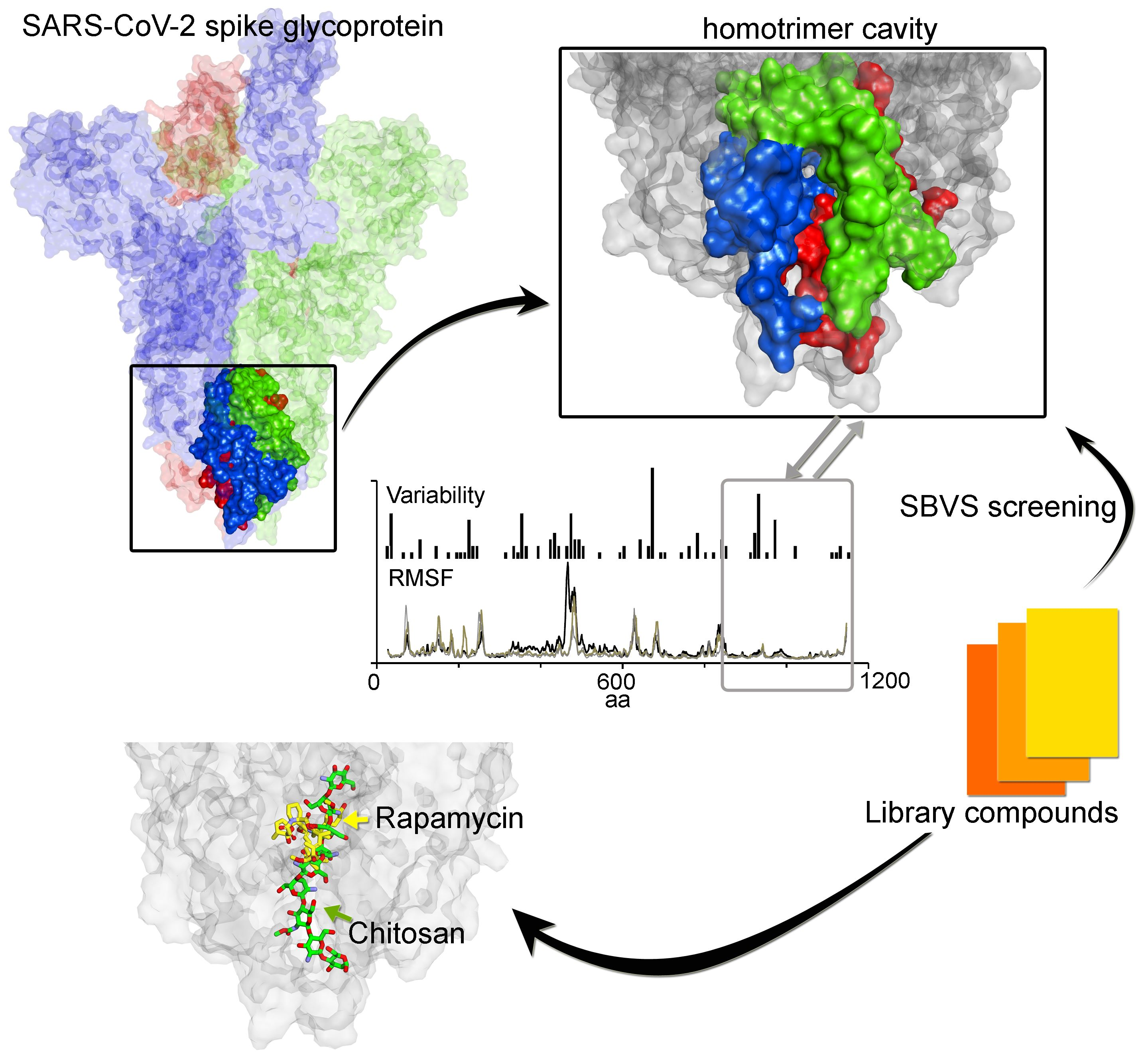
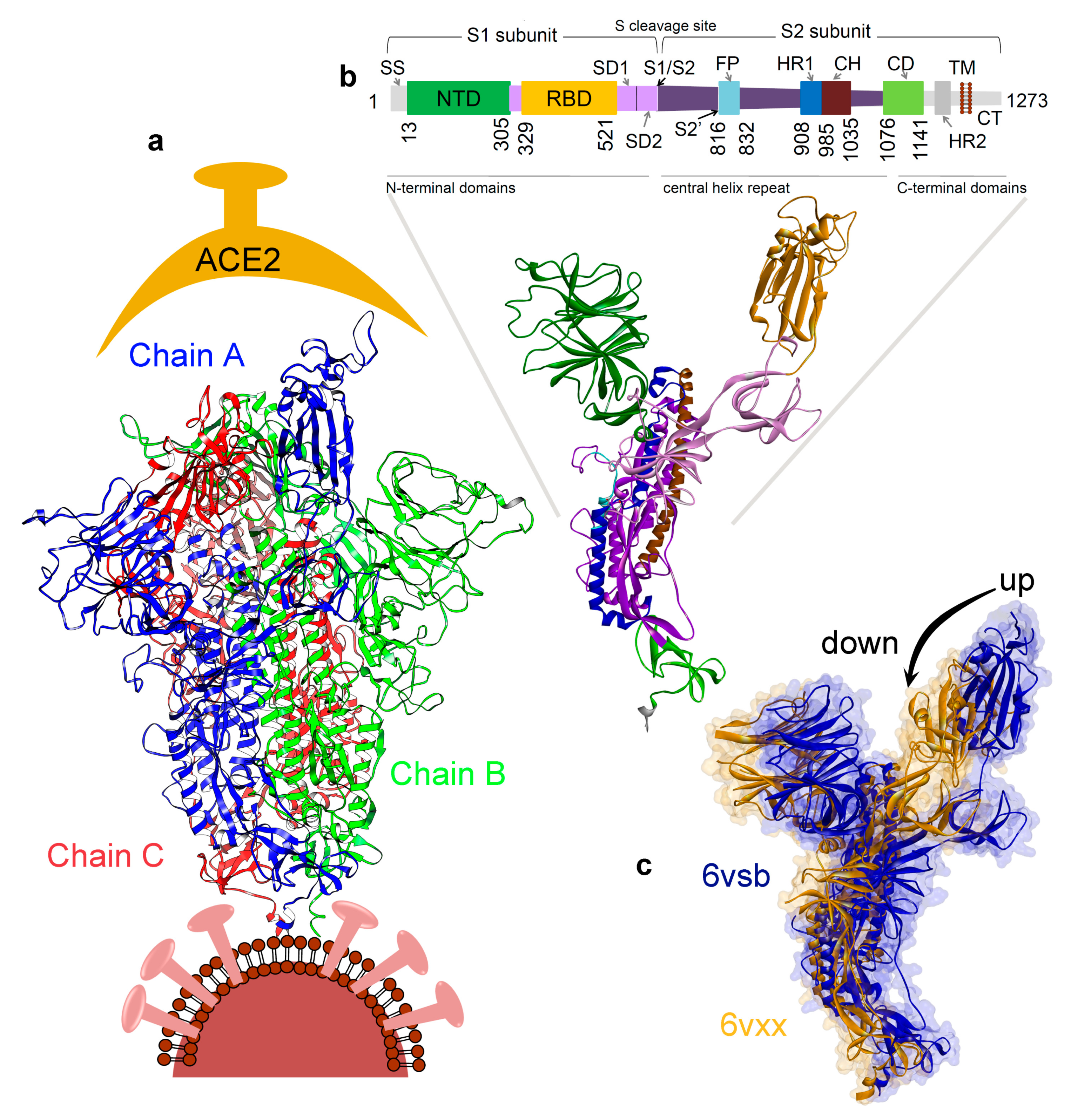
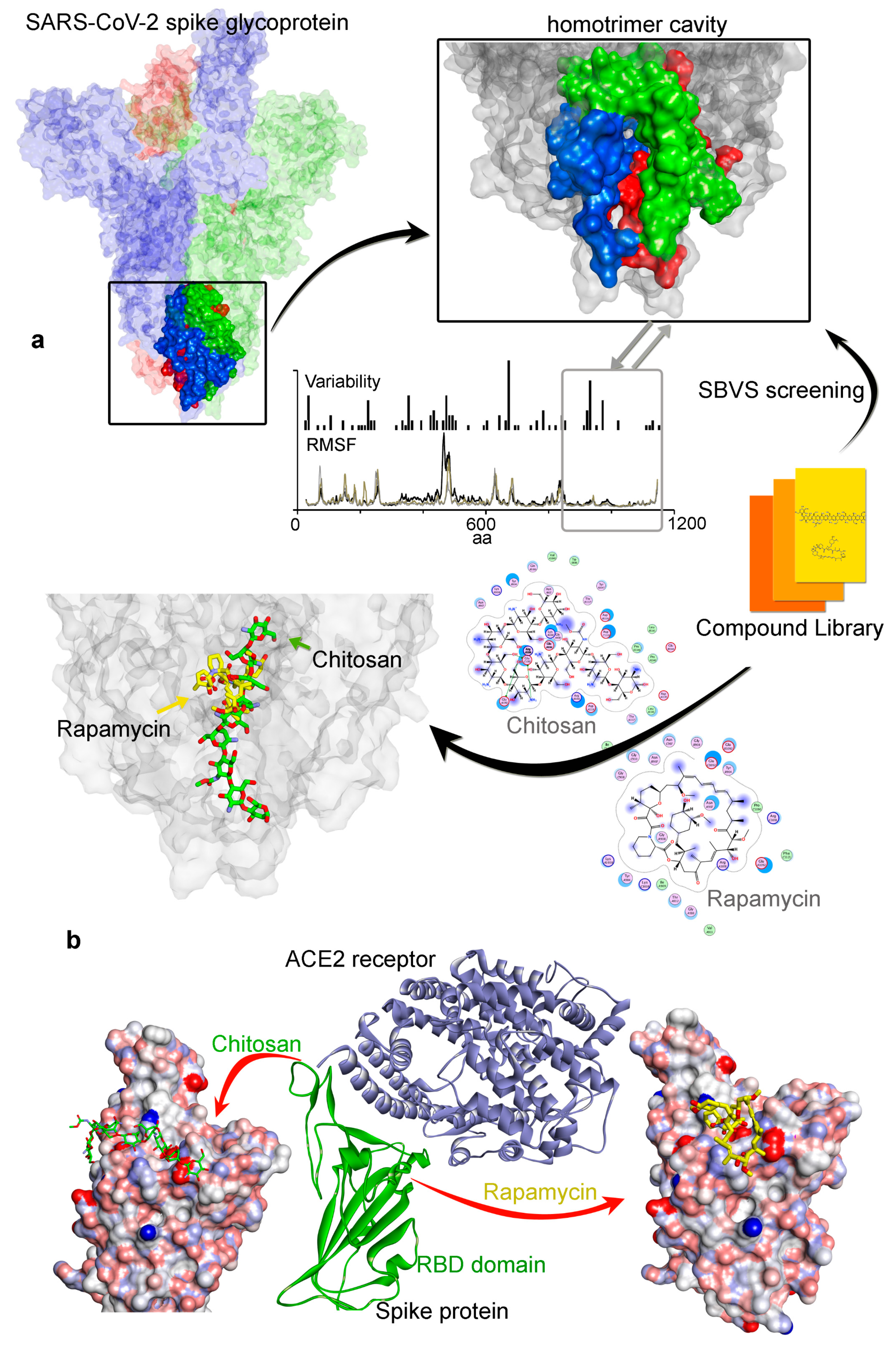
3. Results
3.1. Investigating the SARS-CoV-2 Spike Glycoprotein Reveals a Cavity with Potential Utility as a Drug-Binding Pocket
3.1.1. Variability in the Spike Glycoprotein
3.1.2. Molecular Properties and the Dynamics of the Spike Protein
3.2. Potential Druggability of the Homotrimer Cavity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chan, J.F.W.; To, K.K.W.; Tse, H.; Jin, D.Y.; Yuen, K.Y. Interspecies transmission and emergence of novel viruses: Lessons from bats and birds. Trends Microbiol. 2013, 21, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Cheng, V.C.C.; Lau, S.K.P.; Woo, P.C.Y.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 2007, 20, 660–694. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.W.; Lau, S.K.P.; To, K.K.W.; Cheng, V.C.C.; Woo, P.C.Y.; Yuen, K.Y. Middle east respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.W.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.W.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Woo, P.C.Y.; Yip, C.C.Y.; Tse, H.; Tsoi, H.W.; Cheng, V.C.C.; Lee, P.; Tang, B.S.; Cheung, C.H.; Lee, R.A.; et al. Coronavirus HKU1 and other coronavirus infections in Hong Kong. J. Clin. Microbiol. 2006, 44, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Shanmugaraj, B.; Siriwattananon, K.; Wangkanont, K.; Phoolcharoen, W. Perspectives on monoclonal antibody therapy as potential therapeutic intervention for coronavirus disease-19 (COVID-19). Asian Pac. J. Allergy Immunol. 2020, 38, 10–18. [Google Scholar]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Peihan, W.; Liu, D.; Jing, Y.; Edward, C.H.; et al. A novel bat coronavirus reveals natural insertions at the S1/S2 cleavage site of the Spike protein and a possible recombinant origin of HCoV-19. BioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, X.; Wei, X.; Li, J.; Yang, J.; Tan, H.; Zhu, J.; Zhang, Q.; Wu, J.; Liu, L. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 2020, 92, 595–601. [Google Scholar] [CrossRef]
- Smith, R.D. Responding to global infectious disease outbreaks: Lessons from SARS on the role of risk perception, communication and management. Soc. Sci. Med. 2006, 63, 3113–3123. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Chu, C.M.; Chan, K.H.; Tsoi, H.W.; Huang, Y.; Beatrice, H.L.W.; Rosana, W.S.P.; James, J.C.; Wei-kwang, L.; et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2004, 79, 884–895. [Google Scholar] [CrossRef]
- Peiris, J.S.; Lai, S.T.; Poon, L.L.; Guan, Y.; Yam, L.Y.; Lim, W.; Nicholls, J.; Yee, W.K.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef]
- Yeung, M.L.; Yao, Y.; Jia, L.; Chan, J.F.; Chan, K.H.; Cheung, K.F.; Chen, H.; Poon, V.K.; Tsang, A.K.; To, K.K.; et al. MERS coronavirus induces apoptosis in kidney and lung by upregulating Smad7 and FGF2. Nat. Microbiol. 2016, 1, 16004. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Hill, C.S.; Leist, S.R.; Alexandra, S.; Kenneth, H.D.; Stephanie, A.M.; Maria, L.A.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 and multiple endemic, epidemic and bat coronavirus. Sci. Transl. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; O’meara, M.J.; Guo, J.Z.; Swaney, D.L.; Tummino, T.A.; Hüttenhain, R.; et al. A SARS-CoV-2-Human protein–protein interaction map reveals drug targets and potential drug-repurposing. BioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Stephan, B.; Katharina, R.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Qi, Z.; Xuanling, S.; Qisheng, W.; Linqi, Z.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020. [Google Scholar] [CrossRef]
- Tchesnokov, E.; Feng, J.; Porter, D.; Götte, M. Mechanism of inhibition of Ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses 2019, 11, 326. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Zhou, G.; Zhao, Q. Perspectives on therapeutic neutralizing antibodies against the novel coronavirus SARS-CoV-2. Int. J. Biol. Sci. 2020, 16, 1718–1723. [Google Scholar] [CrossRef]
- Utomo, R.Y.; Ikawati, M.; Meiyanto, E. Revealing the potency of citrus and galangal constituents to halt SARS-CoV-2 infection. Preprints 2020, 2020030214. [Google Scholar] [CrossRef]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Ann. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; Zee, R.V.D.; Haan, C.A.M.D.; Rottier, P.J.M. The coronavirus spike protein is a Class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Quadeer, A.A.; Mckay, M.R. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Lee, S.-Y.; Kim, S.; Kim, M.J.; Kim, H.G.; Jun, S.; Seung, K.; Bum, T.K.; Park, D. Spike protein binding prediction with neutralizing antibodies of SARS-CoV-2. BioRxiv 2020. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Krüger, N.; Müller, M.; Drosten, C.; Pöhlmann, S. The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Wu, A.; Niu, P.; Wang, L.; Zhou, H.; Zhao, X.; Wang, W.; Jingfeng, W.; Chengyang, J.; Xiao, D.; Xianyue, W.; et al. Mutations, recombination and insertion in the evolution of 2019-nCoV. BioRxiv 2020. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Guo, Y.; Xia, L.; Zhou, Q. Structural basis for the recognition of the 2019-nCoV by human ACE2. BioRxiv 2020. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [PubMed]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017, 27, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, 7348–7357. [Google Scholar] [CrossRef]
- Walls, A.C.; Xiong, X.; Park, Y.J.; Tortorici, M.A.; Snijder, J.; Quispe, J.; Cameroni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A.; et al. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell 2019, 176, 1026–1039. [Google Scholar] [CrossRef]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 15092. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Feifei, Q.; Linlin, B.; Lanying, D.; Shuwen, L.; et al. Inhibition of SARS-CoV-2 infection (previously 2019-nCoV) by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- He, J.; Tao, H.; Yan, Y.; Huang, S.Y.; Xiao, Y. Molecular mechanism of evolution and human infection with the novel coronavirus (2019-nCoV). BioRxiv 2020. [Google Scholar] [CrossRef]
- Peng, C.; Zhu, Z.; Shi, Y.; Wang, X.; Mu, K.; Yang, Y.; Xinben, Z.; Zhijian, X.; Zhu, W. Exploring the binding mechanism and accessible angle of SARS-CoV-2 spike and ACE2 by molecular dynamics simulation and free energy calculation. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Othman, H.; Bouslama, Z.; Brandenburg, J.-T.; Rocha, J.D.; Hamdi, Y.; Ghedira, K.; Najet-Srairi, A.; Hazelhurst, S. Interaction of the spike protein RBD from SARS-CoV-2 with ACE2: Similarity with SARS-CoV, hot-spot analysis and effect of the receptor polymorphism. BioRxiv 2020. [Google Scholar] [CrossRef]
- Smith, M.; Smith, J.C. Repurposing therapeutics for COVID-19: Supercomputer-based docking to the SARS-CoV-2 viral spike protein and viral spike protein-human ACE2 interface. BioRxiv 2020. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Senathilake, K.; Samarakoon, S.; Tennekoon, K. Virtual screening of inhibitors against spike glycoprotein of 2019 novel corona virus: A drug repurposing approach. Preprints 2020, 2020030042. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Gotte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020, 295, 4773–4779. [Google Scholar] [CrossRef]
- Delmas, B.; Laude, H. Assembly of coronavirus spike protein into trimers and its role in epitope expression. J. Virol. 1990, 64, 5367–5375. [Google Scholar] [CrossRef]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef]
- Assessment of Evidence for COVID-19-Related Treatments: Updated 3/27/2020. Available online: https://www.ashp.org/Pharmacy-Practice/Resource-Centers/Coronavirus (accessed on 27 March 2020).
- Zhavoronkov, A. Geroprotective and senoremediative strategies to reduce the comorbidity, infection rates, severity, and lethality in gerophilic and gerolavic infections. Aging 2020, 12, 6492–6510. [Google Scholar] [CrossRef]
- Faccenda, E.; Armstrong, J.F.; Harding, S.D.; Pawson, A.J.; Davies, J.A. Coronavirus Information. IUPHAR/BPS Guide to Pharmacology. Available online: https://www.guidetopharmacology.org/coronavius.jsp (accessed on 22 March 2020).
- U.S. National Library of Medicine. Clinical Trials.gov. Available online: https://clinicaltrials.gov (accessed on 24 March 2020).
- Chen, H.; Zhang, Z.; Wang, L.; Huang, Z.; Gong, F.; Li, X.; Chen, Y.; Wu, J.J. First Clinical study using HCV protease inhibitor danoprevir to treat naive and experienced COVID-19 patients. MedRxiv 2020. [Google Scholar] [CrossRef]
- Heiser, K.; McLean, P.F.; Davis, C.T.; Fogelson, B.; Gordon, H.B.; Jacobson, P.; Hurst, B.; Miller, B.; Alfa, R.W.; Earnshaw, B.A.; et al. Identification of potential treatments for COVID-19 through artificial intelligence-enabled phenomic analysis of human cells infected with SARS-CoV-2. BioRxiv 2020. [Google Scholar] [CrossRef]
- Milewska, A.; Chi, Y.; Szczepanski, A.; Barreto-Duran, E.; Liu, K.; Liu, D.; Xiling, G.; Yiyue, G.; Jingxin, L.; Lunbiao, C.; et al. HTCC as a highly effective polymeric inhibitor of SARS-CoV-2 and MERS-CoV. BioRxiv 2020. [Google Scholar] [CrossRef]
- Milewska, A.; Kaminski, K.; Ciejka, J.; Kosowicz, K.; Zeglen, S.; Wojarski, J.; Maria, N.; Krzysztof, S.; Krzysztof, P. HTCC: Broad range inhibitor of coronavirus entry. PLoS ONE 2016, 11, e0156552. [Google Scholar] [CrossRef] [PubMed]
- Milewska, A.; Ciejka, J.; Kaminski, K.; Karewicz, A.; Bielska, D.; Zeglen, S.; Karolak, W.; Nowakowska, M.; Potempa, J.; Bosch, B.J.; et al. Novel polymeric inhibitors of HCoV-NL63. Antivir. Res. 2013, 97, 112–121. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The european molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlic, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The RCSB Protein Data Bank: Redesigned web site and web services. Nucleic Acids Res. 2011, 39, 392–401. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, 296–303. [Google Scholar] [CrossRef]
- Padariya, M.; Kalathiya, U.; Houston, D.R.; Alfaro, J.A. Recognition dynamics of cancer mutations on the ERp57-Tapasin interface. Cancers 2020, 12, 737. [Google Scholar] [CrossRef]
- Kalathiya, U.; Padariya, M.; Pawlicka, K.; Verma, C.S.; Houston, D.; Hupp, T.R.; Alfaro, J.A. Insights into the effects of cancer associated mutations at the UPF2 and ATP-binding sites of NMD master regulator: UPF1. Int. J. Mol. Sci. 2019, 20, 5644. [Google Scholar] [CrossRef]
- Kalathiya, U.; Padariya, M.; Baginski, M. Structural, functional, and stability change predictions in human telomerase upon specific point mutations. Sci. Rep. 2019, 9, 8707. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM force field in GROMACS: Analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Gunsteren, W.F.V.; Berendsen, H.J.C. A Leap-frog algorithm for stochastic dynamics. Mol. Simulat. 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) 2011.10; Chemical Computing Group: Montreal, QC, Canada, 2011.
- Wojciechowski, M.; Lesyng, B. Generalized Born model: Analysis, refinement, and applications to proteins. J. Phys. Chem. B 2004, 108, 18368–18376. [Google Scholar] [CrossRef]
- Edelsbrunner, H. The union of balls and its dual shape. Discrete Comput. Geom. 1995, 13, 415–440. [Google Scholar] [CrossRef]
- Lill, M. Virtual screening in drug design. In Silico Models for Drug Discovery; Kortagere, S., Ed.; Humana Press: Totowa, NJ, USA, 2013; Volume 993, pp. 1–12. [Google Scholar]
- Al-Motawa, M.; Abbas, H.; Wijten, P.; Fuente, A.D.L.; Xue, M.; Rabbani, N.; Thornalley, P.J. Vulnerabilities of the SARS-CoV-2 virus to proteotoxicity-opportunity for repurposed chemotherapy of COVID-19 infection. BioRxiv 2020. [Google Scholar] [CrossRef]
- Fan, H.-H.; Wang, L.-Q.; Liu, W.-L.; An, X.-P.; Liu, Z.-D.; He, X.-Q.; Li-Hua, S.; Tong, Y.-G. Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus (2019-nCoV) related coronavirus model. Chin. Med. J. 2020, 133, 1051–1056. [Google Scholar] [CrossRef]
- Nisha, M.; Mark, A. Targeted therapy and promising novel agents for the treatment of advanced soft tissue sarcomas. Expert Opin. Investig. Drug 2015, 24, 1409–1418. [Google Scholar]
- Squillace, R.M.; Miller, D.; Wardwell, S.D.; Wang, F.; Clackson, T.; Rivera, V.M. Synergistic activity of the mTOR inhibitor ridaforolimus and the antiandrogen bicalutamide in prostate cancer models. Int. J. Oncol. 2012, 41, 425–432. [Google Scholar] [CrossRef][Green Version]
- Iyer, G.; Hanrahan, A.J.; Milowsky, M.I.; Al-Ahmadie, H.; Scott, S.N.; Janakiraman, M.; Pirun, M.; Sander, C.; Socci, N.D.; Ostrovnaya, I.; et al. Genome sequencing identifies a basis for everolimus sensitivity. Science 2012, 338, 221. [Google Scholar] [CrossRef]
- Kean, T.; Roth, S.; Thanou, M. Trimethylatedchitosans as non-viral gene delivery vectors: Cytotoxicity and transfection efficiency. J. Control. Release 2005, 103, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Dryden, M.W.; Payne, P.A.; Smith, V.; Berg, T.C.; Lane, M. Efficacy of selamectin, spinosad, and spinosad/milbemycin oxime against the KS1 Ctenocephalidesfelis flea strain infesting dogs. Parasites Vectors 2013, 6, 80. [Google Scholar] [CrossRef]
- Aldonza, M.; Ku, J.; Hong, J.Y.; Kim, D.; Yu, S.J.; Lee, M.S.; Prayogo, M.C.; Tan, S.; Kim, D.; Han, J.; et al. Prior acquired resistance to paclitaxel relays diverse EGFR-targeted therapy persistence mechanisms. Sci. Adv. 2020, 6, eaav7416. [Google Scholar] [CrossRef]
- Ferlini, C.; Cicchillitti, L.; Raspaglio, G.; Bartollino, S.; Cimitan, S.; Bertucci, C.; Mozzetti, S.; Gallo, D.; Persico, M.; Fattorusso, C.; et al. Paclitaxel Directly Binds to Bcl-2 and Functionally Mimics Activity of Nur77. Cancer Res. 2009, 69, 6906–6914. [Google Scholar] [CrossRef]
- Zeldin, R.K. Pharmacological and therapeutic properties of ritonavir-boosted protease inhibitor therapy in HIV-infected patients. J. Antimicrob. Chemother. 2003, 53, 4–9. [Google Scholar] [CrossRef]
- Deutsch, M.; Papatheodoridis, G.V. Danoprevir, a small-molecule NS3/4A protease inhibitor for the potential oral treatment of HCV infection. Curr. Opin. Investig. Drugs 2010, 11, 951–963. [Google Scholar]
- Zhang, G.; Pomplun, S.; Loftis, A.R.; Loas, A.; Pentelute, B.L. The first-in-class peptide binder to the SARS-CoV-2 spike protein. BioRxiv 2020. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Qibin, G.; Ashley, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020. [Google Scholar] [CrossRef]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.C.D.; So, R.T.Y.; Lv, H.; Chris, K.P.M.; Wilson, I.A. A highly conserved cryptic epitope in the receptor-binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds Against SARS-CoV-2 | GBVI/WSA dG (kcal/mol) | MW g/mol | Previous Target | |
|---|---|---|---|---|
| Trimer Cavity | RBD Domain | |||
| Chitosan [54,55,56] | −67.49 | −37.30 | 161.16 * | Antibacterial [84] |
| Rapamycin (Sirolimus) [47,48,49] | −49.28 | −25.81 | 914.17 | mTOR [47] |
| Paclitaxel [79] | −45.84 | −32.42 | 853.92 | Bcl-2, Microtubule Associated [86,87] |
| SelaMeerin (Selamectin) [80] | −44.24 | −32.35 | 769.96 | Antiparasitic [85] |
| Everolimus (RAD001) [49] | −41.80 | 0.29 | 958.22 | mTOR [83] |
| Ritonavir [48,50,51,52] | −37.92 | −24.11 | 720.94 | HIV Protease [88] |
| Danoprevir (ITMN-191) [52] | −35.09 | −30.80 | 731.83 | Proteasome, HCV, Protease [89] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalathiya, U.; Padariya, M.; Mayordomo, M.; Lisowska, M.; Nicholson, J.; Singh, A.; Baginski, M.; Fahraeus, R.; Carragher, N.; Ball, K.; et al. Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site. J. Clin. Med. 2020, 9, 1473. https://doi.org/10.3390/jcm9051473
Kalathiya U, Padariya M, Mayordomo M, Lisowska M, Nicholson J, Singh A, Baginski M, Fahraeus R, Carragher N, Ball K, et al. Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site. Journal of Clinical Medicine. 2020; 9(5):1473. https://doi.org/10.3390/jcm9051473
Chicago/Turabian StyleKalathiya, Umesh, Monikaben Padariya, Marcos Mayordomo, Małgorzata Lisowska, Judith Nicholson, Ashita Singh, Maciej Baginski, Robin Fahraeus, Neil Carragher, Kathryn Ball, and et al. 2020. "Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site" Journal of Clinical Medicine 9, no. 5: 1473. https://doi.org/10.3390/jcm9051473
APA StyleKalathiya, U., Padariya, M., Mayordomo, M., Lisowska, M., Nicholson, J., Singh, A., Baginski, M., Fahraeus, R., Carragher, N., Ball, K., Haas, J., Daniels, A., Hupp, T. R., & Alfaro, J. A. (2020). Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site. Journal of Clinical Medicine, 9(5), 1473. https://doi.org/10.3390/jcm9051473